


Glycogen storage diseases (GSDs) are inherited disorders due to deficiency of enzymes involved in glycogen metabolism.Reference Ozen1 There are over 12 types of GSDs classified based on the specific enzyme deficiency and affected tissues.Reference Ozen1 GSD type IXa (MIM 306000) is an X-linked condition caused by an enzymatic defect in phosphorylase kinase (PhK) and associated with pathogenic variants in the gene PHKA2.Reference Beauchamp, Taybert and Champion2,Reference Lau, Hui and Fong3
GSD type IXa is characterized by early childhood onset of hepatomegaly and growth retardation. The disease severity is usually mild, and the prognosis is favorable in the vast majority of cases, as both clinical and biochemical abnormalities tend to improve or disappear with age. Neurological manifestations are exceptionally rare.Reference Hodax, Uysal, Quintos and Phornphutkul4 Although brain magnetic resonance imaging (MRI) abnormalities affecting both the gray and white matter have been documented in several other GSDs,Reference Ozen1 no cerebral or cerebellar findings have been described in patients with GSD type IX to our knowledge.
We combined whole-exome sequencing (WES) and MRI data to uncover a PHKA2 mutation that leads to a complex GSD type IXa phenotype accompanied with cerebellar involvement.
All participants signed an informed consent form, including consent to the publication of clinical, genetic, and MRI findings, approved by the institutional ethics committee of the Montreal Neurological Institute.
Genomic DNA was extracted from peripheral blood cells using standard methods. Exome sequencing was performed on the genomic DNA of individual II:1 and II:2 using the Sure Select exome capture kit v.5 (Agilent Technologies, Santa Clara, CA, USA). The obtained libraries were sequenced on an Illumina HiSeq 2000 (Illumina, San Diego, CA, USA) with paired-end 100-bp reads at the McGill University and Genome Quebec Innovation Center (Montreal, Quebec, Canada). Sequences were aligned to the human reference genome (UCSC hg19) using the Burrows–Wheeler Aligner algorithm, variant calling was performed using sequence alignment/map tools, and annotation was done using ANNOVAR and custom strips, as previously described.Reference Tetreault, Fahiminiya and Antonicka5 For Sanger sequencing, polymerase chain reaction (PCR) was used to amplify exon 15 of PHKA2 (NM_000292). The PCR products were sequenced at McGill University and Genome Quebec Innovation Center (Montreal, Quebec, Canada) using a 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Mutation detection analysis was performed using SeqMan v.4.03 (DNASTAR Inc., Madison, WI, USA). Enzymatic PHKA2 function was performed on blood at the MUHC Clinical Laboratory, Montreal, Canada, on a clinical basis.
Imaging studies were performed on a 1.5 Tesla MRI scanner at the Montreal Neurological Institute following standard clinical protocol.
An overview of the medical history of patient 1 and patient 2 is presented in Table 1. Patient 1 (Figure 1, subject II:1 and Figure 2) is a 37-year-old French Canadian male, the first son of non-consanguineous parents. He was born at term after an uneventful pregnancy. At the age of 6 months, during a viral infection complicated by encephalopathy, the patient was discovered to have hepatosplenomegaly, osteoporosis, sideropenic anemia, and global psychomotor development delay. At that time, extensive laboratory testing was performed including: beta-galactocerebrosidase, beta-glucosidase, exosaminidase, neuraminidase, alpha-fucosidase, beta-galactosidase, mannosidase, sphingomyelinase, very long chain fatty acids, and plasmatic catecholamine. Liver and bone marrow biopsies were performed. The hepatosplenomegaly remained unexplained and was documented to regress over time. He developed pharmaco-sensitive epilepsy in childhood with last clinical seizure before the age of 5. We have no documentation, and the mother did not recall the antiepileptic drugs (AEDs) used nor the time of exposure. A bilateral sensorineural hearing loss was first documented at age 12.
Table 1: Outline of the clinical and paraclinical findings of patients 1 and 2
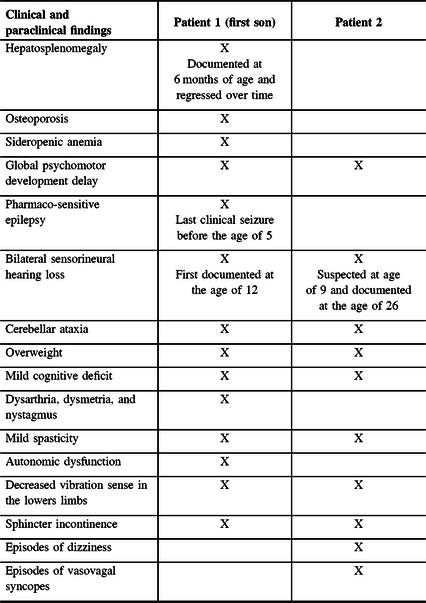

Figure 1: Pedigree of the family and chromatograms showing the PHKA2 variant present in both affected siblings.

Figure 2: Chronological appearance of symptoms and signs in patients 1 and 2.
He came to our attention at the age of 28 for slowly progressive cerebellar ataxia. We performed extensive investigations: repeat expansion and gene testing for known forms of recessive and dominant cerebellar ataxias, isoelettrofocusing of sialotransferrin, sequencing of mtDNA, and comparative genomic hybridization array. At age 37, his examination showed significant overweight, cerebellar features (ataxia, dysarthria, dysmetria, and nystagmus), mild spasticity, autonomic dysfunction, decreased vibration sense in the lower limbs, and a mild cognitive deficit. He has had progressive sphincter incontinence.
The younger brother, patient 2 (Figure 1, subject II:2 and Figure 2), presented a similar clinical history. The patient had a few episodes of uncomplicated febrile seizures. He never received any antiepileptic medications. In addition, he has had episodes of dizziness mainly related to change in position and a few presumed episodes of vasovagal syncopes. He has had a sensorineural hearing deficit since the age of 26. He has had sphincter incontinence for the past few years. On examination at age 34, the neurological examination overlaps his brother’s, with moderate overweight, mild cognitive deficit, mild spasticity, cerebellar ataxia, and decreased vibration sense in the lower extremities.
The mother of the two siblings (subject I:2) was known to have a neurosensory hearing deficit as well, discovered in adult age; her cognitive function and neurological exam were normal. The father (I:1) died in 2011 of pharyngeal cancer but reportedly did not present any neurological symptom. The family history is otherwise negative for neurological disorders.
The brain MRI documented shared abnormalities in patients 1 and 2 (Figure 3). Both brothers present cerebellar and vermian atrophy, specifically of the superior vermis. Mild, non-specific white matter T2 hyperintensities are also evident in the posterior periventricular regions.

Figure 3: Brain MRI findings in the two affected siblings. Sagittal fluid attenuated inversion recovery T2-weighted image (A) in Subject II.2 and sagittal T2-weighted image (B) in subject II.1 showing superior vermian atrophy and enlargement of the fourth ventricle. Of note, the brainstem and corpus callosum are normal in size and shape. Axial T2-weighted image (C) in subject II.1 demonstrating mild white matter hyperintense signal in the centrum semiovale bilaterally.
WES was performed on patients 1 and 2. We first searched for variants in genes known to be associated with cerebellar ataxias and/or cognitive deficit but did not identify potential causative variants. Then, based on a predicated recessive or X-linked mode of transmission, we looked for homozygous, non-synonymous, splicing, and coding indel variants with a minor allele frequency <5% in 1000 Genomes, EVS, and ExAC browsers. We also applied a pathogenicity filter using sorting intolerant from tolerant (SIFT), PolyPhen-2, and combined annotation dependent depletion (CADD).Reference Kumar Prateek6,Reference Kircher, Witten, Jain, O’Roak, Cooper and Shendure7 A single variant (c. 561G>A; p. Thr521Ala; rs137958637) in the gene PHKA2 (NM_000292) located on the X chromosome segregated with the disease in the family. The variant is predicted to be pathogenic by SIFT and Mutation Taster, probably damaging by PolyPhen-2 and highly deleterious by CADD (c-score = 27). It is reported at a frequency of 0.000284 by EVS and is absent in 1000 Genomes. It is present at a highly conserved position by genomic evolutionary rate profiling. Exon 15 is part of the transcript encoding an alpha subunit of phosphorylase kinase.
At age 37 and 34, respectively, both patient 1 and patient 2 had a normal level of blood PhK activity, respectively, 0.9 and 0.6 µmol/min/gram (reference >0.1). These results do not exclude liver PhK deficiency as they are reported to normalize over the years.Reference Rodriguez-Jimenez, Santos-Simarro and Campos-Barros8
We recruited two French Canadian brothers with identical infancy-onset neurological and systemic presentations sharing the same PHKA2 variant (c. 561G>A; p. Thr521Ala). There was a delay of more than 30 years in the diagnosis of GSD IX. The long delay in diagnosis is not unexpected in adult GSD patients, since hepatosplenomegaly and laboratory abnormalities often regress with age. We presume that the delay was partly related to the fact that the patients presented in infancy at a time when PhK activity testing was not as readily available. In addition, our patients presented a more complex clinical picture than the more classical hepatic GSD type IXa phenotype. Specifically, the presence of developmental delay and finally cognitive deficit is very uncommon,Reference Beauchamp, Taybert and Champion2 and cerebellar features have never been reported before.
Our description of vermian atrophy linked to GSD type IX is the first reported to our knowledge. Mild brain abnormalities are not unusual in GSD, especially in GSD type II, more commonly in the form of secondary effects of prolonged hypoglycemia or white matter involvement.Reference Chien9 Interestingly, long-term MRI follow-up enabled the identification of central nervous system involvement in adult subjects.Reference Ebbink Berendine10 However, the GSD type IXa and cerebellar ataxia may be unrelated conditions and the ataxia may have a distinct etiology. Alcohol-related cerebellar degeneration can be excluded since the brothers denied any alcohol consumption, either in the past or present time. Furthermore, the cerebellar atrophy is unlikely related to long-term use of AEDs because of the presence of it in the younger brother who never received AEDs and showed cerebellar signs since the age of 9 years. Other causes, such as Q10 coenzyme deficiencyReference Alcázar-Fabra, Trevisson and Brea-Calvo11 and vitamin E deficiency,Reference Schuelke12 have been ruled out in our cases but need to be taken into account in the clinical work-up of patients with ataxia, since these are treatable conditions and patients may benefit from specific therapies. Finally, although we cannot completely exclude that another mutated gene might be responsible for the neurological presentation, our report underlines that long-term follow-up of adult GSD type IXa patients will lead to a better characterization of its evolution over time.
Acknowledgments
We thank the patients who accepted to participate in this study.
Funding
This work was supported by the Groupe Monaco and the Care4Rare Canada Consortium.
Conflict of Interest
The authors have no conflicts of interest to declare.
Statement of Authorship
CS contributed to the data collection, data interpretation, and drafted the manuscript. M-JD contributed to the data analysis. BB contributed to the study concept and design, data interpretation, and revised the manuscript. RLP contributed to the study concept and design, data collection and interpretation, and revised the manuscript.




