Introduction to metabolic-inflammation
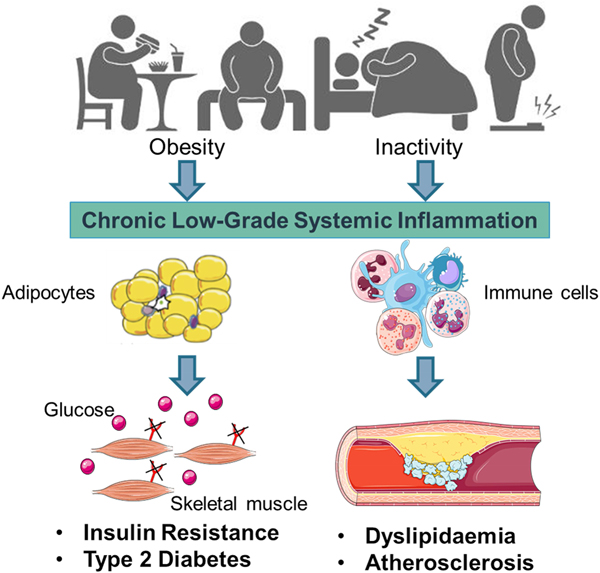
Early work suggested that sub-acute chronic inflammation that typified diet-related diseases including obesity, type-2 diabetes (T2D) and atherosclerosis disrupted metabolism, wherein immune cell mediators, particularly cytokines, impeded a range of metabolic pathways such as insulin signalling, glucose homoeostasis, lipid metabolism, reverse cholesterol transport, etc.(Reference Gregor and Hotamisligil1–Reference O'Reilly, Dillon and Guo3). The negative impact of inflammatory mediators on metabolism has been very well characterised and is relevant within a range of metabolic tissues, including adipose, skeletal muscle, liver, pancreas and the brain(Reference Osborn and Olefsky4). Thus sub-acute chronic inflammation is a classical hallmark of obesity, T2D and CVD (Fig. 1).
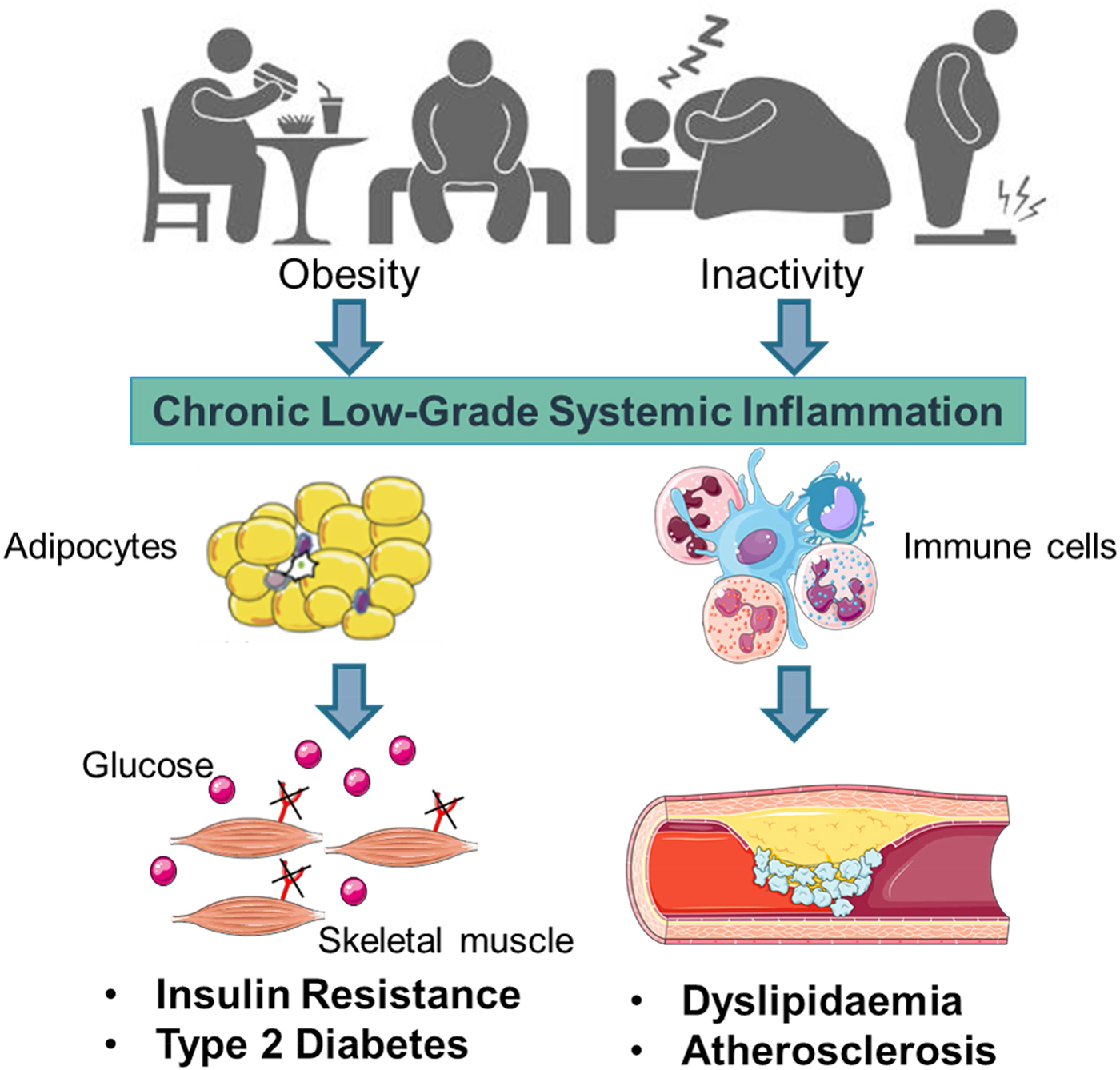
Fig. 1. (Colour online) Inflammation a classic hallmark of obesity, type-2 diabetes (T2D) and CVD. Excess energy intake and inactivity lead to the development of obesity, which is typified by sub-acute, chronic systemic inflammation. This pro-inflammatory phenotype develops in conjunction with expanding adipose tissue and immune cell activation. A range of immune cell mediators, including IL-1β, disrupts adipogenesis, insulin signalling, glucose homoeostasis, lipid and cholesterol metabolism. Thus contributing to obesity related insulin resistance, T2D and atherosclerosis. Figure created using Servier Medical Art, https://smart.servier.com/
Interestingly more recent evidence shows that there is a much more dynamic reciprocal regulatory relationship between metabolism and inflammation, wherein the nature of cellular metabolism actually may determine immune cell functionality and thus inflammation. This review will explore the newer aspects of the inter-relationships between metabolism and inflammation, with a view to understanding potential nutritional relevance. We will focus on IL-1β mediated inflammation because of the strong inter-relationship between dietary fats, metabolic stressors and Nod-like receptor family, pyrin domain-containing three protein (NLRP3) inflammasome mediated inflammation. In terms of exploring the impact of dietary modulation of energy homoeostasis and metabolic-inflammation we will draw upon some simple examples to anticipate advances within this emerging and rapidly advancing field.
Metabolic stress and NLRP3 mediated IL-1β inflammation
Metabolic stress, in the form of obesity, SFA, cholesterol, reactive oxygen species and/or uric acid, promote IL-1β signalling(Reference Tack, Stienstra and Joosten5). It is now widely recognised that IL-1β signalling plays a key role in obesity and T2D risk. Regulatory aspects of IL-1β mediated inflammation are a very interesting aspect of metabolic-inflammation, by virtue of its two stage priming and activation process. As outlined in our recent reviews(Reference Ralston, Lyons and Kennedy6, Reference Kirwan, Lenighan and O'Reilly7), both stages of IL-1β priming and NLRP3 mediated IL-1β activation may be affected by the nutritional environment. As illustrated in Fig. 2, mature IL-1β is produced and secreted by immune cells, following the initial process of priming, wherein the nascent form of pro-IL-1β is formed, followed by the activation stage, wherein pro-IL-1β is converted into mature IL-1β by the NLRP3 inflammasome(Reference Netea, Nold-Petry and Nold8). Structurally, the NLRP3 inflammasome is a multi-protein cytosolic complex composed of the regulatory subunit NLRP3, the effector subunit procaspase-1 and the adaptor molecule apoptosis-associated speck-like protein containing a caspase recruitment domain. Recruitment and activation of caspase-1 to the complex appears to be required for NLRP3 activation. Fig. 2 shows that both priming and activation stages of IL-1β and NLRP3 biology are sensitive to nutritional, metabolic, as well as inflammatory, stress signals stemming from exogenous or endogenous sources, including fatty acids, glucose, reactive oxygen species, ATP and uric acid. For example, SFA induced Toll-like receptor (TLR)4 mediated metabolic stress, as well as inflammatory cytokines, prime pro-IL-1β. Also in terms of the IL-1β activation stage, a variety of metabolic factors, including SFA derivatives, ceramides, reactive oxygen species and ATP activate the NLRP3 inflammasome promoting the conversion of pro-IL-1β into mature IL-1β.
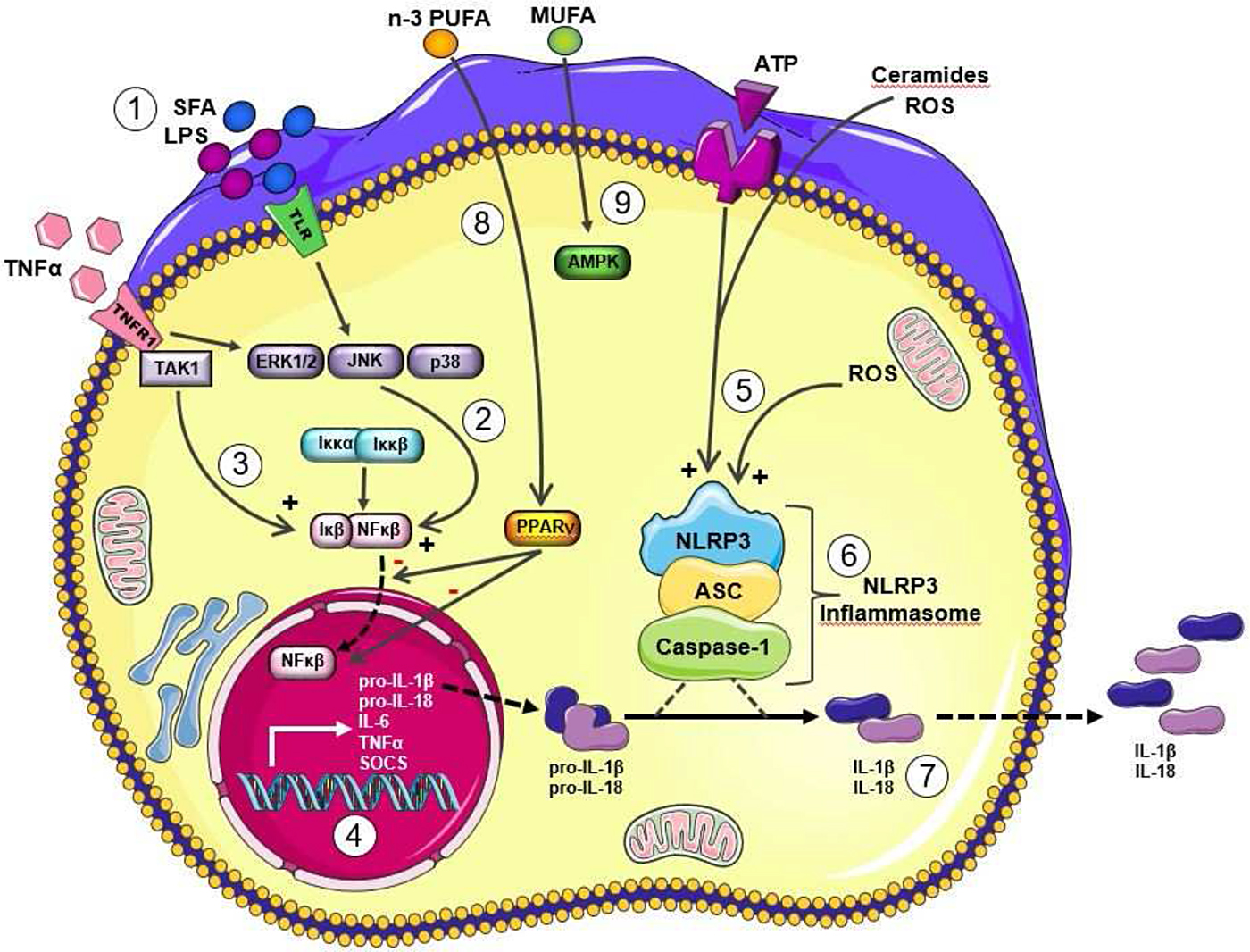
Fig. 2. (Colour online) Metabolic and inflammatory priming and activation of IL-1β via the Nod-like receptor family, pyrin domain-containing three protein (NLRP3) Inflammasome. Adapted from Ralston et al.(Reference Ralston, Lyons and Kennedy6). Metabolic and inflammatory challenges that prime and activation IL-1β via NLRP3 inflammasome activation to modulate IL-1β secretion lipopolysaccharide (LPS) and SFA signal directly and indirectly via Toll-like receptor (TLR)4 (1) to activate stress kinases (2), which in turn activate NF-κB with its translocation to the nucleus. TNFα also stimulates to NF-κB activation (3). Upon translocation to the nucleus, NF-κB stimulates transcription of a range of inflammatory cytokines, including the nascent form of pro-IL-1β (4). This stage represents the priming of pro-IL-1β. Then the activation of pro-IL-1β activation is dependent upon NLRP3 inflammasome activation. A wide range of metabolic stressor molecules including ATP, reactive oxygen species (ROS) and ceramides (derived from SFA) (5) have the ability to activate the NLRP3 inflammasome (6). A key stage of NLRP3 inflammasome activation includes the cleavage of pro-caspase-1 to active caspase-1. The resultant active IL-1β is secreted from the cell (7). IL-18 has a very analogous biological pathway, wherein it is initially formed in a pro-IL-18 form and also requires NLRP3 mediated activation. Other dietary fatty acids may also modulate this process, for example MUFA maintain pAMPK activity, which in turn dampens NLRP3 inflammasome activation and thus reduces the IL-1β/NLRP3 inflammasome mediated inflammatory response (8). Long chain n-3 PUFA may also attenuate these inflammatory pathways, possibly via PPARγ activation, which in turn attenuates NF-κB activation (9). This figure was prepared using the Servier medical art website (http://www.servier.fr). ASC, apoptosis-associated speck-like protein containing a caspase-recruitment domain;TAK 1, transforming growth factor β activated kinase 1; ERK 1/2, extracellular signal-regulated kinase 1/2; JNK c-Jun N-terminal kinase; SOCS, suppressor of cytokine signaling.
A substantial body of evidence has established that IL-1β is key inflammatory signal involved in the development of obesity, insulin resistance and T2D(Reference Stienstra, Joosten and Koenen9). IL-1β disrupts cellular metabolism by interrupting a range of signalling pathways, including insulin sensitivity, lipid metabolism and adipogenesis(Reference Lagathu, Yvan-Charvet and Bastard10, Reference McGillicuddy, Harford and Reynolds11). We have demonstrated that IL-1R1 knockout (IL-1R1−/−) mice are protected from high-fat diet (HFD)-induced insulin resistance, which was ascribed to attenuated adipose inflammation, early in life(Reference McGillicuddy, Harford and Reynolds11). Similarly, other groups have demonstrated that disrupting different components of the NLRP3 inflammasome complex, which regulates IL-1β activation, also protects against HFD-induced insulin resistance and inflammation(Reference Wen, Gris and Lei12, Reference Stienstra, van Diepen and Tack13). Nevertheless the impact of NLRP3 mediated IL-1β signalling on obesity and insulin sensitivity is probably more complex. Later in life IL-1R1−/− mice ultimately develop mature-onset obesity, are profoundly insulin resistance in response to feeding HFD(Reference McGillicuddy, Reynolds and Finucane14). We speculate that IL-1β biology may play a key role in adipogenesis which determines adipose morphology and functionality, which we discuss in the next section.
It is well accepted that SFA are potent pro-inflammatory fatty acids that promote obesity and insulin resistance. Initial lines of evidence suggested that SFA activate TLR, specifically TLR2 and TLR4(Reference Nguyen, Favelyukis and Nguyen15), which in turn activates the transcription factor NF-κB, to promote and prime pro-IL-1β production(Reference Wen, Gris and Lei12, Reference Reynolds, McGillicuddy and Harford16). Interestingly more recent evidence shows that SFA are not in fact TLR4 ligands(Reference Lancaster, Langley and Berglund17). This very recent study showed that TLR4 is not a receptor for the common SFA palmitic acid (PA), which is derived from palm oil. PA does not bind to the TLR4 receptor to induce TLR4 dimerisation and activation of inflammation via NF-κB. Instead PA mediates its pro-inflammatory effect by priming and re-configuring macrophage lipid metabolism, such that the cell is more responsive to SFA induced inflammatory and metabolic insults(Reference Lancaster, Langley and Berglund17).
Energy excess, metabolic-inflammation and adipose tissue health
Energy excess leads to obesity, with expansion of adipose tissue. Feeding HFD of course induce obesity, with concomitant sub-acute chronic inflammation and insulin resistance. However, it is important to appreciate that obese adipose tissue is a highly dynamic organ, not only reflecting energy storage potential. Adipose tissue expansion occurs through enlargement of adipocyte size (hypertrophy) or increased adipocyte number (hyperplasia). Interestingly there is a growing body of evidence that suggests that adipose morphology, hypertrophic v. hyperplasia, may determine adipose functionality and subsequent risk of T2D. Loss of white adipose tissue hyperplastic potential, resulting in hypertrophic adipose, is associated with increasing insulin concentrations and greater risk of insulin resistance and developing T2D(Reference Arner, Westermark and Spalding18, Reference Kim, Lun and Wang19). Therefore there has been great interest in determining the extent to which adipose tissue can be re-programmed such that its functional capacity is optimised to preserve insulin sensitivity, attenuate T2D risk, despite obesity.
Metabolic-inflammation plays a key role in adipogenesis. Adipose morphology reflects complex interplay between adipocyte programming and immune cell activation in adipose tissue. Dietary/metabolic stressors, including SFA prime pro-IL-1β, which is then activated by the NLRP3 inflammasome complex. Several groups have shown that many cytokines, including IL-1β, potently impedes adipogenesis and insulin sensitivity in vitro (Reference Lagathu, Yvan-Charvet and Bastard10). It is possible that the induction of IL-1β associated with feeding a HFD may prevent preadipocyte maturation and promote hyperplastic adipose morphology that typifies HFD-induced obesity. In contrast, mice lacking IL-1R1 signalling exhibit hyperplastic adipose morphology, with attenuated adipose inflammation and insulin resistance, despite obesity, in response to SFA-enriched HFD(Reference McGillicuddy, Reynolds and Finucane14). However the adipogenic potential of adipose tissue is lost with age and indeed the protective effect afforded by lack of IL-1β signalling to preserve hyperplastic adipose morphology was also lost with age.
Interestingly, our recent work showed that obesogenic diets with different fatty acid composition induce different adipose morphology(Reference Finucane, Lyons and Murphy20). Feeding SFA-rich HFD induced hypertrophic adipose morphology with greater NLRP3 mediated inflammation in adipose tissue concomitant with insulin resistance. In contrast, feeding a MUFA HFD was associated with a hyperplastic adipose morphology coincident with improved insulin sensitivity, despite equal adipose tissue weight gain. Detailed analysis of the adipogenic potential of the adipose stroma vascular fraction revealed that the MUFA HFD stroma vascular fraction had greater adipogenic potential, the potential of which was ascribed to lower NLRP3 mediated IL-1β inflammation. This is only one potential example, it is likely that there is more to understand in relation to if and/how nutritional factors may modulate the metabolic-inflammation within the context of adipogenesis, which in turn may determine the physiological, metabolic and health impact of obesity.
Cellular energy homoeostasis and metabolic-inflammation
In the past number of years, the immunology field has demonstrated that metabolic reprogramming defines immune cell functionality(Reference Jha, Huang and Sergushichev21), as opposed to the initial understanding that inflammation disrupted metabolism. At the simplest level, cellular energy balance wherein cells rely upon oxidative phosphorylation (OxPHOS) or glucose dependent glycolytic metabolism to generate cellular ATP, defines the functional nature of immune cells and their inflammatory response. The metabolic pathway selected reflects the requirements and functionality of the immune cell. Activated pro-inflammatory immune cells require a shift wherein ATP is generated from anaerobic glycolysis, rather than deriving cellular energy from more efficient OxPHOS processes utilising the tricarboxylic acid cycle(Reference Jha, Huang and Sergushichev21, Reference MacIver, Michalek and Rathmell22). Jha et al. completed a very elegant combined transcriptomic and metabolomic analysis of the macrophage polarisation process(Reference Jha, Huang and Sergushichev21). Briefly, this systems approach showed that pro-inflammatory macrophages are less reliant upon mitochondrial OxPHOS, due to a disrupted tricarboxylic acid cycle, but depend upon glycolysis as an ATP energy source. In contrast, anti-inflammatory M2 macrophage or alternatively activated macrophage, are almost completely reliant upon OxPHOS with minimal glycolytic rates. Interestingly the nature of the energy substrates, glucose, fatty acids or amino acids, may affect the immune response(Reference O'Neill and Hardie23). For example, there is an interesting switch in fatty acid metabolism, according to macrophage polarisation state. The ability and requirement to synthesise fatty acids, and probably also cholesterol, is a requirement of classically activated, pro-inflammatory M1 macrophage(Reference Carroll, Zaslona and Galvan-Pena24). In contrast, mitochondrial fatty acid β-oxidation is the predominant fuel source used by M2 macrophages.
A range of studies have demonstrated that disrupting different points within metabolic pathways can alter the nature of the inflammatory response. For example, impeding macrophage lipid metabolism by inhibiting fatty acid synthase, which catalyses the endogenous synthesis of SFA, suppresses the pro-inflammatory response, via NLRP3-inflammasome mediated IL-1β activation(Reference Moon, Nakahira and Choi25). In contrast, inhibiting glycolysis via pyruvate kinase inhibition promotes resolution of inflammation(Reference Palsson-McDermott, Curtis and Goel26). These represent only two of several other points in metabolism that can be modulated, in order to affect the inflammatory response. Furthermore, it is also evident that metabolites, as well as metabolic derivatives of altered metabolism, can also re-configure the nature of the inflammatory phenotype. For example, the classical inflammatory insult, lipopolysaccharide which disrupts the tricarboxylic acid cycle, leads to the production of tricarboxylic acid intermediates, including citrate and succinate, which in turn play a functional biological role(Reference Williams and O'Neill27). Citrate promotes lipogenesis and is pro-inflammatory(Reference Iacobazzi and Infantino28). Succinate acts as a pro-inflammatory metabolic signal by increasing Hif-1α dependent IL-1β expression(Reference Tannahill, Curtis and Adamik29). The above examples represent a brief snapshot wherein immune cells utilise different energy substrates and metabolic pathways depending on their pro-inflammatory, anti-inflammatory or resolving role.
Dietary modulation of metabolic-inflammation
Going forward, it will be interesting to determine the extent to which dietary fatty acids, carbohydrate and protein sources may modulate the above processes given that the diet is an important exogenous source of some endogenous metabolites and/or substrates for the metabolic pathways eluded to above. Essentially the extent to which diet can (or cannot) re-programme metabolic-inflammation in adipose tissue is relatively unknown. Kratz et al. demonstrated that metabolic insults such as the SFA PA, glucose and insulin; induced more complex inflammatory-metabolic phenotypes in adipose tissue macrophage compared to the classical lipopolysaccharide agonist, with greater lipid transport protein configuration in association with IL-1β activation(Reference Kratz, Coats and Hisert30). Very recently, Febbraio's team confirmed that PA induces distinct inflammatory pathways different to lipopolysaccharide, not as a TLR4 agonist, but through TLR4 priming and altered macrophage lipid metabolism(Reference Lancaster, Langley and Berglund17). Thus metabolic re-programming is required for SFA induced inflammation in vitro. It will be interesting over the next number of years to determine the extent to which this paradigm hold true in vivo and also translates to man.
In a recent study we investigated the impact of different dietary fatty acids on metabolic-inflammation in an obese setting(Reference Finucane, Lyons and Murphy20). This work provided interesting data with respect to the interaction and reciprocal regulation of NLRP3 mediated IL-1β activation and AMP-activated protein kinase (AMPK). Targeting AMPK mediated immuno-metabolism in obesity is attractive because it is a critical hub of both metabolism and IL-1β inflammation(Reference Herzig and Shaw31). AMPK is primarily known for its critical role in cellular energy homoeostasis, regulating metabolism(Reference Herzig and Shaw31). AMPK activation promotes glucose-sparing, OxPHOS to generate maximal ATP from cellular energy substrates, a process utilised by most quiescent immune cells. This is converse to the rapid glucose uptake and glycolysis typical of activated immune cells. On the inflammatory side, AMPK activation attenuates IL-1β inflammation via NLRP3(Reference O'Neill and Hardie23, Reference Jeong, Hsu and Lee32). Briefly, we showed that feeding a SFA enriched HFD to induce obesity was associated with significant NLRP3 mediated IL-1β inflammation, insulin resistance and adipose hypertrophy(Reference Finucane, Lyons and Murphy20). In contrast, a MUFA-enriched HFD induced equivalent obesity, but adipose IL-1β inflammation and insulin resistance was markedly attenuated compared to the SFA-HFD group. Interestingly we observed that adipose AMPK expression was not reduced following the MUFA-HFD, a common phenomenon associated with obesity. Given AMPK dual role linking metabolism and inflammation, we explored whether the different fatty acids mediated their pro- v. anti-inflammatory effects in an AMPK dependent fashion. Using a combination of AMPK activators, inhibitors and shRNA approaches we were able to show that the MUFA oleic acid not only failed to prime IL-1β but also blocked NLRP3 mediated IL-1β activation in an AMPK dependent manner(Reference Finucane, Lyons and Murphy20). This was opposed to the SFA PA which both primed pro-IL-1β and promoted NLRP3-mediated activation, via down-regulation of AMPK.
Conclusions
Nutritional modulation of metabolic-inflammation is a fast evolving area. No doubt our understanding of the complex interactions between what were considered two independent pathways, but now very much inter-connected, will be advanced. The real challenge will be the extent to which these cellular processes translate to man, so that we can fully appreciate if/how/when dietary constituents either promote or attenuate metabolic-inflammation.
Acknowledgements
The author thanks Marcella O'Reilly who designed Fig. 1.
Financial Support
The research presented in this review work was supported by Science Foundation Ireland principal investigator award (11/PI/1119); Joint Programming Healthy Life for a Healthy Diet (JPI HDHL) funded EU Food Biomarkers Alliance ‘FOODBALL’ (14/JP-HDHL/B3076) and the Irish Department of Agriculture, Food and the Marine, ‘ImmunoMet - dietary manipulation of microbiota diversity for controlling immune function’ (14/F/828) programmes.
Conflict of Interest
None.
Authorship
The author had sole responsibility for all aspects of preparation of this paper.