


16.1 Introduction
The form, fate, and biogeochemical cycling of carbon in subsurface environments impacts and reflects microbial activity and has important implications for global elemental fluxes. Photosynthetically derived organic matter (OM) is transported to a depth where it can continue to fuel life far from solar inputs. Alternative energy-yielding reactions such as the oxidation of minerals and reduced gases can fuel life in the rocky subsurface of both the ocean and continents, altering the distribution and characteristics of carbon compounds. Nonbiological reactions such as the precipitation of calcium carbonate influence the availability of dissolved inorganic carbon for lithoautotrophs and, simultaneously, the carbon cycle over geologic time. The abundances, characteristics, and distributions of carbon in the subsurface can therefore provide an integrated history of biotic and abiotic processes and a template for interpreting similar patterns from other planetary bodies.
The goal of this chapter is to compile insights from disparate environments in order to build a mechanistic understanding of the controls on carbon abundance and distribution in the subsurface. The sections below summarize what is known from the oceanic and continental subsurface, realms that are often studied separately. We synthesize commonalities across these environments, highlight what remains unknown, and propose ideas for future directions.
One challenge with working across the marine–continental divide is that the terminology used to describe organic carbon varies between the two. We will use the following terms and abbreviations: particulate organic carbon (POC), dissolved organic carbon (DOC), and dissolved inorganic carbon (DIC). Another discrepancy between communities is in the use of units, with ppm or mg/L dominating the continental literature and μM or mM in the marine literature. We will use molar units throughout for comparison’s sake. Finally, while the soil community has moved away from the terms “refractory” and “recalcitrant” OM, they are still common in the marine community. Here, these terms refer to OM that has escaped remineralization due to its inherent molecular structure, physical associations with minerals, energetically unfavorable conditions, or the lack of a specific microbial community adapted to carry out the necessary degradative processes.
16.2 Oceanic Sedimentary Subsurface
Approximately 1.68 × 1014 g of organic carbon per year are buried in marine and estuarine sediments (Reference Martiny, Bohannan, Brown, Colwell, Fuhrman and Green1). Burial of organic carbon in sediments represents a transfer of reducing equivalents from Earth’s surface to the subsurface, thereby allowing persistence of oxidized compounds such as O2 at the surface (Reference Hedges2). The rate of burial of organic carbon in marine sediments therefore has an important influence on the redox state, and thus habitability, of Earth’s surface.
Broadly, marine sediments can be divided into river and estuarine delta systems, continental shelves and slopes, and abyssal plains (Figure 16.1). Sediments may be more finely divided into provinces based on microbial community composition, grain size, OM content, and benthic communities, among other variables (Reference Schrenk, Huber and Edwards3).

Figure 16.1 Deep biosphere locations on the continents and in the ocean.
The oxidation of organic carbon in sediments is carried out by a series of heterotrophic organisms. Macrofauna have their greatest influence on the surface sediments of continental shelfs, while the role of meiofauna and microorganisms increases with depth and where oxygen is limited (Reference Rex, Etter, Morris, Crouse, McClain and Johnson4). Remineralization within anoxic sediments is dominated by microorganisms and is most prevalent at temperatures below ~80°C, constituting ~75% of Earth’s total sediment volume of 3.01 × 108 km3 (Reference LaRowe, Burwicz, Arndt, Dale and Amend5). The composition, abundance, and activity of heterotrophic microorganisms in marine sediments therefore has a strong influence on the burial rate and chemical nature of organic carbon. While these reactions are catalyzed by enzymes, they are ultimately controlled by thermodynamics. This section will briefly review the chemical and biological factors that regulate organic carbon oxidation and burial rates, as well as some of the models that can be constructed to describe and predict those rates.
The burial rate of organic carbon in marine sediments is controlled by a range of biological and geological processes, including sedimentation rate, primary productivity, biological activity, sediment organic carbon content, chemical and physical form of organic molecules, and concentrations of oxidants (electron acceptors), as described below and in several reviews and syntheses (Reference Burdige6–Reference Middelburg13). These factors are interrelated: rapid sedimentation rates influence the quality of OM delivered to the sediment surface, which in turns affects oxidation rates, oxygen exposure time (OET), quantity and composition of heterotrophic microbial communities, and concentrations of potential electron acceptors.
16.2.1 Chemical Composition
OM is delivered to marine sediments from marine sources such as sinking plankton and consumers and from terrigenous sources such as plant litter and soil OM. The chemical composition of fresh biomass is relatively well constrained and consists predominately of carbohydrates, proteins, and lipids. The composition of terrestrial material transferred by fluvial or aeolian processes ranges from fresh biomass to highly degraded and altered material. Lignin phenols synthesized solely by vascular plants have long been used to track terrestrial inputs into the ocean (Reference Hedges, Keil and Benner14). Ancient and recycled petrogenic carbon can also be remobilized from the weathering of sedimentary rocks (Reference Blair, Leithold, Ford, Peeler, Holmes and Perkey15). This suite of compounds is subject to biotic and abiotic alteration en route to marine deposition, which further diversifies the range of organic compounds present. Physical processes within the catchment are a major control on the composition and reactivity of OM delivered to the ocean by rivers (Reference Galy, Peucker-Ehrenbrink and Eglinton16), with larger inputs of both recently synthesized and ancient petrogenic organic carbon delivered in regions of higher erosional rates such as small mountainous streams (Reference Blair and Aller10,Reference Hilton, Galy, Hovius, Horng and Chen17–Reference Bao, Lee, Huang, Feng, Dai and Kao19) and some Arctic rivers (Reference Opsahl, Benner and Amon20–Reference Feng, Vonk, van Dongen, Gustafsson, Semiletov and Dudarev23).
Within the sediments, heterotrophic organisms and abiotic processes such as condensation reactions or sulfurization can alter the chemical structures of OM. In general terms, the heterotrophic remineralizaiton of larger organic molecules under anoxic conditions proceeds by the breakdown of polymers into monomers and oligomers, followed by smaller alcohols and organic acids, and finally methane and CO2 (Figure 16.2) (Reference Blair, Carter and Boehme24,Reference Schmitz, Daniel, Deppenmeier and Gottschalk25). As a result, small organic molecules such as acetate, ethane, propane, and methane build up in the porewaters of anaerobic sediments, with additional contributions from acetogenesis and hydrogenotrophic methanogenesis (Reference Blair, Carter and Boehme24,Reference Blair, Martens and Desmarais26–Reference Heuer, Pohlman, Torres, Elvert and Hinrichs29).
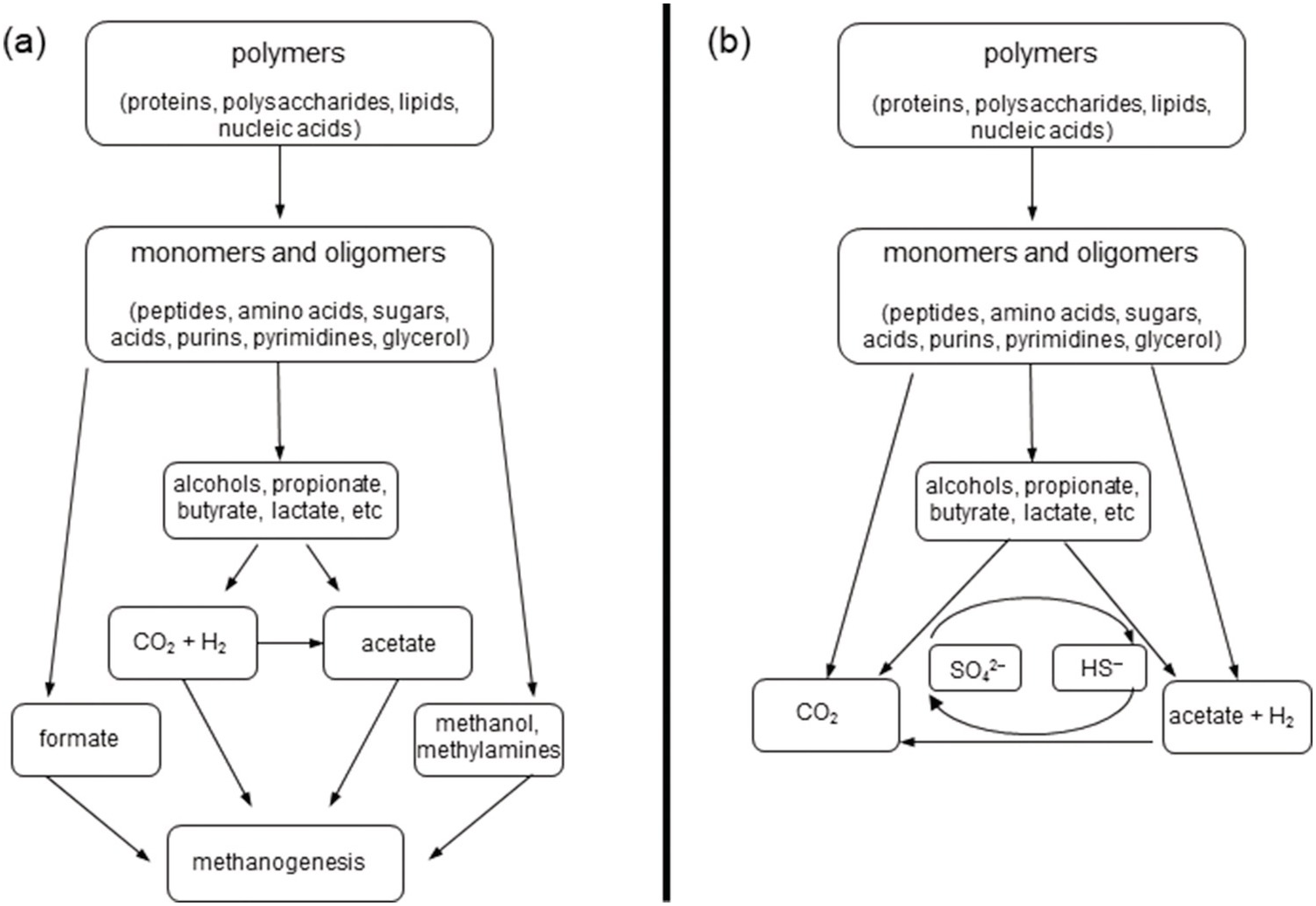
Figure 16.2 Anaerobic breakdown of OM by microorganisms via (a) methanogenesis and (b) sulfidogenesis.
Ultimately, the vast majority of OM produced in the upper water column is respired, with only 1% of gross primary production escaping remineralization to be buried in the deep sediments (Reference Burdige6–Reference Hedges and Keil8,Reference Suess30). Some molecules survive due to chemical structures that are inherently recalcitrant, a process called “selective preservation.” The role of this pathway is disputed. While some compounds such as highly cross-linked macromolecules are inherently less bioavailable than others, microorganisms are capable of metabolizing even ancient and highly altered OM relatively quickly under favorable conditions. Molecules may become less bioavailable due to nonbiological alteration. Abiotic sulfurization of organic molecules, deamination of peptides, and condensation of nitrogen-containing heterocyclic molecules all appear to promote the preservation of organics in sediments (Reference Burdige7,Reference Abdulla, Burdige and Komada31). Random recombination of molecules or the production of altered metabolites by heterotrophic microorganisms can also rapidly convert labile organic carbon into far less reactive material (Reference Jiao, Herndl, Hansell, Benner, Kattner and Wilhelm32,Reference Lechtenfeld, Kattner, Flerus, McCallister, Schmitt-Kopplin and Koch33). Temperature can promote some of these transformations, as discussed in detail in Section 16.4.
16.2.2 Bulk Controls on OM Preservation
In locations with relatively rapid sediment accumulation rates such as river deltas and continental shelf sediments, greater OM preservation is most closely associated with higher mineral surface areas and shorter OETs (Reference Burdige7,Reference Hartnett, Keil, Hedges and Devol34). These factors have proven broadly predictive of organic carbon distributions, although they do little to reveal the underlying mechanisms of preservation, nor do they allow for predictions of future responses to changing environmental conditions.
A prevailing paradigm is that microorganisms access POC only after it has been solubilized into DOC (Reference Hee, Pease, Alperin and Martens35). Organic molecules enter cells via general uptake porins, which can only accommodate molecules in the size range of 600–1000 Da (Reference Benz and Bauer36). Organic molecules in seawater, sediment porewater, and soils that are larger than 1000 Da are, however, more bioavailable than small molecules on average (Reference Burdige and Gardner37–Reference Walker, Beaupre, Guilderson, McCarthy and Druffel40), apparently because smaller molecules tend to be more extensively modified than larger molecules (Reference Kelleher and Simpson41). Therefore, microbial extracellular enzymes appear necessary for the uptake and utilization of the most bioavailable organic carbon in sediments. Consistent with this paradigm, extracellular enzyme activity has been observed in deep, old sediments, including 217,000-year-old Mediterranean sapropels (Reference Coolen and Overmann42,Reference Coolen, Cypionka, Sass, Sass and Overmann43) and Baltic Sea sediments that are up to 10,000 years old (Reference Schmidt44).
Several findings complicate the view that extracellular enzymes catalyze the rate-limiting step in biological organic carbon oxidation. Extracellular enzyme activities can outstrip the ability of sediment microbes to take up hydrolysate on timescales of days to years, leading to accumulations of apparently bioavailable low-molecular-weight DOM (Reference Robador, Bruchert, Steen and Arnosti45). Further, cells do not exclusively take up organic compounds via general uptake porins. Active transporters, for instance, use energy gradients to pass specific molecules through the cell membrane. These can be extremely large: for instance, certain TonB-dependent transporters can import intact proteins up to 69 kDa (Reference Noinaj, Guillier, Barnard and Buchanan46). Additionally, in seawater and in cow rumen, some cells are able to take up larger oligosaccharides into their periplasm, store them over extended periods, and then metabolize them when conditions are right (Reference Arnosti and Repeta47,Reference Cuskin, Lowe, Temple, Zhu, Cameron and Pudlo48). The extent to which these mechanisms are important in sediments is not known, but temporal decoupling between macromolecule hydrolysis and metabolism could have implications for the dynamics of sediment OM oxidation.
16.2.3 Sorption
It has been observed for nearly 40 years that the volume-specific quantity of mineral surface area in sediments is correlated with organic carbon content (Reference Tanoue and Handa49). The mechanism underlying this relationship is not precisely understood. Sediments tend to accumulate quantities of organic carbon that are roughly equivalent to the amount that would be required to cover minerals in an organic monolayer (Reference Mayer50,Reference Mayer51). Sedimentary organic carbon, however, exists in discrete “blebs” (Figure 16.3), so the fact that the average quantity of OM per unit of mineral surface area is roughly monolayer equivalent appears to be essentially coincidental (Reference Mayer52).

Figure 16.3 (a) Scanning transmission X-ray microscope image and (b) optical density map of the organic carbon distribution of sediments from 1.75 m below seafloor at Integrated Ocean Drilling Program Site 1231 Hole B, Peru Basin. The optical density map was generated by subtracting a pre-edge X-ray image from a post-edge X-ray image; brighter pixels correspond to higher concentrations of organic carbon. OM associated with particles is not distributed evenly over the surface.
Several mechanisms appear responsible for the protection of OM by mineral surfaces. First, OM may be occluded between mineral grains, within minerals themselves, or even within a matrix of more recalcitrant sorbed organic compounds (Reference Knicker and Hatcher53,Reference Moore, Nunn, Goodlett and Harvey54). Encased OM represents a sterile microenvironment in which biological oxidation is impossible. Second, even when sorbed OM is physically accessible to microorganisms, sorption slows or halts the diffusion of organic compounds to cell membranes (Reference Wu and Gschwend55). Finally, sorption distorts the physical structure of extracellular enzymes, preventing them from functioning normally, while simultaneously protecting enzymes from degradation and thereby substantially extending their active lifetimes (Reference Espeland and Wetzel56,Reference Tietjen and Wetzel57). Associations with iron oxides, which include chelation, coprecipitation, and noncovalent bonding to oxide surfaces, accounts for an average of 20% of organic carbon in sediments (Reference Lalonde, Mucci, Ouellet and Gelinas58,Reference Barber, Brandes, Leri, Lalonde, Balind and Wirick59). A full understanding of the mechanisms of microbial OM oxidation in sediments requires consideration of both the interactions between organic carbon and sediment minerals and the effects of mineral surfaces on the metabolisms of microorganisms.
16.2.4 Oxygen Exposure Time
Typical marine sediments underlying oxygenated seawater contain oxic porewater near the sediment–water interface, which becomes anoxic with increasing depth due to heterotrophic organic carbon oxidation. The depth of the oxic layer can vary dramatically, from millimeters or less in rapidly accumulating, organic-rich sediments to meters in ocean gyres. The presence of oxygen enhances the remineralization of organic molecules (Reference Cowie and Hedges60–Reference Keil, Hu, Tsamakis and Hedges62), and the term “oxygen exposure time” was coined to quantify the average time that sedimentary OM is exposed to “oxic” conditions, which can range from days to thousands of years (Reference Hedges and Keil8,Reference Hartnett, Keil, Hedges and Devol34,Reference Keil, Hu, Tsamakis and Hedges62,Reference Hedges, Hu, Devol, Hartnett, Tsamakis and Keil63). Organic carbon oxidation is substantially faster in oxic sediments than anoxic sediments because the greater free energy of reaction of organic carbon with oxygen allows for a denser microbial community capable of catalyzing faster oxidation and because specific reactions (e.g. the oxidation of lignin via an oxygen radical intermediate) are not possible, or are vastly slower, in the absence of molecular oxygen (Reference Kirk and Farrell64). Thus, shorter exposure times are associated with higher organic carbon burial efficiencies and the preservation of less degraded materials (Reference Burdige7,Reference Hedges and Keil8,Reference Middelburg13,Reference Hartnett, Keil, Hedges and Devol34,Reference Hedges, Hu, Devol, Hartnett, Tsamakis and Keil63,Reference Cowie, Hedges, Prahl and Delange65,Reference Cowie, Calvert, Pedersen, Schulz and von Rad66). This correlation is not absolute, however. Large provinces of ocean sediment underlying gyres are oxic to the basement, representing as much as 86 million years of OET (Reference Røy67). In such sediments, sedimentation rates are exceedingly slow and the sediments are very organic poor, and most oxidation apparently occurs directly at the sediment–water interface. Organic carbon oxidation in “rich” anoxic systems such as rapidly accumulating estuarine sediments can exceed 100 µM C day–1, primarily via sulfate reduction, compared with ~3 × 10–6 µM C day–1 in oxic gyre sediments.
16.2.5 Models of Organic Carbon Diagenesis
Due to the chemical complexity of sedimentary OM, sedimentary diagenetic models have focused on the transformation of bulk organic carbon to CO2. One common class of models assumes the following form:
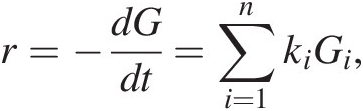 (16.1)
(16.1)where r is the bulk rate of CO2 production, equivalent to the rate disappearance of bulk organic carbon (G), which in turn is the sum of the oxidation rates of different carbon pools (Gi), each of which is oxidized according to a different, characteristic rate constant (ki) (Reference Westrich and Berner68). Frequently, these “multi-G” models only include two or three reactivity classes of OM: usually a fast-reacting “labile” pool, an unreactive “recalcitrant” pool, and sometimes an intermediate “semi-labile” pool. Related models include the reactivity-continuum model, which assumes an infinite number of reactivity pools (Reference Boudreau and Ruddick69,Reference Tarutis70), and that of Middelburg (Reference Middelburg71), in which a single time-dependent reactivity rate constant is assumed. These models are mathematically straightforward, but they are somewhat mechanistically disconnected from the reality of sediment OM, which is tremendously chemically complex (Reference Burdige7,Reference Hedges, Eglinton, Hatcher, Kirchman, Arnosti and Derenne72).
Recently, models that include a broader set of parameters, such as microbial biomass, enzyme substrate specificity, and temperature–rate relationships, have been successfully employed in soils and sediments (Reference Wieder, Bonan and Allison73,Reference Bradley, Amend and LaRowe74). By including a wider range of processes, these models have the capacity to both quantitatively fit bulk organic carbon concentration data and make reasonable predictions about systems’ likely responses to changing environments.
16.3 Oceanic Rocky Subsurface
Below ocean sediments, the igneous ocean crust hosts ~2% of the total volume of the ocean, making it the largest aquifer system on Earth (Reference Johnson and Pruis75). Seawater actively circulates through this aquifer and drives the transfer of heat and elements between fluids and rocks with ramifications for ocean chemistry (Reference Edmond, Measures, McDuff, Chan, Collier and Grant76–Reference German, Casciotti, Dutay, Heimburger, Jenkins and Measures79) and for the thermal, physical, and geochemical structure of the crust and mantle (Reference Stein and Stein80,Reference Alt81). Microbial life is widespread in the rocky oceanic subsurface and both exploits and influences these exchanges (Reference Summit and Baross82–Reference Orcutt, Sylvan, Knab and Edwards84), altering the abundance and form of carbon. Fluid flow through the rocky subsurface is ultimately driven by a source of heat such as cooling magma or hot rocks (Reference Stein and Stein80,Reference Sclater, Jaupart and Galson85). Heated fluids rise buoyantly and ultimately exit the sub-seafloor, drawing cool seawater into the crust to replace it.
The carbon characteristics of the fluids and rocks in hydrothermal systems and the igneous basement differ greatly depending on the type of host rock, the temperature of the system, and the presence or absence of sediments. Some systems are further influenced by factors such as phase separation, magma injections, seismic activity, extent of subduction, and even tides (Reference Butterfield, McDuff, Mottl, Lilley, Lupton and Massoth86–Reference Seewald, Cruse and Saccocia89). Below, carbon transformations are described in some of the primary types of hydrothermal circulation systems (Figure 16.4).
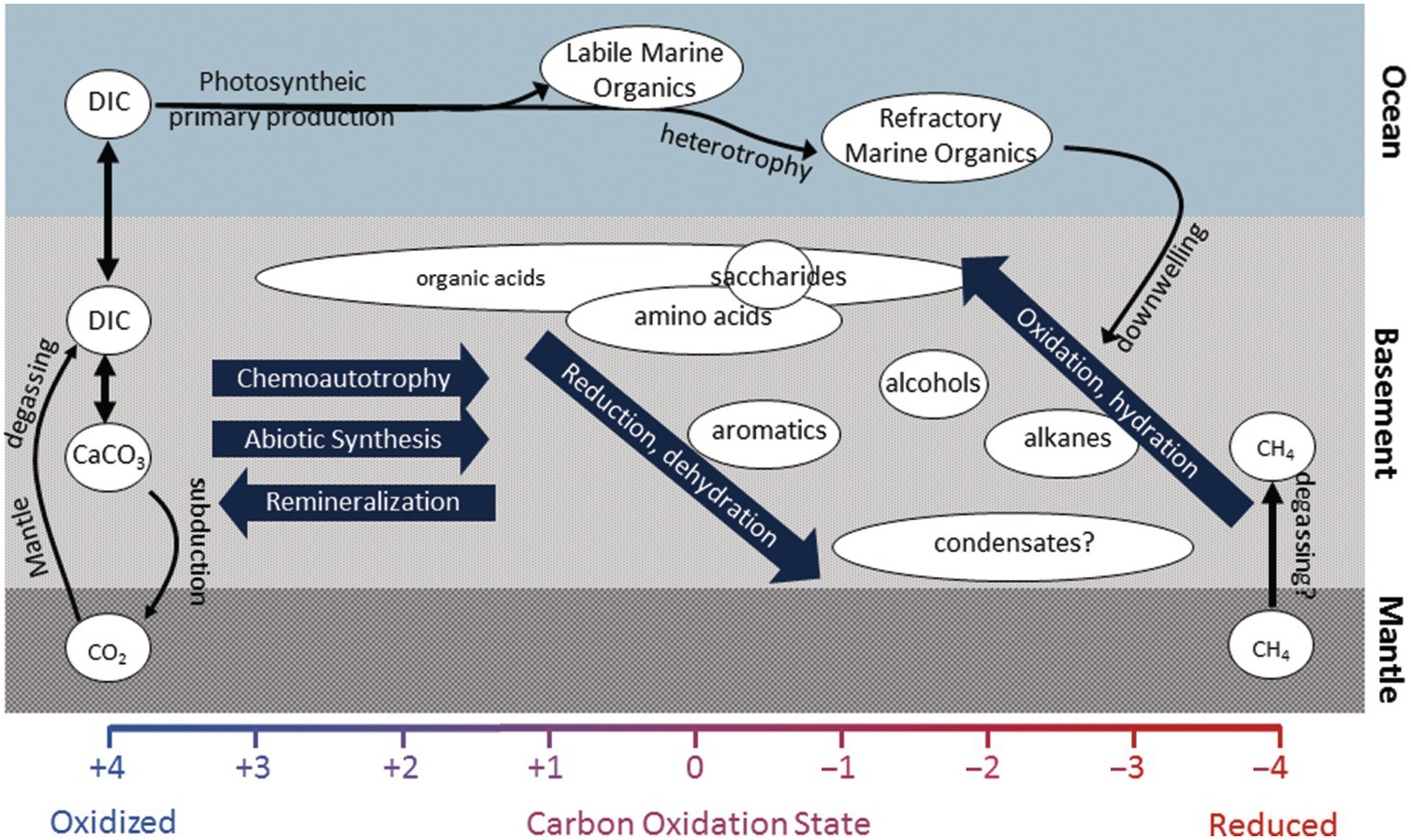
Figure 16.4 The abundance and composition of organic molecules in hydrothermal fluids will reflect a complex reaction history. While chemoautotrophy and abiotic synthesis involve the reduction of inorganic carbon into organic molecules, remineralization will do the reverse. Oxidation and dehydration reactions produce smaller, more polar compounds that are generally more labile and more easily consumed by heterotrophic microorganisms. Reduction and dehydration reactions may produce larger and more apolar material that is more resistant to microbial degradation and may be sequestered in the subsurface or persist for long periods of time in the deep ocean.
16.3.1 Characteristics of Recharge Water
The chemical composition of the seawater that enters into the rocky subsurface has a strong influence on subsequent water–rock and microbial reactions. Deep seawater carries DIC in concentrations of 2.1–2.3 mM (Reference Sarmiento and Gruber90) and DOC in concentrations of ~34–48 µM (Reference Hedges2,Reference Hansell, Carlson, Repeta and Schlitzer91). DOC is composed of a complex set of molecules, some of which turnover rapidly on timescales of hours to years. The majority of DOC, however, is slow to remineralize and has the potential to be stored for millennia in the ocean’s interior (see (Reference Hansell92) for review). Refractory DOC is highly degraded, has few recognizable biomarkers, and has a 14C age of 4000–6000 years, substantially longer than the mixing time of the ocean (Reference Druffel, Williams, Bauer and Ertel93). DOC isolated from seawater and subjected to nuclear magnetic resonance and Fourier-transform ion cyclotron resonance mass spectrometry is composed primarily of carboxyl-rich aliphatic matter (Reference Hertkorn, Benner, Frommberger, Schmitt-Kopplin, Witt and Kaiser94), acylated polysaccharides (Reference Aluwihare, Repeta and Chen95), and carotenoid degradation products (Reference Arakawa, Aluwihare, Simpson, Soong, Stephens and Lane-Coplen96) (see (Reference Repeta and Hansell97) for review).
16.3.2 Axial High Temperature, Basalt Hosted
The most widely recognized hydrothermal systems are close to axial spreading centers, where new injections of magma maintain high temperatures (Figure 16.1). The host rock is mafic and fluids exit through chimney structures at temperatures that can reach >400°C (Reference Campbell, Palmer, Klinkhammer, Bowers, Edmond and Lawrence98). Exiting fluids are rich in dissolved metals that, upon mixing with cold seawater, precipitate the sulfide minerals that give them the name “black smokers.” In the water column, the hot fluids mix further with seawater, cool, reach neutral buoyancy, and spread away from the vent field. The chemical signatures from these plumes of water can be detected thousands of kilometers away from the field (Reference Conway and John99,Reference Resing, Sedwick, German, Jenkins, Moffett and Sohst100).
The majority of high-temperature vent fluids have DIC concentrations equal to or greater than deep seawater due to inputs of magmatic CO2 (Figure 16.5 and Table 16.1) (Reference McCollom, Lowell, Seewald, Metaxas and Perfit101). DIC concentrations are generally 3–30 mM, or they can be higher when fluids are impacted by phase separation, fresh inputs of magma, or sedimentary degradation (see (Reference McCollom, Lowell, Seewald, Metaxas and Perfit101) for review). Additions of magmatic CO2 are identified by δ13C isotopic signatures (–9‰ to –4‰) (Reference Pineau and Javoy102,Reference DesMarais and Moore103) that are markedly different from deep seawater DIC (–0.5‰ to 1.0‰) (Reference Kroopnick104). The lack of 14C in CO2 from some hydrothermal fluids demonstrates that the DIC carried with recharge water can be fully removed during sub-seafloor circulation in some cases (Reference Proskurowski, Lilley and Brown105). Calcium carbonate veins in basalts and gabbros have isotope values consistent with precipitation of marine DIC at the relatively low temperatures of seawater recharge (Reference Alt and Teagle106,Reference Eickmann, Bach, Rosner and Peckmann107).
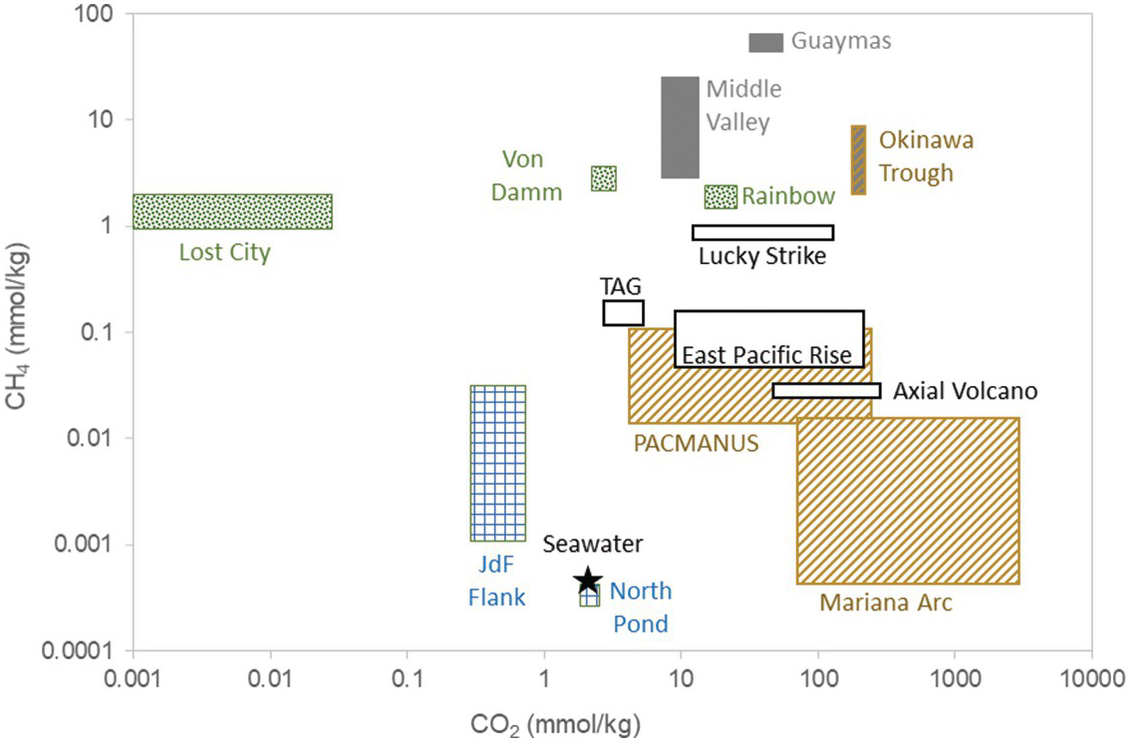
Figure 16.5 Range of methane and CO2 concentrations in basalt-hosted high-temperature (black outline; Axial Volcano, Trans-Atlantic Geotraverse (TAG), 9°N East Pacific Rise, Lucky Strike), ultramafic-hosted (green diamonds; Lost City, East Summit of Von Damm, Rainbow), ridge flank (blue checkers; Juan de Fuca ridge flank, North Pond), back-arc basins (orange diagonal; PACMANUS, Mariana Arc, Okinawa Trough), and sedimented systems (gray boxes; Guaymas, Middle Valley, Okinawa Trough). Seawater composition is included for comparison. Methane concentrations at North Pond are plotted at the reported detection limit of the analysis (0.5 µM).
| System type | Seawater | Sedimentary porewater | Basalt hosted, unsedimented, high temperature | Basalt hosted, unsedimented, diffuse/mixed fluids | Basalt hosted, sedimented, high temperature | Ridge Flank (“warm”) | Ridge Flank (“cool”) | Ultramafic influenced, high temperature | Ultramafic dominated | Silicic back-arc | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Example system | Below 1500 ma | Cascadia Marginb 0–65 mbsf | Cascadia Marginb 65–189 mbsf | East Pacific Rise (9°50'N)c | Axial Volcanod | TAGe | East Pacific Rise diffuse (9°50'N)f | Axial Volcanog | Guaymas Basinh | Middle Valleyi | Juan de Fuca Ridge Flankj | North Pond Basementk | Von Damm (East Summit)l | Rainbowm | Lost Cityn | PACMANUSo | Okinawa Trough (sedimented)p |
| Temperatures (°C) | 2–5 | ≤9 | ~8–12 | 275–371 | 217–328 | 290–321 | 23–55 | 3–78 | 100–315 | 40–281 | 64 | 3.1–3.8 | 226 | 350–367 | 30–91 | 152–358 | >220–320 |
| pH (at 23°C) | 7.8 | 7.7–8.0 | 7.7–8.4 | 3.5–4.2 | 3.5–4.4 | 3.1–3.5 | 5.8–6.4 | 4.6–5.8 | 4.5–6.1 | – | 7.5 | 7.4–7.6 | 5.6 | 2.8–3.4 | 9–11 | 2.3–4.7 | 4.7–5.4 |
| H2 (mM) | 0.0003 | – | – | 0.27–8.4 | <0.1–0.8 | 0.10–0.37 | BDL–0.0058 | <0.5 | 0.52–3.30 | 1.9–8.2 | 0.3–0.7 | – | 18.2–19.2 | 12.3–16.5 | 1–14 | 0.0084–0.306 | 0.05 |
| Dissolved iron (µM) | <0.001 | – | – | 8–5150 | 12–1065 | 1640–5170 | <2–277 | 35–400 | 17–180 | – | 0.6–1.1 | – | – | 23,700–24,050 | <3.5 µM | 76–14,600 | – |
| ΣCO2 (mM) | 2.1–2.3 | 7–24 | 18–29 | 9.4–219 | 50–285 | 2.9–5.0 | 3.0–11.8 | – | 35–54 | 8.2–13 | 0.2–0.6 | 2.0–2.4 | 2.80 | 16.0–24.6 | 0.0001–0.026 | 4.4–274 | 198–200 |
| δ13CCO2 (‰) | –0.6 to 0.4 | –24.4 to 25.6 | 27.9–33.6 | –4.2 to –3.7 | – | –13.0 to –6.9 | –4.2 to –2.2 | – | –9.4 | –34.6 to –20.7 | –9.7 to –1.3 | –0.16 to 0.67 | 0.8–0.9 | –3.15 to –2.5 | ~–9 | –5.7 to –2.3 | –5.0 to –4.7 |
| F14CCO2 | 0.7511–0.9677 | – | – | – | – | – | – | – | 0.056 | – | 0.083–0.233 | 0.595–0.865 | 0.0251–0.0373 | – | – | – | – |
| CH4 (mM) | 0.0003 | BDL–69 | 71–236 | 0.05–0.16 | 0.025 | 0.12–0.16 | 0.003–0.500 | <0.6 | 44.2–58.8 | 3.0–22.6 | 0.001–0.030 | <0.0005 | 2.81 | 1.6–2.5 | 0.9–2.0 | 0.014–0.085 | 2.4–7.1 |
| δ13CCH4 (‰) | – | –59.7 ± 7.1 | –46.7 to –41.7 | –34.6 to –16.8 | – | –9.5 to –8.0 | – | – | –43.8 | –55.5 to –50.8 | –58 to –23 | – | –15.6 to –15.3 | –17.7 to –15.8 | –13.6 to –9.3 | –20.8 to –7.4 | –41.2 to –36.1 |
| F14CCH4 | – | – | – | – | – | – | – | – | 0.077 | – | – | 0.0056–0.0064 | – | 0.0017–0.0062 | – | – | |
| Σ(C2H6–C4H8) (µM) | BDL | 0–35 ppmvq | 0.5–7318 ppmvq | – | – | – | – | – | – | 14–310 | – | – | 695 | 0.84 | 1.0–2.0 | – | – |
| δ13CC2H6,C4H8 (‰) | – | – | – | – | – | – | – | – | – | –25.3 to –18.7 | – | – | –12.9 to –9.8 | – | –16.0 to –13.0 | – | – |
| CO (µM) | BDL | – | – | BDL–2.0 | – | BDL | 5.7–17.8 | – | 27–92.4 | – | – | – | n.d. | 5.0–7.4 | BDL | 0.006–0.17 | – |
| CH3SH (nM) | BDL | – | – | 2.4–4.9 | – | 12 | – | – | 11–10,000 | – | – | – | 22 | 7.4–10.3 | 1.4–1.9 | – | – |
| DOC (µM) | 35–45 | 400–3200 | 1700–5100 | – | 8–24 | – | – | 34–71 | 111–2112 | – | 11–18 | 18–33 | – | – | 68–106 | – | – |
| δ13CDOC (‰) | –20 to –22 | –23.6 to –22.1 | –20.2 ± 0.4 | – | – | – | – | –18.6 | – | – | –34.5 to –24.8 | –26.6 to –23.9 | – | – | –21.0 to –10.5 | – | – |
| F14CDOC | 0.444–0.767 | – | – | – | – | – | – | 0.481r | – | – | 0.166–0.230q 0.186–0.204 | 0.352–0.472 | – | – | – | – | – |
| Formate (µM) | BDL | – | – | – | – | – | – | – | <40 | – | – | – | 88.2 | – | 36–158 | – | – |
| Acetate (µM) | BDL | 5–57 | 14–89 | – | – | – | – | – | BDL–295 | – | – | – | – | – | 1–35 | – | – |
| Hydrolizable amino acids (µM) | 80–160 | – | – | – | – | – | – | – | 5.2 | – | 0.043–0.089 | – | – | – | 0.7–2.3 | – | – |
“–” is used where no reports available in the literature.
BDL = below detection limit; mbsf = neters below seafloor; TAG = Trans-Atlantic Geotraverse.
a (Reference Druffel, Williams, Bauer and Ertel93, Reference Kroopnick104, Reference Beaupre and Druffel108, Reference Reeves, McDermott and Seewald109).
b (Reference Heuer, Pohlman, Torres, Elvert and Hinrichs29, Reference Riedel, Collett, Malone and Scientists110).
c (Reference Reeves, McDermott and Seewald109, Reference Von Damm and Lilley111–Reference Yucel and Luther113).
d (Reference Butterfield, Massoth, McDuff, Lupton and Lilley114–Reference Lang, Butterfield, Lilley, Johnson and Hedges116).
e (Reference Campbell, Palmer, Klinkhammer, Bowers, Edmond and Lawrence98, Reference Reeves, McDermott and Seewald109, Reference Charlou and Donval117–Reference Charlou, Donval, Konn, Ondreas, Fouquet, Jean-Baptiste and Rona119).
f (Reference Reeves, McDermott and Seewald109, Reference Von Damm and Lilley111, Reference Proskurowski, Lilley and Olson120).
g (Reference Butterfield, McDuff, Mottl, Lilley, Lupton and Massoth86, Reference Butterfield, Massoth, McDuff, Lupton and Lilley114, Reference Lang, Butterfield, Lilley, Johnson and Hedges116, Reference McCarthy, Beaupre, Walker, Voparil, Guilderson and Druffel121).
h (Reference Reeves, McDermott and Seewald109, Reference Haberstroh and Karl122–Reference McKay, Klokman, Mendlovitz, LaRowe, Hoer and Albert126).
j (Reference Lang, Butterfield, Lilley, Johnson and Hedges116, Reference Wheat, Mottl, Fisher, Kadko, Davis and Baker128–Reference Lin, Repeta, Xu and Rappe133).
k (Reference Meyer, Jaekel, Tully, Glazer, Wheat and Lin134, Reference Walter, Jaekel, Osterholz, Fisher, Huber and Pearson135).
m (Reference Reeves, McDermott and Seewald109, Reference Charlou, Donval, Konn, Ondreas, Fouquet, Jean-Baptiste and Rona119, Reference Douville, Charlou, Oelkers, Bienvenu, Colon and Donval137–Reference Jean-Baptiste, Fourre, Charlou, German and Radford-Knoery139).
n (Reference Reeves, McDermott and Seewald109, Reference Proskurowski, Lilley, Seewald, Früh-Green, Olson and Lupton112, Reference Kelley, Karson, Früh-Green, Yoerger, Shank and Butterfield140–Reference Seyfried, Pester, Tutolo and Ding143).
p (Reference Sakai, Gamo, Kim, Tsutsumi, Tanaka and Ishibashi145, Reference Ishibashi, Sano, Wakita, Gamo, Tsutsumi and Sakai146).
q Headspace gas concentrations in equilibrium with sediments.
r F14C of ultrafiltrated DOC (>1000 Da).
Methane concentrations in sediment-free, high-temperature axial fluids (~7–200 µM) are higher than those of seawater (0.0003 µM), but generally low when compared to sedimented or ultramafic-influenced systems (Figure 16.5 and Table 16.1). For example, vent fluid CH4 concentrations range from 7 to 213 µM from high-temperature vents from along the East Pacific Rise (Reference Von Damm and Lilley111,Reference Von Damm123,Reference Welhan, Craig and Rona147–Reference Merlivat, Pineau and Javoy150), while those from along the Mid-Atlantic Ridge (MAR) range from 8 to 147 µM (Reference Campbell, Palmer, Klinkhammer, Bowers, Edmond and Lawrence98,Reference Charlou and Donval117–Reference Charlou, Donval, Konn, Ondreas, Fouquet, Jean-Baptiste and Rona119,Reference Jeanbaptiste, Charlou, Stievenard, Donval, Bougault and Mevel151–Reference Lein, Grichuk, Gurvich and Bogdanov153). Concentrations can spike as a result of volcanic eruptions and due to outgassing after a dike injection (Reference Lilley, Butterfield, Lupton and Olson88,Reference Seewald, Cruse and Saccocia89,Reference Yucel and Luther113).
The majority of the DOC carried with deep seawater is destroyed during circulation through mafic hydrothermal systems. The first evidence for this removal came from a study of amino acids in the sediment-covered Guaymas Basin (Reference Blair, Martens and Desmarais26,Reference Haberstroh and Karl122). Concentrations of dissolved free amino acids in high-temperature fluids (>150°C) were below detection limits and below deep ocean concentrations, with the losses attributed to the instability of organic compounds at high temperatures (Reference Haberstroh and Karl122,Reference Martens154). The DOC content of black smoker vents on the unsedimented portions of the Juan de Fuca spreading center is less than half that of deep seawater (<17 versus 36 µM) (Reference Lang, Butterfield, Lilley, Johnson and Hedges116). Concentrations of DOC that can be isolated onto solid-phase extraction (SPE-DOC) phases are ~92% lower in unsedimented black smokers from Juan de Fuca and the MAR than in deep seawater (Reference Hawkes, Rossel, Stubbins, Butterfield, Connelly and Achterberg155). It is possible to experimentally reproduce losses of OM by heating (Reference Lin, Koch, Feseker, Ziervogel, Goldhammer and Schmidt125,Reference Hawkes, Rossel, Stubbins, Butterfield, Connelly and Achterberg155–Reference Seewald, Seyfried and Thornton157), though this does not conclusively rule out alternative removal mechanisms such as sorption onto mineral surfaces or heterotrophy.
16.3.3 Axial Diffuse Vents, Basalt Hosted
Adjacent to axial, high-temperature systems, local seawater enters the crust, creating “diffuse vents.” The mixing of oxygenated seawater and reduced hydrothermal fluids results in chemical disequilibria that microorganisms can exploit for metabolic energy (Reference McCollom and Shock158). Due to mixing and conductive cooling of fluids, temperatures are often well below the upper temperature limits of life (122°C) (Reference Takai, Nakamura, Toki, Tsunogai, Miyazaki and Miyazaki159). As a result, these zones are thriving sub-seafloor microbial habitats (Reference Schrenk, Huber and Edwards3,Reference Summit and Baross82,Reference Orcutt, Sylvan, Knab and Edwards84,Reference Karl, Wirsen and Jannasch160,Reference Edwards, Becker and Colwell161). Microbial activity can alter fluid chemistry, resulting in losses of H2S and H2 and gains of CH4 relative to high-temperature fluids (Reference Von Damm and Lilley111,Reference Proskurowski, Lilley, Seewald, Früh-Green, Olson and Lupton112,Reference Butterfield, Roe, Lilley, Huber, Baross and Embley115,Reference Wankel, Germanovich, Lilley, Genc, DiPerna and Bradley162).
In diffuse vents on the Juan de Fuca Ridge, DOC is elevated over local deep seawater (~47 versus 36 µM), attributed in part to sub-seafloor autotrophic production (Reference Lang, Butterfield, Lilley, Johnson and Hedges116). This DOC has a lower 14C content and a more positive δ13C value than local seawater, consistent with a contribution of chemolithoautotrophs incorporating a pre-aged carbon source such as mantle CO2 (Reference McCarthy, Beaupre, Walker, Voparil, Guilderson and Druffel121).
16.3.4 Ridge Flanks
Fluid continues to flow through the rocky subsurface far from the ridge axis, as rocks cool in the absence of new magma injections (Figure 16.1). The extent of advective flow through these “ridge flank” systems can be determined from discrepancies between modeled conductive heat loss and heat flow measurements that indicate the convective flow of water in crust that is 0–65 Ma (Reference Johnson and Pruis75,Reference Stein and Stein80). Sediment cover precludes fluid transport into and out of the crust; bare-rock seamounts are therefore the primary locations of advective transport (Reference Wheat, Mottl, Fisher, Kadko, Davis and Baker128). Even in regions with thick sedimentary layers, however, exchange of water, carbon, elements, and nutrients continues between deep sedimentary porewater and basement fluids.
Based on magnesium budgets, fluid fluxes through “cool” ridge flank systems (<45°C) are substantially larger than those through warmer systems (Reference Mottl and Wheat77,Reference Mottl, Halbach, Tunnicliffe and Hein163). Cool basement fluids (<20°C) have been accessed by Integrated Ocean Drilling Program drilling in the North Pond sedimented basin on the MAR (Reference Edwards, Becker and Colwell161). Dorado Outcrop on the Cocos Plate has also been confirmed to vigorously vent large quantities of water at temperatures of 10–20°C (Reference Wheat, Fisher, McManus, Hulme and Orcutt164). The “warm” ridge flank system on the Juan de Fuca ridge has been intensely studied for decades, including via series of Ocean Drilling Program boreholes that have been drilled perpendicular to the ridge to allow direct access to the basement (Reference Elderfield, Wheat, Mottl, Monnin and Spiro165).
DIC is substantially lower in Juan de Fuca ridge flank fluids than in seawater (0.1–0.9 versus 2.6 mmol/kg; Table 16.1), likely due to precipitation of calcium carbonate in the subsurface (Reference Walker, McCarthy, Fisher and Guilderson130,Reference Mottl, Wheat, Baker, Becker, Davis and Feely166,Reference Sansone, Mottl, Olson, Wheat and Lilley167). In contrast, fluids from the lower-temperature Dorado and North Pond systems have DIC concentrations that are similar to seawater (Reference Meyer, Jaekel, Tully, Glazer, Wheat and Lin134,Reference Wheat, Fisher, McManus, Hulme and Orcutt164). In many cases, the δ13C values of DIC are lower than that of seawater, suggesting an input from remineralization of organic carbon or CO2 trapped in basaltic vesicles (Reference Walker, McCarthy, Fisher and Guilderson130,Reference Lin, Repeta, Xu and Rappe133,Reference Walter, Jaekel, Osterholz, Fisher, Huber and Pearson135). The apparent 14C age of DIC is often used as a measure of fluid residence time, although this must be treated with caution, as mixing with older water masses, remobilization of calcium carbonate, input of basalt vesicle CO2, and remineralization of 14C-depleted OM can influence these signatures (Reference Lin, Repeta, Xu and Rappe133,Reference Maloszewski and Zuber168–Reference Bethke and Johnson170).
Methane concentrations are low but detectable in Juan de Fuca ridge flank fluids (1–32 µmol/kg) (Reference Lin, Cowen, Olson, Amend and Lilley131,Reference Lin, Cowen, Olson, Lilley, Jungbluth and Wilson132). The isotopic signatures of methane (–58.0‰ to –22.5‰) indicate a mixture of processes, including biogenic production and oxidation (Reference Lin, Cowen, Olson, Lilley, Jungbluth and Wilson132). Methane concentrations at North Pond were below detection (Reference Meyer, Jaekel, Tully, Glazer, Wheat and Lin134).
DOC concentrations are lower than seawater in ridge flank fluids on the Juan de Fuca ridge and at North Pond (Reference Lang, Butterfield, Lilley, Johnson and Hedges116,Reference Lin, Cowen, Olson, Amend and Lilley131,Reference Lin, Repeta, Xu and Rappe133–Reference Walter, Jaekel, Osterholz, Fisher, Huber and Pearson135). In both cases, this DOC has a lower 14C content and δ13C signatures that are more negative than those of starting seawater (Reference McCarthy, Beaupre, Walker, Voparil, Guilderson and Druffel121,Reference Lin, Repeta, Xu and Rappe133,Reference Walter, Jaekel, Osterholz, Fisher, Huber and Pearson135). This pattern was initially attributed to a complete removal of seawater DOC, followed by an input of chemosynthetically derived organic material (Reference McCarthy, Beaupre, Walker, Voparil, Guilderson and Druffel121). New data suggest that the isotopic signatures could instead be attributed to the selective oxidation and removal of portions of the seawater DOC pool (Reference Lin, Repeta, Xu and Rappe133,Reference Walter, Jaekel, Osterholz, Fisher, Huber and Pearson135). Diffusion of porewater from the sediments covering the ridge flank may also contribute some organic compounds to the fluids (Reference Lin, Amend, LaRowe, Bingham and Cowen124), as this exchange impacts the inorganic chemistry (Reference Wheat, Hulme, Fisher, Orcutt and Becker129,Reference Lin, Cowen, Olson, Amend and Lilley131,Reference Elderfield, Wheat, Mottl, Monnin and Spiro165).
16.3.5 Ultramafic Influenced
Systems hosted on ultramafic rocks undergo water–rock reactions that are distinct from those of mafic environments. Ultramafic systems can be located on spreading centers and influenced by magmatic injections, but they can also be far from the spreading center or along ultra-slow-spreading centers with little to no magmatic influence. The compositional differences between ultramafic rocks derived predominantly from Earth’s mantle and mafic rocks such as basalt and gabbro give rise to fluids with distinct chemical signatures. Fluids that have reacted peridotites are strongly enriched in H2 and CH4 and, in some cases, have drastically lower metal contents (Figure 16.5 and Table 16.1).
The earliest recognitions of an ultramafic hydrothermal signature in the ocean came from high ratios of CH4 to Mn and suspended particulate matter in the water column on the MAR (Reference Rona, Widenfalk and Bostrom171–Reference Rona, Bougault, Charlou, Appriou, Nelsen and Trefry173). Subsequently, the Logatchev, Rainbow, Menez Gwen, Ashadze, and Nibelungen hydrothermal fields were identified along the MAR, with fluid chemistries that exhibit a mixture of magmatic influences such as high temperatures (200–372°C), acidic pHs (2–4 at 25°C), and high metal contents (e.g. millimolar concentrations of Fe and hundreds of micromolar concentrations of Mn), but also ultramafic influences such as millimolar concentrations of CH4 and H2 (Table 16.1; for reviews, see Reference Charlou, Donval, Konn, Ondreas, Fouquet, Jean-Baptiste and Rona119,Reference Schrenk, Brazelton, Lang, Hazen, Jones and Baross174). Peridotite-influenced systems have since been identified on the Mid-Cayman Rise (Reference McDermott, Seewald, German and Sylva136,Reference German, Bowen, Coleman, Honig, Huber and Jakuba175) and Marianas Forearc (Reference Mottl, Halbach, Tunnicliffe and Hein163,Reference Haggerty176). The ultramafic-dominated system in the Lost City Hydrothermal field has minimal interaction with magmatic processes, resulting in lower fluid temperatures (40–91°C), alkaline pHs (9–11 at 23°C), and low metal contents (<100 nM of Fe and <50 nM of Mn) (Reference Kelley, Karson, Früh-Green, Yoerger, Shank and Butterfield140,Reference Seyfried, Pester, Tutolo and Ding143,Reference Kelley, Karson, Blackman, Früh-Green, Butterfield and Lilley177). A magmatic influence is still evident, however, in elevated the 3He content of fluids (Reference Proskurowski, Lilley, Seewald, Früh-Green, Olson and Lupton112). Ultramafic-dominated, low-temperature, alkaline systems are also present in the shallow waters of Prony Bay in New Caledonia, fed by meteoric water (Reference Monnin, Chavagnac, Boulart, Menez, Gerard and Gerard178), and on the Southern Mariana Forearc at the Shinkai Seep Field (Reference Ohara, Reagan, Fujikura, Watanabe, Michibayashi and Ishii179).
The inorganic carbon concentration in ultramafic-influenced systems is highly dependent on pH and magmatic inputs. In low-pH ultramafic systems, concentrations of ΣCO2 can reach as high as those observed in magmatic systems, at ~4–20 mM (Table 16.1; see (Reference Charlou, Donval, Konn, Ondreas, Fouquet, Jean-Baptiste and Rona119) for a review). The δ13C values of this CO2 display “typical” mid-ocean ridge values of –4‰ to –2‰ in some cases such as the Rainbow vent field (Reference Charlou, Donval, Konn, Ondreas, Fouquet, Jean-Baptiste and Rona119). In other locations such as the Logatchev field, it is unusually positive, up to +9.5‰, even in fluids with ΣCO2 concentrations higher than seawater (Reference Charlou, Donval, Konn, Ondreas, Fouquet, Jean-Baptiste and Rona119,Reference Konn, Charlou, Holm and Mousis180). In alkaline ultramafic systems such as Lost City, the high pHs lead to the rapid precipitation of calcium carbonate and therefore vanishingly low concentrations of ΣCO2 in end-member fluids (Reference Proskurowski, Lilley, Seewald, Früh-Green, Olson and Lupton112,Reference Kelley, Karson, Früh-Green, Yoerger, Shank and Butterfield140). This removal likely occurs throughout the fluid circulation pathway. Carbonate mineralization is common in ultramafic rocks (Reference Bonatti, Lawrence, Hamlyn and Breger181), and isotope signatures indicate precipitation occurs both at cold seawater temperatures and at warmer (65–95°C) temperatures, where δ13C values indicate that the source ΣCO2 has a substantial mantle component (Reference Eickmann, Bach, Rosner and Peckmann107).
Methane concentrations in ultramafic systems are frequently an order of magnitude higher than those in unsedimented, basalt-hosted systems (Figure 16.5 and Table 16.1), and substantial methane anomalies along the MAR have been attributed to exports from these systems (Reference Charlou, Donval, Konn, Ondreas, Fouquet, Jean-Baptiste and Rona119,Reference Rona, Widenfalk and Bostrom171–Reference Rona, Bougault, Charlou, Appriou, Nelsen and Trefry173). Estimates from mantle 3He exports suggest serpentinization of ultramafic rocks could account globally for about 75% of the methane flux from mid-ocean ridge systems (Reference Keir182). Isotopic signatures point to a nonbiological source for this methane (Reference Proskurowski, Lilley, Seewald, Früh-Green, Olson and Lupton112,Reference Charlou, Donval, Konn, Ondreas, Fouquet, Jean-Baptiste and Rona119,Reference McCollom and Seewald183), although in most systems more CH4 is present than would be expected in thermodynamic equilibrium with CO2 (for reviews, see (Reference McCollom, Lowell, Seewald, Metaxas and Perfit101,Reference McCollom and Seewald183)). One possibility is that the methane was formed long ago, at higher temperatures than the present day, and is subsequently stripped from vesicles in the rocks (Reference McDermott, Seewald, German and Sylva136,Reference Wang, Reeves, McDermott, Seewald and Ono184), which contain high CH4 and CO2 contents (Reference Kelley and Früh-Green185,Reference Kelley and Früh-Green186). Biologically derived methane from methanogenesis may also contribute (Reference Bradley and Summons187), albeit at relatively low levels when compared to the dominant nonbiological signature.
Short-chain hydrocarbons such as ethane, propane, and butane have been found in the low-micromolar concentrations in a wide range of ultramafic systems (Table 16.1) (Reference Proskurowski, Lilley, Seewald, Früh-Green, Olson and Lupton112,Reference McDermott, Seewald, German and Sylva136,Reference Konn, Charlou, Holm and Mousis180). Isotopic values that decrease with increasing chain length have been used to demonstrate that these species are not derived from the decomposition of sediments and could have a nonbiological origin such as Fischer–Tropsch-type reactions (Reference Proskurowski, Lilley, Seewald, Früh-Green, Olson and Lupton112). At the Lost City and Von Damm hydrothermal fields, the concentrations of these compounds increase in conjunction with methane concentrations (Reference Proskurowski, Lilley, Seewald, Früh-Green, Olson and Lupton112,Reference McDermott, Seewald, German and Sylva136), indicating similar processes may lead to their formation and/or cycling.
Formate and acetate have been reported in elevated concentrations in multiple ultramafic systems including Lost City (formate: 36–158 µM; acetate: 1–35 µM) (Reference Lang, Butterfield, Schulte, Kelley and Lilley141), Von Damm (formate: below detection to 669 µM) (Reference McDermott, Seewald, German and Sylva136), and Prony Bay (formate: ~4 µM; acetate: ~70 µM) (Reference Pisapia, Gerard, Gerard, Lecourt, Lang and Pelletier188). At Lost City, the isotopic composition of formate indicates it is synthesized by two pathways: abiotic synthesis in the subsurface that results in 14C-free formate with a δ13C signature (–13.0‰ to –8.9‰) similar to methane and short-chain hydrocarbons; and near-surface biological synthesis that incorporates modern DIC, resulting in formate with substantial 14C and a more positive δ13C signature (–9.1‰ to –4.3‰) (Reference Lang, Früh-Green, Bernasconi, Brazelton, Schrenk and McGonigle189). At the Von Damm vent field, higher concentrations of formate are found in hot mixed fluids than in pure end-member hydrothermal fluids, demonstrating that this species forms abiotically on timescales of hours to days (Reference McDermott, Seewald, German and Sylva136). At Lost City, the δ13C of acetate (–27‰ to –17‰) could be attributed to a mixture of anaerobic fermentation and acetogenesis (Reference Lang, Butterfield, Schulte, Kelley and Lilley141,Reference Lang, Früh-Green, Bernasconi, Brazelton, Schrenk and McGonigle189). Given the high abundances of microorganisms in the chimneys, the acetate could also be due to a thermocatalytic breakdown of complex organics in the biomass (Reference Lang, Butterfield, Schulte, Kelley and Lilley141,Reference Lang, Früh-Green, Bernasconi, Brazelton, Schrenk and McGonigle189).
Hydrolyzable amino acids are present in high abundances in Lost City fluids and chimneys (Reference Lang, Früh-Green, Bernasconi and Butterfield142). In the fluids, the highest concentrations were observed in locations where concentrations of H2 had been drawn down by sulfate reducers living in the sub-seafloor or chimney. The 13C of amino acids isolated from the chimneys had fractionation patterns consistent with synthesis by a chemolithoautotrophic source (Reference Lang, Früh-Green, Bernasconi and Butterfield142). In high-temperature fluids (>300°C) from Rainbow and Ashadze, dissolved free amino acids were detected in the picomolar concentration range, with tryptophan, phenylalanine, and leucine detected in the fluids but not in deep seawater (Reference Konn, Charlou, Holm and Mousis180). Tryptophan and phenylalanine contain aromatic rings that may assist in molecular stability at high temperatures (Reference Kawka and Simoneit190).
16.3.6 Fluxes between the Ocean and Crust
Hydrothermal circulation is the primary means of transferring materials between the crust and ocean (Reference Elderfield and Schultz78,Reference Kadko, Baross, Alt, Humphris, Zierenberg, Mullineaux and Thomson191). The net flux of constituents includes both input and removal processes, though these may be geographically and temporally distinct. The impact of hydrothermal circulation on the carbon budget of the ocean remains unconstrained in many ways.
Inorganic carbon is transferred to the deep ocean via magma degassing and removed by carbonate precipitation in the sub-seafloor at roughly similar rates (Figures 16.4 and 16.5). Degassing of mantle volatiles through high-temperature venting is estimated to input ~1 × 1012 mol C yr–1 of ΣCO2 into the ocean (Reference Kadko, Baross, Alt, Humphris, Zierenberg, Mullineaux and Thomson191,Reference Saal, Hauri, Langmuir and Perfit192). Carbonate precipitation is estimated to remove 1–3 × 1012 mol C yr–1 in ridge flanks (Reference Sansone, Mottl, Olson, Wheat and Lilley167,Reference Staudigel, Hart, Schmincke and Smith193,Reference Alt and Teagle194). Seawater passing through peridotites results in a loss of 0.4–2.0 × 1011 mol C yr–1, although stable isotope signatures indicate that approximately half of the carbon sequestered into the rock is in the form of organic carbon (Reference Schwarzenbach, Lang, Frah-Green, Lilley, Bemasconi and Mehay195,Reference Alt, Schwarzenbach, Frueh-Green, Shanks, Bernasconi and Garrido196).
Export and removal fluxes of DOC can be estimated by combining changes in concentrations with water fluxes through different types of hydrothermal systems (Reference Johnson and Pruis75,Reference Stein and Stein80,Reference Mottl, Halbach, Tunnicliffe and Hein163). If high-temperature vents remove an average of ~20 µmol of seawater DOC per liter, approximately 0.7–1.4 × 1010 g C yr–1 would be lost globally (Reference Lang, Butterfield, Lilley, Johnson and Hedges116). A similar scale loss of 1.4 ± 0.7 × 1010 g C yr–1 has been estimated based on changes of SPE-DOC concentrations (Reference Hawkes, Rossel, Stubbins, Butterfield, Connelly and Achterberg155). Ridge flank regions where crustal temperatures are “warm” (>45°C) have more substantial chemical changes in circulating fluids but smaller fluid fluxes than regions where crustal temperatures are “cool” (Reference Mottl and Wheat77,Reference Mottl, Halbach, Tunnicliffe and Hein163). If concentrations from the “warm” Juan de Fuca ridge flank system are typical of such systems, 2–13 × 1010 g C yr–1 would be removed (Reference Lang, Butterfield, Lilley, Johnson and Hedges116). Due to the larger water fluxes, if DOC concentrations through the “cool” crust at North Pond are globally representative, losses would be an order of magnitude higher at ~9–14 × 1011 g C yr–1 or ~5% of the total annual deep oceanic DOC loss (Reference Walter, Jaekel, Osterholz, Fisher, Huber and Pearson135).
16.4 Sedimented Hydrothermal Systems
Where spreading centers occur under thick sediment packages, hot water rapidly alters the OM fueling heterotrophic communities (e.g. (Reference Lin, Koch, Feseker, Ziervogel, Goldhammer and Schmidt125,Reference Wellsbury, Goodman, Barth, Cragg, Barnes and Parkes156,Reference Kawka and Simoneit190)), releasing inorganic carbon (Reference Seewald, Cruse and Saccocia89), influencing local physiochemical conditions, or forming complex oil-like materials (e.g. (Reference Simoneit and Lonsdale197,Reference Kvenvolden and Simoneit198)). The form and fate of carbon in heated sediments depend on its origin (terrigenous versus marine versus chemoautotrophic), temperature, and flow rate. Upon heating, a series of reactions similar to those that give rise to petroleum proceeds, with important differences due to the more water-rich conditions. The production of petroleum is generally considered to begin at ~50–70°C (Reference Tissot and Welte199). Weak bonds that sorb organic molecules onto surfaces break most easily, followed by bonds involving oxygen, sulfur, or nitrogen. Carbon–carbon bonds require the most energy – and therefore greater temperature or time – to break (Reference Tissot and Welte199).
Small polar compounds can be mobilized through enhanced desorption and the destruction of noncovalent bonds. The most labile material is removed from the solid phase due to microbial activity, pyrolysis, and/or desorption (Reference Wellsbury, Goodman, Barth, Cragg, Barnes and Parkes156). Over time, the amount of OM transferred into the aqueous phase decreases as the material is physically transported out of the system or biodegraded by microorganisms (Reference Lin, Koch, Feseker, Ziervogel, Goldhammer and Schmidt125,Reference Wellsbury, Goodman, Barth, Cragg, Barnes and Parkes156,Reference Seewald, Seyfried and Thornton157).
Unlike the dry “cracking” reactions that dominate petroleum reservoirs, breaking carbon–carbon bonds in the presence of water results in more oxidized products. Cracking reactions proceed at temperatures above ~100°C and result in CH4 and low-molecular-weight hydrocarbons (Reference Tissot and Welte199–Reference Lewan, Winters and McDonald201). In contrast, in the presence of water and minerals, n-alkanes will instead degrade to oxygenated products such as alcohols, ketones, carboxylic acids, and, ultimately, CO2 and CH4 (Reference Seewald, Cruse and Saccocia89,Reference Seewald202). Sediments heated in aqueous environments produce copious amounts of acetate in particular. The reaction temperature impacts the products, with higher temperatures favoring more oxidized products such as CO2 over CH4 and propanol over propane (Reference Shock, Canovas, Yang, Boyer, Johnson and Robinson203).
Reduction, condensation, and dehydration reactions proceed at higher temperatures to form macromolecules and aromatics, causing compounds to revert to their most stable states (Figure 16.4) (Reference Simoneit204,Reference Didyk and Simoneit205). Polycyclic aromatic hydrocarbons and cyclic polysulfides, major components of some hydrothermal oils, form only under very high heat (>~300°C) and are signatures of elevated temperatures (Reference Kawka and Simoneit190,Reference Simoneit and Lonsdale197,Reference Kvenvolden and Simoneit198). Polypeptides form through dehydration and reduction, while lipids crack and recombine (Reference Rushdi and Simoneit206).
Water washing will selectively transport more soluble components from the subsurface to the surface and leave behind larger condensates (Reference Lin, Koch, Feseker, Ziervogel, Goldhammer and Schmidt125,Reference Kawka and Simoneit190,Reference Tissot and Welte199,Reference Simoneit, Lein, Peresypkin and Osipov207). Smaller alkanes (<C10), aromatic volatiles, compounds containing C–N–S bonds, oligosaccharides, and oligopeptides are often missing in sediments subjected to “water washing,” while fluids and plumes contain higher concentrations of these compounds (Reference Kawka and Simoneit190,Reference Simoneit204,Reference Simoneit, Lein, Peresypkin and Osipov207).
These released compounds are highly biodegradable and fuel heterotrophic organisms. The labile amino acids released from sterilized sediments, for example, are utilized and reworked by microorganisms in parallel, nonsterilized experiments (Reference Lin, Koch, Feseker, Ziervogel, Goldhammer and Schmidt125). In general, the low-molecular-weight organic acids that are primary breakdown products of heating sediments in the presence of water, particularly acetate, are important substrates for anaerobic microorganisms (Reference Parkes, Taylor and Jorckramberg208).
The residual OM that is not removed with water washing is enriched in less soluble material, leading to “hydrothermal petroleum.” Cooling near the sediment–water interface can help trap less soluble compounds through differential condensation and solidification (Reference Kawka and Simoneit190,Reference Simoneit, Lein, Peresypkin and Osipov207,Reference Simoneit, Kawka and Brault209). The distribution of compounds and the maturity of these oils are highly variable.
16.5 Continental Subsurface
Geological heterogeneity produced through plate tectonics diversifies and segments the continental deep subsurface and its constituent biospheres differently from in the marine realm. Mountain and basin formation juxtaposes reactive rocks and minerals and creates new hydrological flow paths. Rock and water ages on the continents range from modern to billions of years (Reference Murdoch, Germanovich, Wang, Onstott, Elsworth and Stetler210,Reference Holland, Lollar, Li, Lacrampe-Couloume, Slater and Ballentine211). Terrestrial vegetation supplies vast quantities of organic carbon, although this influence is attenuated with increasing depth. The water age, hydrological connectivity, and major element chemistry of continental subsurface sites dramatically impact carbon cycling and the nature of in situ biospheres.
The continental deep subsurface extends downward from the base of the critical zone (Reference Pedersen212,Reference Colwell and D’Hondt213), although specific depths and thresholds have yet to be defined, particularly on the upper boundary. The penetration of life into the continental crust appears to be limited not strictly by depth, but rather by temperature, permeability, and perhaps aridity, with clear life detection in even the deepest boreholes and mines. Sites lacking identifiable life are few and far between and appear to be limited by temperature (e.g. German continental deep drilling program (KTB) cores in the Black Forest (Reference Huber, Huber, Lüdemann and Stetter214)) or aridity (Reference Colwell and D’Hondt213).
Estimates of the size of the continental deep biosphere are large (ranging from 2.3 × 1015 to 1017 g C), mirroring similar estimates of the marine deep biosphere (4.1 × 1015 g C) and rivaling terrestrial soils (2.6 × 1016 g C) (Reference Whitman, Coleman and Wiebe215–Reference McMahon and Parnell217). The uncertainty in these calculations spans orders of magnitude and has not changed significantly since the original estimates by Whitman (Reference Whitman, Coleman and Wiebe215; Chapter 17, this volume), although the trend is downward (Chapter 17, this volume). However, increasing levels of inquiry applied globally using advanced methodologies have identified abundant, taxonomically diverse communities within the continental deep subsurface, giving credence to vast amounts of carbon contained and being cycled by these ecosystems (Reference Fredrickson and Onstott218–Reference Probst, Castelle, Singh, Brown, Anantharaman, Sharon, Hug, Burstein, Emerson, Thomas and Banfield222).
16.5.1 Types of Continental Deep Subsurface Environments
Continental deep subsurface environments can be broadly divided between sedimentary and crystalline host rocks, but even within this framework they range significantly in carbon content, isolation from the surface, and dominant carbon cycling processes (Figure 16.1). The best-studied sites are found in shallow sedimentary and igneous aquifers owing largely to their relevance to human water supplies (Reference Stevens and McKinley223–Reference Murphy, Schramke, Fredrickson, Bledsoe, Francis and Sklarew231). Hydrocarbon reservoirs contain vast quantities of organic carbon and have distinct microbiology associated with their formation waters (Reference Means and Hubbard232–Reference Aitken, Jones and Larter234). A recent emphasis on deep coal beds and their constituent carbon cycling has come to the scientific forefront due to their importance in gas extraction via deep fracking technologies (Reference Strapoc, Mastalerz, Dawson, Macalady, Callaghan and Wawrik235). Deep crystalline bedrock sites feature the oldest, deepest, and most isolated deep biosphere environments (Reference Holland, Lollar, Li, Lacrampe-Couloume, Slater and Ballentine211,Reference Pedersen, Fredrickson and Fletcher236–Reference Lollar, Onstott, Lacrampe-Couloume and Ballentine238). Caves, in contrast, sit at the interface between the surface and the deep and are covered more completely in other reviews (Reference Boston, Spilde, Northup, Melim, Soroka and Kleina239–Reference Barton and Northup241). This section will describe the forms, cycling, and fate of organic carbon in each environment.
While the deep subsurface biosphere is pervasive, it is difficult to access reliably. Common access points are wells, boreholes, mines, and caves. Each approach has the potential to impact in situ processes and must be considered when evaluating data sets. Natural springs are often considered as “portals” or “windows” into the deep biosphere, often showing a mix of surface and subsurface communities (Reference Suzuki, Ishii, Wu, Cheung, Tenney and Wanger242,Reference Magnabosco, Tekere, Lau, Linage, Kuloyo and Erasmus243). The last 10 years has seen the establishment of a number of deep subsurface observatories including into permafrost (Permafrost Tunnel Research Facility, AL, USA), deep crystalline bedrock (Deep Mine Microbial Observatory (DeMMO), SD, USA; Coast Range Ophiolite Microbial Observatory (CrOMO), CA, USA; Äspö Hard Rock Laboratory, Sweden; and many others), and sedimentary aquifers (Deep Biosphere in Terrestrial Systems (DEBITS), New Zealand; Savannah River Site, SC, USA).
16.5.2 Continental Carbon Cycling
Organic carbon in the continental deep biosphere may derive from surficial inputs, in situ autotrophic carbon fixation, water–rock reactions, or ancient sedimentary sources. The relative balance of these sources depends sharply on geology, both by surface connectivity and host lithology. The following sections describe this balance in sedimentary and igneous aquifers, hydrocarbon reservoirs, deep coal beds, and deep crystalline bedrock.
Key processes in subsurface carbon cycling depend on the relative recalcitrance of ancient OM, supply of labile organic carbon, input of metabolic oxidants and reductants, and aquifer porosity and permeability. Microbial carbon fixation produces labile organic carbon and methane, whereas heterotrophic microbial processes consume both labile and recalcitrant subsurface organic carbon. The relative importance of these two end members broadly suggests autotrophic processes dominate in crystalline and deep rock aquifers whereas heterotrophic processes are more abundant within sedimentary systems, although numerous counterexamples exist and both processes (e.g. (Reference Stevens251)) must be active for a functioning ecosystem (Reference Fredrickson and Balkwill225). Organic acids and short-chain hydrocarbons are key microbial products and substrates within most continental subsurface settings with typical concentrations in the 10–100 µM range (Table 16.2). Due to rock dissolution and other processes, DIC can be very high, and may get much higher as aquifers are targeted for anthropogenic carbon sequestration (Reference Probst, Castelle, Singh, Brown, Anantharaman, Sharon, Hug, Burstein, Emerson, Thomas and Banfield222).
Table 16.2 Summary of characteristics and carbon contents of different types of continental subsurface systems.
| System type | Shallow sedimentary aquifers | Shallow igneous aquifers | Hydrocarbon reservoir formation waters | Coal beds | Deep bedrock |
|---|---|---|---|---|---|
| Example systems | Lower Saxony, Germanya | Columbia River flood basaltsb | Palo Duro Basinc | German lignite depositsd | South African gold minese |
| Defining characteristics | Shallow confined and unconfined aquifers with abundant sedimentary OM, fresh waters | Thick basalt deposits with confined aquifers, interbedded sediments, fracture-based porosity, and relatively fresh NaCl-dominated fluids | Sedimentary hydrocarbons interfacing with aqueous brines, organic alkalinity may exceed bicarbonate alkalinity | Extremely organic-rich sediments at varying stages of thermal maturity, limited porosity and permeability | Deep (1.0–3.3 km) fracture-based fluids with thousand- to million-year recharge times |
| Temperatures | Low to moderate | Low to moderate | Moderate to high | Low to moderate | Low to high |
| Recharge timescale | Rapid to moderate | Rapid to moderate | Moderate to long | Moderate to long | Moderate to very long |
| pH | Circumneutral | 7.5–8.5 shallow, 8.0–10.5 deep | 5–8 | 6.8–7.2 | 7.4–9.4 |
| H2 (mM) | – | Up to 0.06 | – | – | 7.4 |
| SO4 (mM) | – | Generally <0.5, but up to 2 | Up to 25 | Up to 0.148 | 0.623 |
| Total DIC (mM) | – | 0.125–2.800 | – | 19.8–43.6 | 0.09–2.40 |
| δ13CCO2 (‰) | –20 | –30 to 20; mostly –10 | – | –14 to 20 | –43 to –5 |
| CH4 (mM) | 0.00089–2.68000 | Up to 160 | Very high | 0.010–0.100 | 0.026–8.800 |
| δ13CCH4 | –110 to 20 (mean –70) | – | Variable | –81 to –71 | –58 to –37 |
| Short-chain hydrocarbons Σ(C2H6–C4H8) (µM) | ~3 (median) | – | High | – | <0.1–201 |
| DOC (mM) | 0.17–0.30 | 0.16–0.39 | 0.05–14.75 | 0.19–0.95 | n.d. to 0.410 |
| Formate | – | – | – | 2.22–31.1 µmol/g sed | 0.44–34.00 µM |
| Acetate | – | – | 6.08 mM | 1.7–8.5 µmol/g sedf | 0.07–28.00 µM |
| Amino acids | – | – | – | – | 0.0133–2.6700 µM |
| Typical cell density (cells/mL) | 103–108 | 103–105 | High | 107 | 10–105 |
Ranges are reported for the example systems, where there are data available. Ranges and qualitative measures are given where there is significant variability reported or differences between reports. “–” is used to indicate no available reports.
sed = sediment.
a (Reference Schloemer, Elbracht, Blumenberg and Illing229, Reference Baker, Valett and Dahm244, Reference Kotelnikova245).
c (Reference Grundger, Jimenez, Thielemann, Straaten, Luders and Richnow228, Reference Means and Hubbard232, Reference Lundegard and Kharaka233).
d (Reference Grundger, Jimenez, Thielemann, Straaten, Luders and Richnow228, Reference Vieth, Mangelsdorf, Sykes and Horsfield247).
16.5.3 Sedimentary and Igneous Aquifers
Both sedimentary (e.g. Atlantic coastal plain) and igneous aquifers (e.g. Columbia River basalt aquifer) have been shown to contain vibrant microbial communities and have been the subject of intensive study due to their economic and social importance as sources of drinking and industrial water, as well as their vulnerability to anthropogenic contamination (Reference Fredrickson and Balkwill225,Reference Lapworth, Gooddy, Butcher and Morris226,Reference Murphy, Schramke, Fredrickson, Bledsoe, Francis and Sklarew231). Recharge timescales of aquifers vary over many orders of magnitude (months to millions of years), controlling the relative supply of exogenous DOC and electron acceptors. In many systems, significant supply of young sedimentary carbon produces relatively high DOC, methane, and organic acid concentrations. The composition of this DOC can be complex, including significant amounts of nitrogen- and sulfur-bearing organic molecules (Reference Longnecker and Kujawinski252). While oligotrophic compared to surface environments, aquifers are relatively carbon rich for the subsurface and can support correspondingly high cell densities (e.g. 105 cells/mL), even in oligotrophic crystalline aquifers (Reference Fry, Fredrickson, Fishbain, Wagner and Stahl246).
Primary productivity within aquifers varies tremendously based on exogenous and sedimentary organic carbon supply, but is significant in some settings. Hydrogen production can be large and may support autotrophic populations, particularly in igneous and ultrabasic host environments, fueling the so-called subsurface lithoautotrophic microbial ecosystems (Reference Stevens and McKinley223,Reference Stevens251,Reference Chapelle, O’Neill, Bradley, Methe, Ciufo and Knobel253). Utilization of iron oxide minerals as terminal electron acceptors for both autotrophic and heterotrophic metabolisms is common, producing high concentrations of dissolved ferrous iron in many groundwaters (Reference Griebler and Lueders254,Reference Flynn, Sanford, Ryu, Bethke, Levine and Ashbolt255). Iron and sulfur oxidative metabolisms are also found where microaerophilic conditions or sufficient nitrate concentrations exist (Reference Griebler and Lueders254,Reference Probst, Ladd, Jarett, Geller-McGrath, Sieber and Emerson256). Sulfate is a less dominant anion in continental settings relative to its ubiquity in the marine realm, but where present, it can fuel significant populations of autotrophic and heterotrophic sulfate-reducing bacteria (Reference Griebler and Lueders254,Reference Flynn, Sanford, Ryu, Bethke, Levine and Ashbolt255).
Heterotrophic processes rely on the input of DOC from the mineralization of sedimentary carbon, aquifer recharge, or in situ microbial activity. In 85% of US aquifers, DOC concentrations were <175 µM (median 42 and 58 µM). These ranges were not significantly different between sedimentary and crystalline aquifers (ranging from 8 to 275 µM, median 42 µM) (Reference Leenheer, Malcolm and McKinley257). A more recent analysis of DOC concentration across UK aquifers showed a range of 15–1550 µM (257 µM average) (Reference Lapworth, Gooddy, Butcher and Morris226), although this sampling includes evidence for significant contamination from agriculture and concomitant microbial respiration that introduced OM. Locally, high concentrations of organic acids (up to 60 µM formate) can be produced by microbial degradation of complex sedimentary OM, particular in shale horizons, which may then diffuse to more porous sediments, driving respiration (Reference McMahon and Chapelle258). For shallow aquifer systems, periodic environmental changes related to seasonal shifts, water table fluctuations, or land use may transport both DOC and oxidants to depth, driving increases in heterotrophic respiration (Reference Baker, Valett and Dahm244,Reference Kusel, Totsche, Trumbore, Lehmann, Steinhauser and Herrmann259).
Methane is a ubiquitous reservoir of organic carbon in sedimentary aquifers. Methane concentrations are extremely variable but sometimes can reach extremely high values (e.g. 0.9 nM to 2.7 mM in the Lower Saxony region of Germany (Reference Schloemer, Elbracht, Blumenberg and Illing229) and 3.1 nM to 293 µM across Great Britain (Reference Bell, Darling, Ward, Basava-Reddi, Halwa and Manamsa230)). Sources of methane vary and include abiotic and biotic sources, including microbial methanogenesis (including hydrogenotrophic, acetoclastic, and methyl fermentation) as well as thermogenic cracking of buried OM (Reference Veto, Futo, Horvath and Szanto224,Reference Schloemer, Elbracht, Blumenberg and Illing229,Reference Bell, Darling, Ward, Basava-Reddi, Halwa and Manamsa230). The isotopic composition of methane and co-occurring short-chain hydrocarbons can be used to assess methane sources and suggest active microbial CO2 reduction as the primary source in both German and British aquifers (Reference Schloemer, Elbracht, Blumenberg and Illing229,Reference Bell, Darling, Ward, Basava-Reddi, Halwa and Manamsa230). High concentrations tend to correlate to organic-rich, low-SO4 geological formations (Reference Schloemer, Elbracht, Blumenberg and Illing229).
16.5.4 Hydrocarbon Reservoirs
Hydrocarbon reservoirs were among the earliest studied continental deep biospheres, with experiments beginning in the 1920s by Colwell and D’Hondt (Reference Colwell and D’Hondt213). These systems are characterized by large accumulations of liquid and gaseous hydrocarbons, providing abundant sources of carbon and electron donors, but they tend to be correspondingly depleted in oxidants and nutrients. Extremely high concentrations of volatile organic acids (particularly acetate) comprise the majority of DOC in the water phases of hydrocarbon-bearing basins, reaching concentrations of up to hundreds of mM (Reference Means and Hubbard232,Reference Head, Jones and Larter260,Reference Nazina, Shestakova, Ivoilov, Kostrukov, Belyaev and Ivanov261).
The most significant metabolisms in hydrocarbon reservoirs are sulfate reduction, methanogenesis, acetogenesis, iron reduction, and fermentation (Reference Head, Jones and Larter260–Reference Magot, Ollivier and Patel262), the balance of which is determined by electron acceptor supply. Spatially, biodegradation of oil is concentrated at the oil–water interface and is limited by reservoir temperature, with limited activity being observed above 80°C (Reference Aitken, Jones and Larter234,Reference Larter, Huan, Adams, Bennett, Jokanola and Oldenburg263). Anthropogenic influence through drilling, water introduction, casing, and fracturing of reservoirs and the introduction of exogenous microbes can significantly change in situ carbon cycling, most notoriously causing reservoir souring by stimulating sulfate-reducing populations in previously methanogenic reservoirs. For more a complete description of carbon cycling and biodegradation in hydrocarbon reservoirs, see the reviews by Larter et al. (Reference Larter, Huan, Adams, Bennett, Jokanola and Oldenburg263), Means and Hubbard (Reference Means and Hubbard232), and Head et al. (Reference Head, Jones and Larter260).
16.5.5 Deep Coal Beds
Coal is formed through the burial and diagenesis of large accumulations of terrestrial plant matter and therefore contains an extremely high organic carbon content. The bioavailablity of this OM to deep subsurface microbes depends on thermal maturity and burial history, which control the form and speciation of OM as well as the sterilization history of resident microbial populations (Reference Strapoc, Mastalerz, Dawson, Macalady, Callaghan and Wawrik235). Low-maturity (rank) coals are the most bioavailable and actively accumulate biogenic methane. Aqueous extracts of low-maturity coals and lignites produce extremely high concentrations of organic acids, including acetate, formate, and oxalate in the range of 0.37–2.5.0 mg/g sediment (Reference Vieth, Mangelsdorf, Sykes and Horsfield247,Reference Zhu, Vieth-Hillebrand, Wilke and Horsfield264). Yields of labile OM decrease significantly with increasing thermal maturity (Reference Vieth, Mangelsdorf, Sykes and Horsfield247,Reference Zhu, Vieth-Hillebrand, Wilke and Horsfield264).
Biogenic methane appears to universally accumulate in coals at <80°C (Reference Strapoc, Mastalerz, Dawson, Macalady, Callaghan and Wawrik235). Microbial processing of coal to methane is a multistep process that requires and supports an ecosystem of microbes. First, organic polymers are fragmented into hydrocarbon intermediates, followed by a secondary fermentation to methanogenic substrates like CO2, H2, organic acids, and alcohols. These substrates then fuel acetoclastic, methylotrophic, and hydrogenotrophic methanogens (Reference Strapoc, Mastalerz, Dawson, Macalady, Callaghan and Wawrik235). The rate and efficiency of these processes in different coal deposits and the accessibility of this methane for extraction are of considerable economic importance. For a more complete review of coal bed biogeochemistry, see Strapoc et al. (Reference Strapoc, Mastalerz, Dawson, Macalady, Callaghan and Wawrik235).
16.5.6 Deep Bedrock
Deep crystalline bedrock-hosted biospheres stand in contrast to the aforementioned settings in their constituent reservoirs and fluxes of carbon and energy. Here, inputs from the surface are limited, with water residence times reaching millions to billions of years (e.g. (Reference Holland, Lollar, Li, Lacrampe-Couloume, Slater and Ballentine211,Reference Onstott, Lin, Davidson, Mislowack, Borcsik and Hall265)) and sedimentary carbon (where present) is recalcitrant to graphitic carbon (Reference Kietavainen, Ahonen, Niinikoski, Nykanen and Kukkonen266). The largest pool of organic carbon is often as methane, although considerable variability is present with depth and lithology (Reference Simkus, Slater, Lollar, Wilkie, Kieft and Magnabosco248,Reference Kietavainen, Ahonen, Niinikoski, Nykanen and Kukkonen266). Porosity and permeability is fracture based, adding a stochastic temporal dynamic to fluxes and mixing (Reference Onstott, Lin, Davidson, Mislowack, Borcsik and Hall265,Reference Lollar, Voglesonger, Lin, Lacrampe-Couloume, Telling and Abrajano267).
Hydrogen, methane, sulfate, and iron cycling drive primary production in deep crystalline bedrock settings. The relative importance of these processes is variable with depth, host lithology, and fluid chemistry (Reference Osburn, LaRowe, Momper and Amend221,Reference Simkus, Slater, Lollar, Wilkie, Kieft and Magnabosco248). Precambrian rocks, which constitute the best-studied deep crystalline biospheres, are prolific producers of hydrogen (up to mM concentrations) (Reference Lollar, Onstott, Lacrampe-Couloume and Ballentine238,Reference Lollar, Voglesonger, Lin, Lacrampe-Couloume, Telling and Abrajano267), which can serve as the terminal electron donor for either sulfate reduction or CO2 reduction-based metabolisms (Reference Lau, Kieft, Kuloyo, Linage-Alvarez, Van Heerden and Lindsay249,Reference Onstott, Lin, Davidson, Mislowack, Borcsik and Hall265,Reference Suzuki, Konno, Fukuda, Komatsu, Hirota and Watanabe268–Reference Magnabosco, Ryan, Lau, Kuloyo, Lollar and Kieft272).
Analysis of subsurface genomes shows that enzymes of hydrogen metabolism are overrepresented, emphasizing the potential dominance of this metabolic strategy (Reference Colman, Poudel, Stamps, Boyd and Spear273). Metagenomic surveys suggest that carbon fixation is performed primarily using the reductive acetyl CoA pathway (Reference Probst, Ladd, Jarett, Geller-McGrath, Sieber and Emerson256,Reference Momper, Jungbluth, Lee and Amend271,Reference Magnabosco, Ryan, Lau, Kuloyo, Lollar and Kieft272). Extreme metabolic flexibility has been observed in the cosmopolitan subsurface dweller Candidatus Desulforudis audaxviator, which can grow in near monoculture in isolated fracture systems (Reference Chivian, Brodie, Alm, Culley, Dehal and DeSantis220) and has been found globally (Reference Osburn, LaRowe, Momper and Amend221,Reference Jungbluth, del Rio, Tringe, Stepanauskas and Rappe274).
Heterotrophic microbes and metabolisms have been found to dominate in some deep crystalline settings, despite the apparent limited availability of exogenous carbon. Abiogenic sources of methane in addition to limited populations of microbial methanogens supply a significant flux of methane to fuel methanotrophic communities reaching tens of mM concentrations (Reference Simkus, Slater, Lollar, Wilkie, Kieft and Magnabosco248,Reference Kietavainen, Ahonen, Niinikoski, Nykanen and Kukkonen266,Reference Lollar, Lacrampe-Couloume, Slater, Ward, Moser and Gihring275,Reference Kadnikov, Frank, Mardanov, Beletskii, Ivasenko and Pimenov276). Methane cycling has been observed to be most active at moderate depths (0.5–1.5 km) rather than in the deepest, most isolated settings (Reference Simkus, Slater, Lollar, Wilkie, Kieft and Magnabosco248,Reference Kietavainen, Ahonen, Niinikoski, Nykanen and Kukkonen266,Reference Momper, Reese, Zinke, Wanger, Osburn and Moser277). Other sources of carbon for heterotrophic communities include biofilm-based small organic compounds (Reference Onstott, Lin, Davidson, Mislowack, Borcsik and Hall265,Reference Wanger, Southam and Onstott278), free organic acids formed through fermentation or abiogenesis, and ancient organic carbon (Reference Purkamo, Bomberg, Nyyssonen, Kukkonen, Ahonen and Itavaara279).
Mineral and biofilm-based metabolisms may be particularly important in deep crystalline settings. Increasing evidence of extensive adaptation to life in biofilms is emerging from these environments in the form of physical adaptations like grappling appendages observed in putative Candidatus “Altiarchaeum” (Reference Probst, Ladd, Jarett, Geller-McGrath, Sieber and Emerson256), as well extracellular electron transport in subsurface isolates (Reference Jangir, French, Momper, Moser, Amend and El-Naggar280). In high-pH settings, autotrophic populations may depend on solid carbonate minerals due to carbon speciation in ultrabasic environments (Reference Suzuki, Kuenen, Schipper, van der Velde, Ishii and Wu269). Differences between the attached and planktonic communities have long been observed in crystalline aquifer settings (Reference Osburn, LaRowe, Momper and Amend221,Reference Momper, Reese, Zinke, Wanger, Osburn and Moser277,Reference Lehman, Colwell and Bala281), often with orders of magnitude higher cell densities present within the biofilms (Reference Wanger, Southam and Onstott278). The net suggestion of these observations is that mineral and biofilm-based lifestyles are the norm for the deep continental subsurface, but are as yet undersampled. Efforts to cultivate and characterize the metabolic capacities of these attached communities are underway.
16.6 Conclusion
16.6.1 Broad Similarities across Systems
The deep biosphere spans an incredible range of physical and chemical conditions. Despite their heterogeneity, some broad similarities are present across systems. Organic carbon concentrations reach their highest levels in regions that have large inputs from primary producers, either presently (continental margins, shallow sedimentary aquifers, diffuse hydrothermal vents) or in the past (hydrocarbon reservoirs, coal beds). In contrast, concentrations are lower in rocky areas with little sedimentary input and low amounts of chemolithoautotrophy in both continental (shallow igneous aquifers, deep bedrock) and marine (ridge flanks, high-temperature hydrothermal vents) systems. Elevated concentrations of methane are related to the anaerobic breakdown of OM and associated methanogenesis (sedimentary porewaters, sedimented hydrothermal systems, hydrocarbon reservoirs, coal beds), but also due to hydrogenotrophic methanogenesis, with the hydrogen supplied by water–rock reactions (basalts) and from mantle inputs (hydrothermal systems). Somewhat surprising is the persistence of some forms of organic carbon that are generally thought to be readily accessible to microorganisms, such as acetate, in many systems (sedimentary porewaters, shallow igneous aquifers, deep bedrock).
16.6.2 Limits to Knowledge and Unknowns
(1) Exchange/transformation of carbon between aqueous and solid phases. A characteristic of subsurface environments is the ubiquitous presence of a solid phase, be it from surface-derived particles or crystalline rocks. The exchange and transformation of carbon between the aqueous and solid phases is therefore a major mechanism for controlling the form and fate of carbon in the subsurface. Major questions remain as to what controls these exchanges and the degree to which they are catalyzed by minerals and microorganisms.
(2) Bioavailability of organic carbon. Microbial respiration has been invoked to account for the oxidation of OM that is millions of years old in sediments (Reference Røy67) and the removal of oceanic dissolved OM that is thousands of years old in the basaltic basement (Reference Walter, Jaekel, Osterholz, Fisher, Huber and Pearson135). Even 365 million-year-old shale carbon can be incorporated into cellular biomass given the right conditions (Reference Petsch, Eglinton and Edwards282). These studies raise the intriguing question of whether all OM is, ultimately, bioavailable give enough time and a favorable setting, or whether there is some pool that will resist remineralization to CO2 under all circumstances. This question ties directly into point (3) below.
(3) Controls on reaction rates of biogeochemical processes. The rate at which carbon is transformed or remineralized is fundamentally important to understanding the short- and long-term controls on the global carbon cycle and to identifying the distribution of subsurface life. The processes occurring in the marine and continental subsurface are inherently difficult to accurately mimic in laboratory experiments. While short-term experiments can address the more reactive portions of the organic pool, our understanding of the transformations that occur over century or millennium timescales, particularly when uncultured microorganisms mediate the reactions, is more challenging but no less important.
(4) Predictive ability. This review describes what types of carbon are present in distinct geological, geochemical, and biological environments. Ultimately, however, the reverse is a major goal: the ability to have such a fundamental grasp of the mechanistic controls on carbon cycling that it is possible to accurately predict what types and abundances of carbon will be present in a given system.
(5) Characterization of OM. Despite decades of effort and major progress on several fronts, the molecular structure of the vast majority of OM in the subsurface remains uncharacterized. This gap in our knowledge will continue to inhibit our understanding of carbon biogeochemical cycling in the subsurface.
Acknowledgments
Funding support for SQL is from C-DEBI (NSF grant OIA-0939564) and National Science Foundation grant OCE-1536702. Funding support for ADS is from C-DEBI and NSF OCE-1357242. Funding support for MRO is from NASA Exobiology (NNH14ZDA001N) and NAI Life Underground (NNH12ZDA002C). This is C-DEBI contribution 475.
Questions for the Classroom
1 Continental and oceanic subsurface crystalline aquifers are similar in many ways. How do the characteristics of carbon in, for example, the oceanic North Pond and the continental South African gold mine systems compare and differ? Why?
2 Are the sites studied thus far representative of globally relevant locations where carbon is processed in the subsurface? What locations or geological systems are missing? Why have these not yet been studied? What are the prospects for studying these locations?
3 What effect are humans having on carbon in the deep biosphere?
4 What are the next steps to improve our ability to computationally model different forms of carbon in the subsurface and how they change in the subsurface?
5 Imagine a hypothetical microorganism that is capable of remineralizing any type of nonbioavailable OM back to CO2. If this microorganism proliferated in subsurface environments, what would the effect be?
6 Ultraviolet radiation can create radical species (compounds that are highly reactive due to the presence of an unpaired electron) that can oxidize organic molecules via random reactions. What is the likely effect of a change in atmospheric ozone concentrations, and therefore ultraviolet flux to Earth’s surface, on organic carbon burial rates?








 Open access
Open access