
5 results
Atomic-scale deformation mechanisms at high-pressure in inderborite, CaMg[B3O3(OH)5]2(H2O)4⋅2H2O
-
- Journal:
- Mineralogical Magazine / Volume 88 / Issue 4 / August 2024
- Published online by Cambridge University Press:
- 22 May 2024, pp. 473-481
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Structures of Intergrown Triclinic and Monoclinic IIb Chlorites from Kenya
-
- Journal:
- Clays and Clay Minerals / Volume 37 / Issue 4 / August 1989
- Published online by Cambridge University Press:
- 02 April 2024, pp. 308-316
-
- Article
-
- You have access
- Export citation
Polytypism of Micas. III. X-Ray Diffraction Identification
-
- Journal:
- Clays and Clay Minerals / Volume 34 / Issue 1 / February 1986
- Published online by Cambridge University Press:
- 02 April 2024, pp. 53-68
-
- Article
-
- You have access
- Export citation
Al-Saturated Phlogopite: Charge Considerations and Crystal Chemistry
-
- Journal:
- Clays and Clay Minerals / Volume 57 / Issue 6 / December 2009
- Published online by Cambridge University Press:
- 01 January 2024, pp. 673-685
-
- Article
-
- You have access
- Export citation
-
Factors controlling the crystal structure of phlogopite have been widely investigated; but the role of electrostatic interactions, for example, has received much less attention than other factors. The purpose of the present study was to peform a single-crystal refinement of an Al-saturated phlogopite and to use that refinement to supplement crystal-chemical analyses. The phlogopite investigated was from the Rumford quadrangle, Maine, and has the following chemistry: \$\end{document}
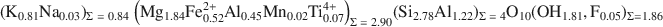 . The sample is a 1M polytype with C2/m symmetry and cell dimensions of a = 5.3220(4), b = 9.2170(7), c = 10.2511(8) Å, and β = 100.081(1)°. Hydrogen atoms were located and the crystal structure was refined to give parameters R1 = 0.0301 and weighted R2 = 0.0887. The octahedral M1 site was larger than the M2 (average M1—O: 2.079 Å, average M2—O: 2.062 Å) and the electron counts were equal (M1 = M2 = 14.8 e−); based on bond distances, which are more accurate than electron counts in determining occupancy; this result is consistent with a slight preference of Mg for M2 and Fe2+ for M1.
. The sample is a 1M polytype with C2/m symmetry and cell dimensions of a = 5.3220(4), b = 9.2170(7), c = 10.2511(8) Å, and β = 100.081(1)°. Hydrogen atoms were located and the crystal structure was refined to give parameters R1 = 0.0301 and weighted R2 = 0.0887. The octahedral M1 site was larger than the M2 (average M1—O: 2.079 Å, average M2—O: 2.062 Å) and the electron counts were equal (M1 = M2 = 14.8 e−); based on bond distances, which are more accurate than electron counts in determining occupancy; this result is consistent with a slight preference of Mg for M2 and Fe2+ for M1.Thirty-five Al-rich, natural phlogopite-1M samples that are of (1) high metamorphic grade, and that have (2) total Al contents ⩾ 1.27 atoms per formula unit (a.p.f.u.), (3) Fe3+ contents ⩽ 0.11 a.p.f.u., and (4) Mn contents ⩽0.10 a.p.f.u. along with the newly described phlogopite, exhibited crystal chemical trends related to increasing Al content. Octahedral substitutions of smaller, high-charge cations (i.e. Al) apparently decrease distortions in the octahedral sites and produce longer M2—O4 distances. In addition, VIFe-F avoidance apparently occurs in high Al-content samples, which are generally high in VIFe. The data set also shows that these samples have limited ordering among M sites (Fe2+ in M1 and Al in M2), an increase in β (99.96° to 100.32°) possibly caused by cation ordering and therefore size differences of M1 and M2, and interlayer (A) sites with A—Oouter distances that increase and A—Oinner distances that decrease with increasing Ti content.
Computer models were used to simulate electrostatic interactions in phlogopite structures with variable Al concentrations utilizing Pauling’s electrostatic valency principle, which considers first-coordination electrostatic interactions. The model results were compared to the maximum Al concentrations in natural and synthetic phlogopite samples. Model results revealed no indications (e.g. a limit reached or a sudden change occurred) that charge saturation/undersaturation of the apical oxygen atoms at Al contents equal to the maximum in natural and/or synthetic samples causes instability that could not be balanced by bond-length variation. However, a cation of higher charge substituting at M1 (or M2) may result in higher electrostatic repulsions between the other octahedral sites. Thus, the Al3+ content in the octahedral sites may reach a maximum, with Fe2+ for Mg substitutions favored.
Redefinition of thalénite-(Y) and discreditation of fluorthalénite-(Y): A re-investigation of type material from the Österby pegmatite, Dalarna, Sweden, and from additional localities
-
- Journal:
- Mineralogical Magazine / Volume 79 / Issue 4 / August 2015
- Published online by Cambridge University Press:
- 02 January 2018, pp. 965-983
-
- Article
- Export citation
