Enteric infections can result in high incidence of morbidity and mortality in humans and animals(Reference Dubreuil, Isaacson and Schifferli1). Enterotoxigenic Escherichia coli (ETEC) is one of leading bacterial causes of intestinal inflammation and diarrhoeal illness(Reference McLamb, Gibson and Overman2,Reference Sheikh, Tumala and Vickers3) . ETEC colonises the intestine cells and then produces enterotoxins that trigger systemic or local immune system, resulting in excessive release of pro-inflammatory cytokines, which leads to a breakdown of intestinal integrity and epithelial function(Reference Sheikh, Tumala and Vickers3). Overproduction of proinflammatory cytokines also leads to cell death or tissue damage and conversely cell death can exacerbate inflammatory response(Reference León, Garrote and Arranz4,Reference Kono and Rock5) .
Long-chain PUFA play critical role in neonatal growth and development. Long-chain PUFA include n-3 PUFA rich in deep sea fish oil and n-6 PUFA rich in vegetable oil. Eicosapentaenoic acid (EPA) (20:5 (n-3)), DHA (22:6 (n-3)) and arachidonic acid (ARA) (20:4 (n-6)) are representative members of the two families. EPA and DHA can be synthesised from linolenic acid, and ARA can be synthesised from linoleic acid. Research has shown n-3 PUFA improved intestinal health or limited intestinal inflammation and reduced intestinal damage in many animal models and clinical trials(Reference López-Pedrosa, Ramirez and Torres6–Reference Xiao, Liu and Qin9). In addition, EPA and DHA have been approved by the FDA for supplementation into infant formulas because of the essential role in fetal intelligence and vision development(Reference Flock, Harris and Kris-Etherton10). Generally, it is regarded that n-3 PUFA is ‘beneficial’ as anti-inflammatory and n-6 PUFA is ‘harmful’ as proinflammatory in many previous research. However, some studies have shown that n-6 PUFA, especially ARA as well as its metabolites, could improve recovery of damaged intestinal mucosa(Reference Ruthig and Meckling-Gill11–Reference Jacobi, Moeser and Corl13). However, the precise mechanisms of PUFA, especially n-6 PUFA in modulating intestinal damage and barrier functions, remain largely unknown.
The occurrence of intestinal diseases is closely related to the cell death. Emerging evidence points to a crucial role of pyroptosis and necroptosis as important modes of programmed cell death in intestinal diseases(Reference Günther, Neumann and Neurath14,Reference Chen, He and Hu15) . Pyroptosis and necroptosis are pro-inflammatory, leading to the spread of inflammation(Reference Patankar and Becker16). Necroptosis, a newly established type of cell death, combines the features of apoptosis and necrosis and is mainly mediated by receptor-interacting protein kinase (RIP) 1, RIP3 and mixed lineage kinase-like protein (MLKL)(Reference Ahn and Prince17). Pyroptosis is mediated by the activation of inflammasomes and gasdermin D (GSDMD)(Reference Shi, Gao and Shao18). Recently, pyroptosis and necroptosis have been shown to play an important role in intestinal injury caused by multiple factors, such as ischaemia reperfusion and inflammation(Reference Pierdomenico, Negroni and Stronati19–Reference Zeng, Duan and Hu22). Until now, there is little research about the effects of long-chain PUFA on pyroptosis and necroptosis signalling pathways. Previous studies have reported that PUFA (such as fish oil and flaxseed oil) could prevent intestinal inflammation and protected intestinal health by toll-like receptors 4 (TLR4) and NOD signalling pathways(Reference Zhu, Wang and Wang7,Reference Liu, Chen and Odle8) ; however, the molecular mechanism was still little known.
Therefore, the goal of this study was to investigate whether EPA or ARA, two representative long-chain n-3 or n-6 PUFA, could alleviate cell inflammation and injury through inhibiting pyroptosis and necroptosis signalling pathways. In the current experiment, enterotoxigenic Escherichia coli (ETEC) K88, the most prevalent ETEC strain implicated in newborn and post weaning diarrhea in piglets, was used to establish a model of cell injury. The intestinal porcine epithelial cell line (IPEC-1), which is highly susceptible to ETEC challenge, was employed to elaborate the effects and underlying mechanisms of EPA and ARA(Reference McLamb, Gibson and Overman2).
Materials and methods
Cell culture
The IPEC-1 cell line was derived from small intestine of a neonatal piglet, which was from Dr. Guoyao Wu’s laboratory at Texas A&M University. The cells were cultured according to the standard protocol as our previous study described(Reference Xiao, Liu and Qin9). EPA (C20:5n-3) or ARA (C20:4n-6) was purchased from Sigma Chemical, USA.
Bacterial strains
ETEC K88 was obtained from the China Veterinary Culture Collection Center and grown in Luria-Bertani (LB) medium (Oxoid, UK). After overnight incubation at 37°C with vigorous shaking, bacteria were diluted 1:100 in fresh LB and grown for about 12 h until reaching mid-log phase for all experiments.
Bacterial adhesion assay
IPEC-1 cells were seeded onto six-well plates (Corning, USA) at a density of 1 × 105 cells/ml and incubated with 12·5 µg/ml EPA (38 µmol/l EPA) or 3·125 µg/ml ARA (10 µmol/l ARA) for 24 h and then treated with PBS or 1 × 108 ETEC K88/ml for 3 h. We chose the bacterial concentration and time points of incubation based on preliminary experiments. The amount of EPA and ARA added were determined according to Willemsen et al. (Reference Willemsen, Koetsier and Balvers23) and Xiao et al.(Reference Xiao, Liu and Qin9) and our preliminary research.
The bacterial adhesion to the epithelial cells to detected on 1, 2 and 3 h post ETEC K88 stimulation. After removing the bacteria that was not adhering to the cells, cells in cultures were washed with PBS, lysed and homogenised with 0·1 % Triton X-100 (Sigma-Aldrich, USA) in PBS and then plated on LB agar after serial dilution. Plates were incubated overnight at 37°C, after which the number of colony-forming units was counted.
In vitro antibacterial activity
Agarose diffusion method was used to detect the in vitro antibacterial effects of EPA and ARA on ETEC K88. After inoculating ETEC K88 into LB medium containing 1 % agar for 1 d, the medium was poured into the culture dish to solidify. Then the oxford cup was taken out, and 38 µmol/l EPA or10 µmol/l ARA or PBS were added into the holes to incubate for 24 h at 37°C.
ELISA assay
IPEC-1 cells were seeded onto 6-well plates at a density of 1 × 105 cells/ml and incubated with 0, 38 µmol/l EPA or 10 µmol/l ARA for 24 h and then treated with PBS or 1 × 108 ETEC K88/ml for 2 h. The concentrations of TNF-α, IL-6, IL-8, endotoxin and HMGB1 in supernatants were determined using commercially available ELISA kits (4A Biotech, China) according to the protocols.
Measurement of cell barrier function
IPEC-1 cells were cultured in the permeable transwell inserts (Corning, USA) at a density of 1 × 105 cells/ml. After becoming confluent and polarised, cells were treated with 0, 38 µmol/l EPA or 10 µmol/l ARA A for 24 h, and then stimulated with PBS or 1 × 108 ETEC K88/ml for 3 h. Transepithelial electrical resistance (TEER) was determined every 24 h as previously described by Xiao et al.(Reference Xiao, Liu and Qin9).
Paracellular permeability was also determined as we previously described(Reference Xiao, Liu and Qin9). The flux of fluorescein isothiocyanate-labelled dextran (FD4) was determined every 12 h after ETEC K88 stimulation. The calculation of FD4 flux was according to our previous study(Reference Xiao, Liu and Qin9).
Confocal immunofluorescence microscopy
Cells were seeded on glass coverslips (Corning, USA) at a density of 1 × 105 cells/ml and cultured with 0, 38 µmol/l EPA or 10 µmol/l ARA for 24 h, and then treated with PBS or 1 × 108 ETEC K88/ml for another 2 h. After fixed and permeabilised, cells were blocked and then treated with primary antibodies including anti-claudin-1 antibody (Invitrogen, USA), occludin (Abcam, UK) and ZO-1 (Biorbyt, UK) according to our previous study described(Reference Xiao, Liu and Qin9). After adding the secondary antibody (Invitrogen, USA) and counterstaining with 4,6-diamidino-2-phenylindole (Sigma-Aldrich, USA), cells were mounted on the confocal laser scanning microscope (Olympus FV101, Japan) for observation.
IncuCyte ZOOM™ assay
IPEC-1 cells were seeded in 24-well plates at a density of 1 × 105 cells/ml and were cultured with 0, 38 µmol/l EPA or 10 µmol/l ARA in the presence or absence of 1 × 108 ETEC K88/ml. The cells were treated in the CO2 incubator of IncuCyte ZOOM™ Live Cell Imaging System (Essen BioScience, USA) for 36 h. This machine can automatically monitor the cell growth and necrosis in real time. The necrosis cells were dyed with red colour by yoyo-3 (Sigma, USA), which was a nucleic acid stain dye. The image and data were analysed by the IncuCyte S3 software (Essen Bioscience, USA).
Quantitative real-time PCR
IPEC-1 cells were seeded into 12-well plates at a density of 1 × 105 cells/ml and incubated with 0, 38 µmol EPA/l or 10 µmol/l ARA for 24 h, and then treated with PBS or 1 × 108 ETEC K88/ml for another 2 h. Then total RNA was extracted using the RNAiso Plus Kit (TaKaRa Biotechnology, China) according to the manufacturer’s guidelines. After purification and quantitation, reverse transcription was performed using the PrimeScript® RT Reagent Kit (TaKaRa Biotechnology, China) following the manufacturer’s instructions. Quantitative analysis of the PCR was performed on the Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, USA) using a SYBR Premix Ex Taqe (Tli RNase H Plus) qPCR kit (TaKaRa Biotechnology, China), as we previous described(Reference Xiao, Liu and Qin9). The gene expression was calculated by the 2-ΔΔCt method according to our previous protocol(Reference Xiao, Liu and Qin9). Expression levels of targeted biological triplicates were normalised to the reference genes GAPDH. Primers used for qPCR analyses are listed in Table 1.
Table 1. Primer sequences used for real-time PCR*

* TLR4, toll-like receptor; IRAK1, IL-1 receptor-associated kinase 1; TRAF6, TNF-α receptor-associated factor 6; RIP1, receptor-interacting protein kinase 1; RIP3, receptor-interacting protein kinase 3; MLKL, mixed-line kinase-like domain protein; LBP, LPS-binding protein; MD2, myeloid differentiation factor-2; CD14, cluster differentiation factor-14; TNF-α, tumour necrosis factor-α; IL-6, interleukin-6; IL-8, interleukin-6; IL-18, interleukin-18; PGAM5, phosphoglycerate mutase 5; FADD, fas-associated death domain protein; TNFR1, tumour necrosis factor receptor 1; DRP1, motility-related protein 1; HMGB1, high mobility protein 1; NLRP3, nucleotide binding oligomerisation domain-like receptor protein 3; NLRC4, nod-like receptors family CARD domain-containing protein.
Western blot
IPEC-1 cells were seeded into 6-well plates at a density of 1 × 105 cells/ml and incubated with 0, 38 µmol/l EPA or 10 µmol/l ARA for 24 h, and then treated with PBS or 1 × 108 ETEC K88/ml for another 2 h. After cell lysed, the membrane proteins and total proteins were extracted according to the manufacturer’s instructions. The bands of the targeted protein were immunoblot as we previously described(Reference Xiao, Liu and Qin9). The primary antibodies were anti-occludin (1:1000, Abcam, UK), claudin-1 (1:1000, Invitrogen, USA), ZO-1 (1:1000, Biorbyt, UK), phospho-RIP1 (1:2000, Cell Signaling Technology, USA), phospho-RIP3 (1:2000, Cell Signaling Technology, USA), phospho-MLKL (1:1000, Cell Signaling Technology, USA), β-actin (1:10 000, Sigma Aldrich, USA) and NaK-ATPase (1:1000, Cell Signaling Technology, USA) and the secondary antibody was HRP-conjugated secondary antibody (1:5000, AntGene Biotech Co., Ltd, China). The results were expressed as the abundance of each target protein relative to β-actin.
Statistical analysis
All data were analysed with ANOVA using the general linear model procedures of SPSS version 22 (SPSS Inc.) for a 2 × 3 factorial design except for the bacterial adhesion and antibacterial activity experiments. The statistical model for the 2 × 3 factorial design included the effects of PUFA (Control, EPA, ARA), ETEC K88 (PBS or ETEC K88) and their interactions (PUFA × ETEC K88). When there was a significant interaction or a trend for interaction, post hoc testing was carried out using Duncan’s multiple comparison tests. The bacterial adhesion and antibacterial activity were analysed by t-test. Data were presented as means with standard errors. P ≤ 0·05 was considered significant, and 0·05 < P ≤ 0·10 was considered a trend.
Results
Effects of EPA and arachidonic acid on ETEC K88 growth, adhesion and endotoxin content in Intestinal porcine epithelial cells 1 cells
EPA or ARA did not influence ETEC K88 growth compared with the control group (online Supplemental Fig. 1).
Cells treated with EPA or ARA had lower bacterial adhesion than control cells at 1 h (P < 0·001) and 2 h (P < 0·01) after ETEC K88 challenge (Fig. 1(a)). There was no difference in bacterial adhesion at 3 h after ETEC K88 challenge among cells treated with EPA or ARA and the control cells.
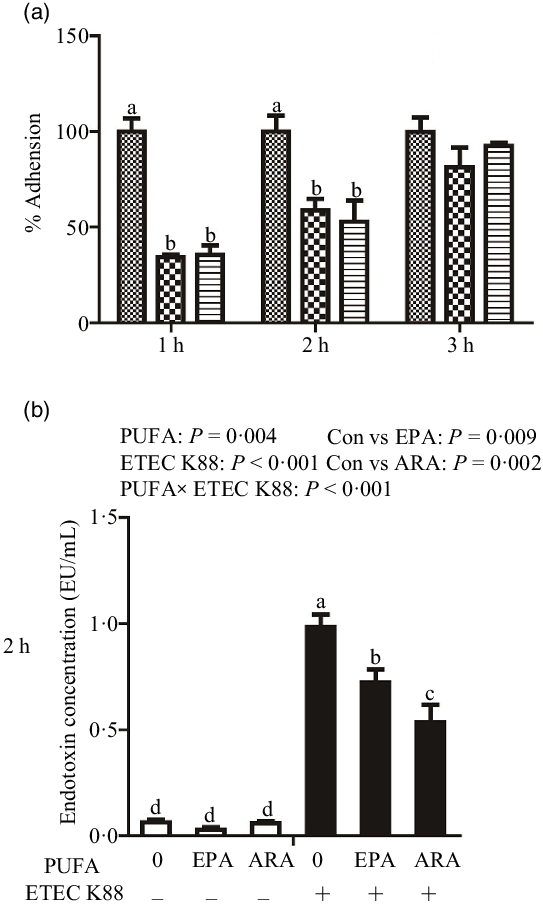
Fig. 1. Effect of EPA and arachidonic acid (ARA) on ETEC K88 adhesion and endotoxin content after ETEC K88 challenge in IPEC-1 cells. (a) Effect of EPA and ARA on ETEC K88 adhesion. Cells were pre-incubated with 0, 38 µmol EPA or 10 µmol ARA for 24 h and then treated with PBS or 1 × 108 ETEC K88/ml for 3 h, ![]() , ETEC K88;
, ETEC K88; ![]() , EPA + ETEC K88;
, EPA + ETEC K88; ![]() , ARA + ETEC K88. (b) Endotoxin content after ETEC K88 challenge. IPEC-1 cells were incubated with 0, 38 µmol EPA or 10 µmol ARA for 24 h and then treated with PBS or 1 × 108 ETEC K88/ml for 2 h. Values are means ± se, n 6. a,b,c,dMeans without a common letter differ, P < 0·05. IPEC-1, intestinal porcine epithelial cell 1.
, ARA + ETEC K88. (b) Endotoxin content after ETEC K88 challenge. IPEC-1 cells were incubated with 0, 38 µmol EPA or 10 µmol ARA for 24 h and then treated with PBS or 1 × 108 ETEC K88/ml for 2 h. Values are means ± se, n 6. a,b,c,dMeans without a common letter differ, P < 0·05. IPEC-1, intestinal porcine epithelial cell 1.
Cells treated with ETEC K88 had higher endotoxin content (P < 0·001) in the supernatant at 2 h (Fig. 1(b)). There was a PUFA × ETEC K88 interaction (P < 0·001) observed for endotoxin content in which cells treated with EPA or ARA had lower endotoxin secretion (P < 0·05) than control cells in the presence of ETEC K88, whereas endotoxin content did not differ among non-ETEC K88-treated cells.
Effects of EPA and arachidonic acid on cell barrier integrity in intestinal porcine epithelial cells 1 cells challenged with ETEC K88
Cells treated with ETEC K88 had lower TEER (P < 0·001) than control cells at 1 h, 2 h, 3 h and 4 h after challenge (Fig. 2(a)–(d)). Cells treated with EPA alone had higher TEER than control cells at 1 h, 2 h, 3 h and 4 h, and cells treated with ARA alone had higher TEER than control cells at 1 h and 2 h. There was a PUFA × ETEC K88 interaction observed for TEER at 2 h and 4 h (P < 0·01) in which cells treated with EPA or ARA had higher TEER (P < 0·001) than control cells in the non-ETEC K88-treated cells at 2 h; however, cells treated with EPA had higher TEER and cells treated with ARA had no difference on TEER in the presence of ETEC K88. Cells treated with EPA or ARA had higher TEER in the presence of ETEC K88 at 4 h post challenge; however, cells treated with EPA had higher TEER and cells treated with ARA had lower TEER. No interaction was observed for TEER at 1 h and 3 h in which cells treated with EPA or ARA had higher TEER than the control cells (P < 0·001).
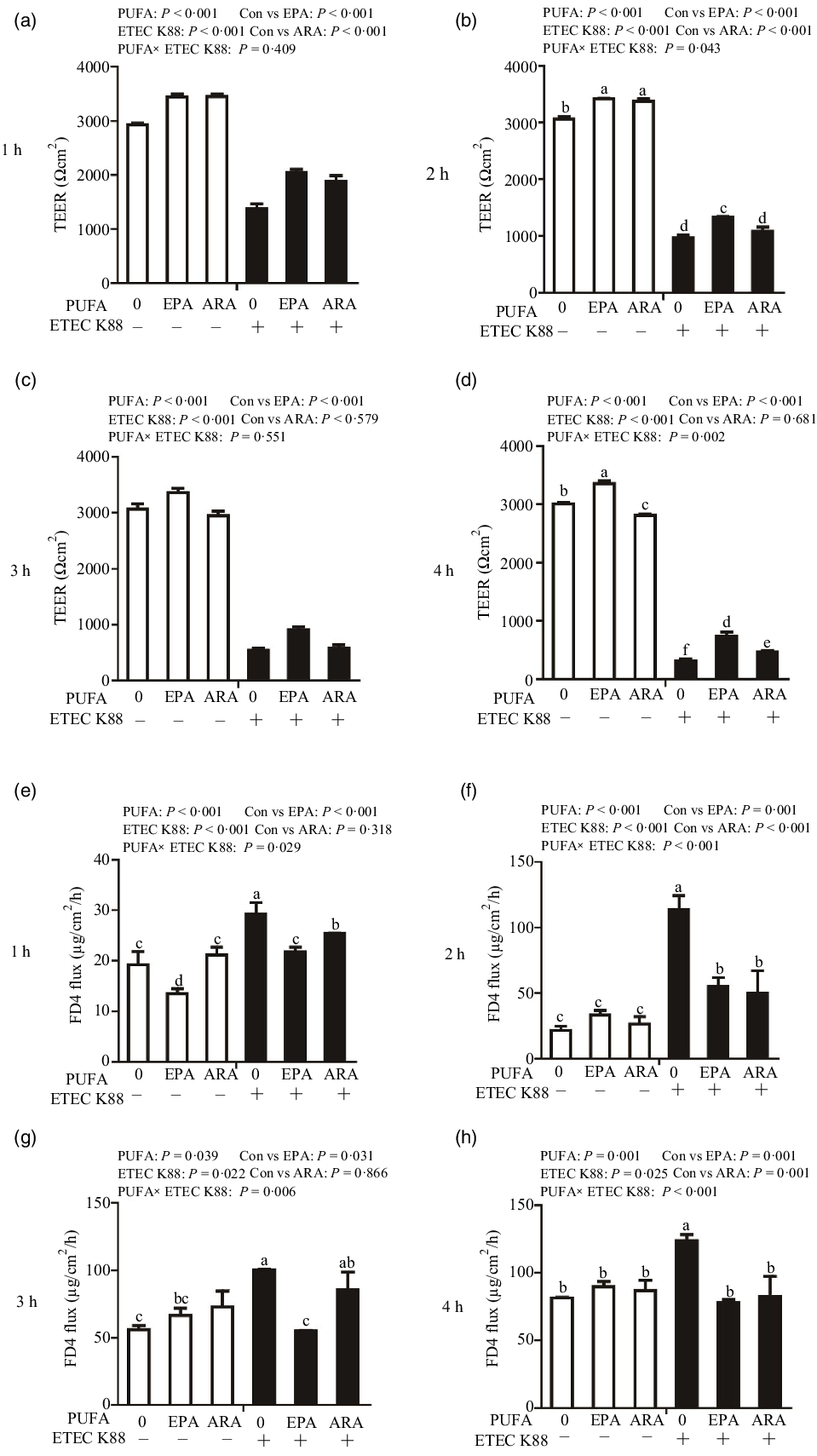
Fig. 2. Effects of EPA and arachidonic acid (ARA) on TEER and FD4 flux after ETEC K88 challenge in IPEC-1 cells. Cells were incubated with or without 38 µmol EPA or 10 µmol ARA for 24 h, followed by exposure with or without 1 × 108 ETEC K88/mL for 1 h, 2 h, 3 h and 4 h. (a–d) Effects of EPA and ARA on TEER. (e–h) Effects of EPA and ARA on FD4 flux. Values are means ± se, n 6. a,b,c,d,e,fMeans without a common letter differ, P < 0·05. IPEC-1, intestinal porcine epithelial cell 1; TEER, transepithelial electrical resistance; FD4, fluorescein isothiocyanate (FITC)-labelled dextran 4 kDa.
Cells treated with ETEC K88 had higher FD4 flux (P < 0·001) compared with the control cells at 1 h, 2 h, 3 h and 4 h after challenge (Fig. 2(e)–(h)). Cells treated with EPA alone also had lower FD4 flux at 1 h. There was no difference in FD4 flux at 1 h, 2 h, 3 h and 4 h in cells treated with ARA alone compared with the control cells. There was a PUFA × ETEC K88 interaction observed for FD4 flux at 1 h, 2 h, 3 h and 4 h (P < 0·05) in which cells treated with EPA or ARA had lower FD4 flux (P < 0·05) than control group among ETEC K88-treated cells, whereas FD4 flux did not differ among PBS-treated cells at 2 h, 3 h and 4 h post challenge. Cells treated with EPA had lower FD4 flux (P < 0·05) than control group among ETEC K88-treated cells at 1 h, 2 h, 3 h and 4 h (P < 0·05), and cells treated with ARA had lower FD4 flux (P < 0·05) than control group among ETEC K88-treated cells at 1 h, 2 h and 4 h.
Effects of EPA and DHA on protein expression and cellular distribution of tight junction proteins in intestinal porcine epithelial cells 1 cells challenged with ETEC K88
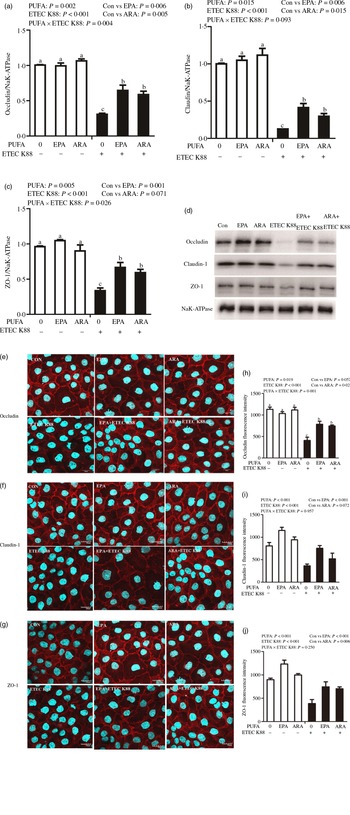
Cells treated with ETEC K88 displayed lower protein expression of membrane occludin (P < 0·001), claudin-1 (P < 0·001) and ZO-1 (P < 0·001) than those cells treated with PBS. There was a PUFA × ETEC K88 interaction (P < 0·05) observed for membrane occludin and ZO-1 expression and a trend for claudin-1 protein (Fig. 3(a)–(d)). Cells treated with EPA or ARA after ETEC K88 stimulation had higher occludin, claudin-1and ZO-1 in the presence of ETEC K88, whereas occludin, claudin-1 and ZO-1 protein did not differ among non-ETEC K88-treated cells.
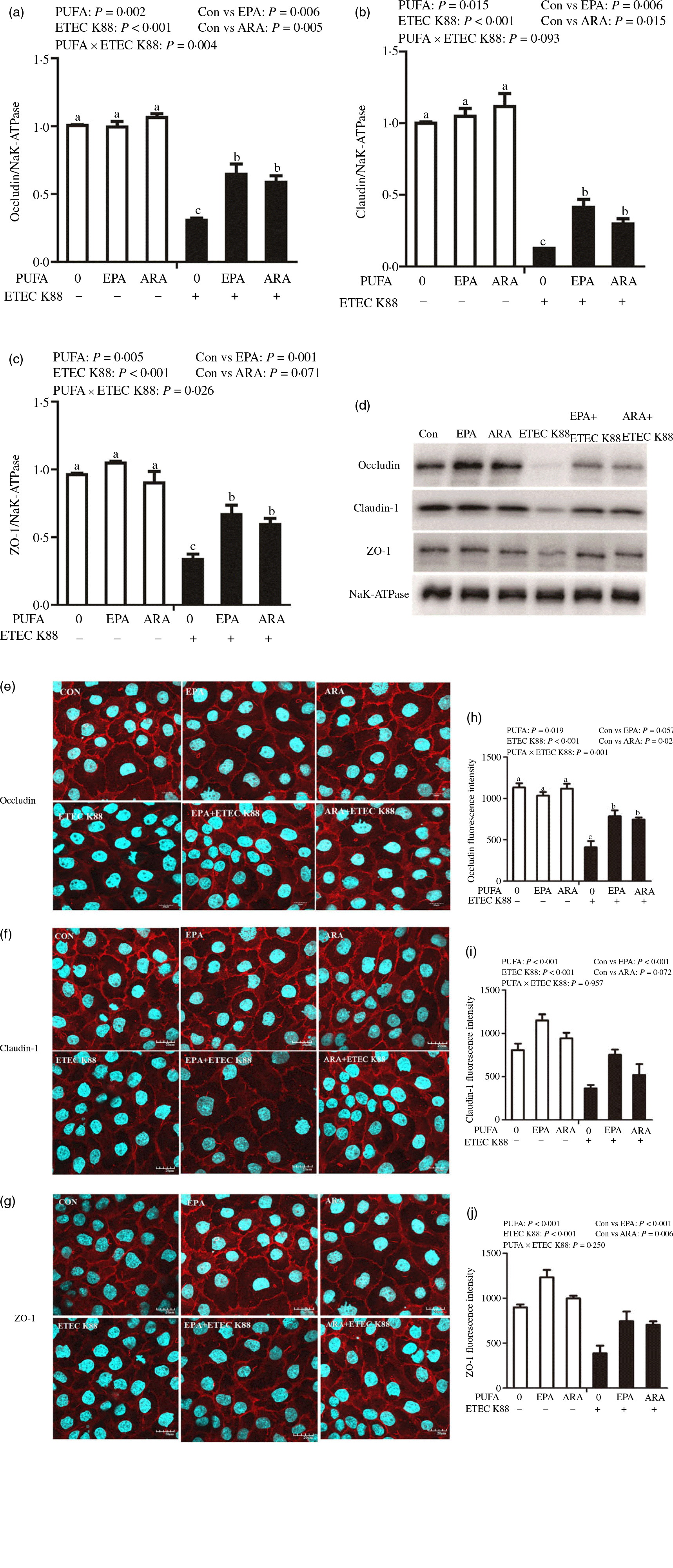
Fig. 3. Effects of EPA and arachidonic acid (ARA) on tight protein expression and distribution after ETEC K88 challenge in IPEC-1 cells. Cells were incubated with or without 38 µmol EPA or 10 µmol ARA for 24 h, and then treated with or without 1 × 108 ETEC K88/ml for 2 h. (a–d) Tight protein expression. (e–j) Tight protein distribution. The distribution of tight junction proteins was visualised by a confocal microscope. Values are means ± se, n 6. a,b,cMeans without a common letter differ, P < 0·05. IPEC-1, intestinal porcine epithelial cell 1; ZO-1, zonula occludens-1.
Cells treated with ETEC K88 exhibited disrupted localisation of occluding, claudin-1 and ZO-1 proteins at plasma membrane compared with the control cells. EPA and ARA incubation prevented the disturbance of occludin, claudin-1 and ZO-1 (Fig. 3(e)–(j)) protein induced by ETEC K88 and promoted the localisation of these three proteins to the plasma membrane.
Effects of EPA and arachidonic acid on cell necrosis in intestinal porcine epithelial cells 1 cells challenged with ETEC K88
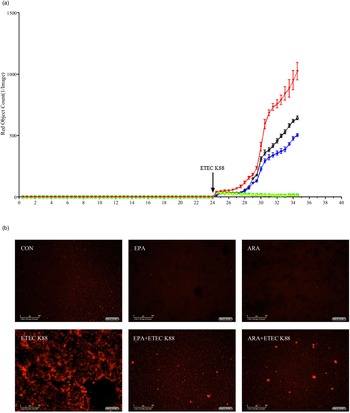
To explore the effect of EPA and ARA on cell necrosis, we used the IncuCyte ZOOM™ Live Cell Imaging System to monitor dynamic changes of cells (online Supplemental videos 1–6). Cells incubated with ETEC K88 had a higher number of necrotic cells than the PBS-control cells from 24 to 28 h (Fig. 4(a)). There was a PUFA × ETEC K88 interaction (P < 0·01)) observed for necrotic cell number at 26 and 28 h in which cells incubated with EPA or ARA had lower cell necrosis (P < 0·001)) among ETEC K88 stimulation groups, whereas there was no difference among non-ETEC K88-treated groups (online Supplemental Fig. 2). The cell necrosis was also verified by images from the system at 26 h after ETEC K88 treatment (Fig. 4(b)).

Fig. 4. Effects of EPA and arachidonic acid (ARA) on cell necrosis in the presence or absence of ETEC K88 in IPEC-1 cells by real-time, automated live-cell imaging and analysis system for 36 h. Cells were pre-incubated with 0 or 38 µmol EPA or 10 µmol ARA for 24 h and then treated with PBS or 1 × 108 ETEC K88/ml for 12 h. (a) Dynamic observation of cell necrosis. (b) Representative images of cell necrosis at 3 h after ETEC K88 treatment (cells marked with red dye are necrotic cells). IPEC-1, intestinal porcine epithelial cell 1. ![]() , CON;
, CON; ![]() , EPA;
, EPA; ![]() , ARA;
, ARA; ![]() , ETEC K88;
, ETEC K88; ![]() , EPA + ETEC K88;
, EPA + ETEC K88; ![]() , ARA ETEC K88
, ARA ETEC K88
Effects of EPA and arachidonic acid on pro-inflammatory cytokine expression in intestinal porcine epithelial cells 1 cells challenged with ETEC K88
Cells incubated with ETEC K88 had higher mRNA expressions of TNF-α, IL-8 and IL-6 (P < 0·001) (Fig. 5(a)–(c)). There was a PUFA × ETEC K88 interaction observed for TNF-α, IL-8 and IL-6 mRNA expression (P < 0·05) in which cells treated with EPA or ARA had lower mRNA expression of TNF-α, IL-8 and IL-6 (P < 0·05) than the control group among ETEC K88-treated cells, whereas TNF-α, IL-8 and IL-6 mRNA expression did not differ among non-ETEC K88-treated cells.
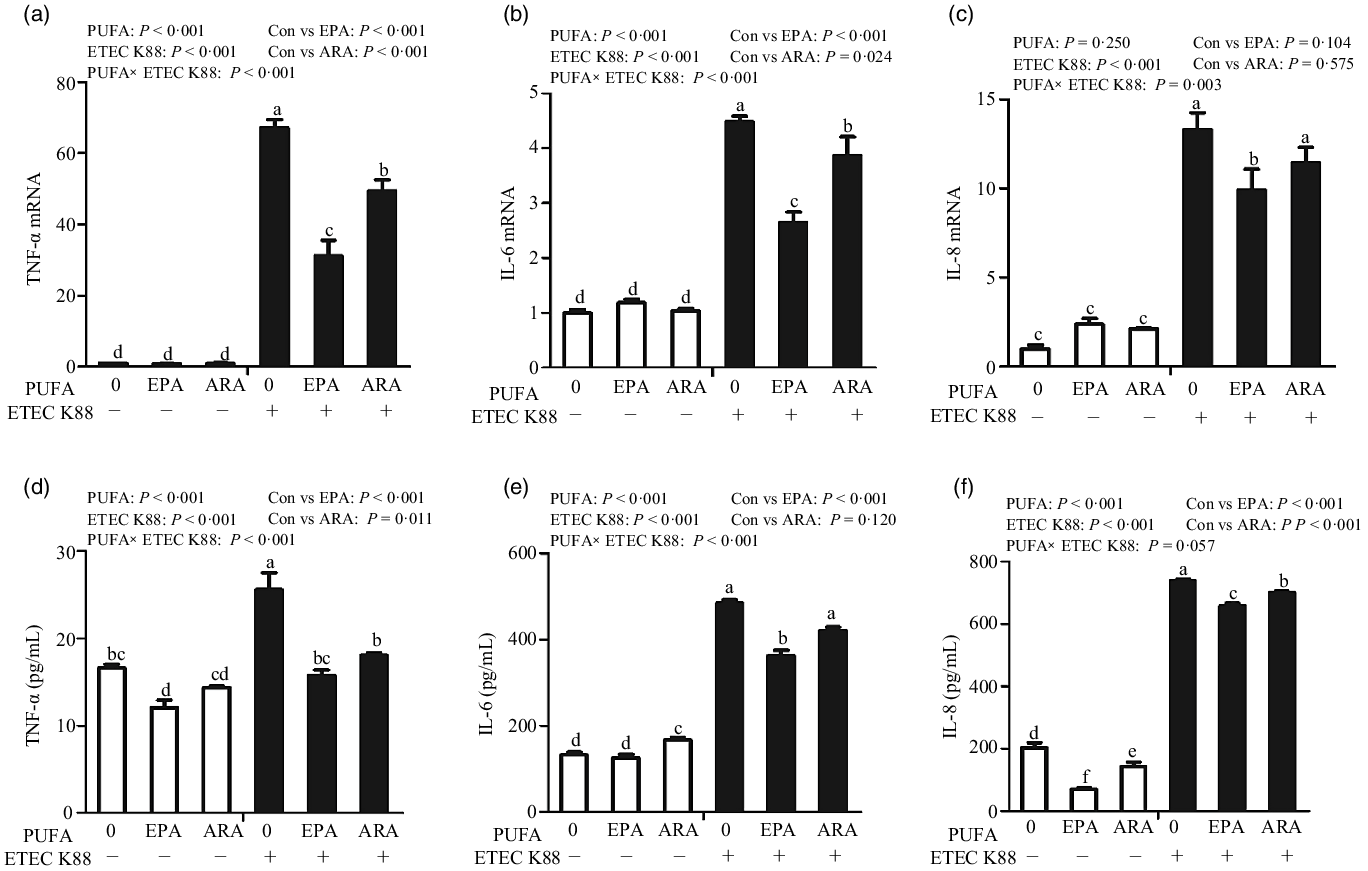
Fig. 5. Effects of EPA and arachidonic acid (ARA) on inflammatory cytokines expression in IPEC-1 cells after ETEC K88 challenge. Cells were pre-incubated with 0, 38 µmol EPA or 10 µmol ARA for 24 h, followed by treatment with PBS or 1 × 108 ETEC K88/ml for another 2 h. (a–c) mRNA expressions of TNF-α, IL-8 and IL-6 after ETEC K88 challenge. (d–f) Protein concentration of TNF-α, IL-8 and IL-6 in supernatant after ETEC K88 challenge. Values are means ± se, n 6. a,b,c,d,e,fMeans without a common letter differ, P < 0·05. IL-6, interleukin-6; IL-8, interleukin-8; TNF-α, tumour necrosis factor-α.
Cells treated with ETEC K88 had higher concentration of TNF-α, IL-8 and IL-6 (P < 0·001) (Fig. 5(d)–(f)). There was a PUFA × ETEC K88 interaction observed for TNF-α and IL-6 concentration in which cells treated with EPA or ARA had lower TNF-α and IL-6 concentration in the presence of ETEC K88, whereas cells treated with EPA had lower TNF-α.
Effects of EPA and arachidonic acid on toll-like receptor 4 signalling pathway in intestinal porcine epithelial cells 1 cells challenged with ETEC K88
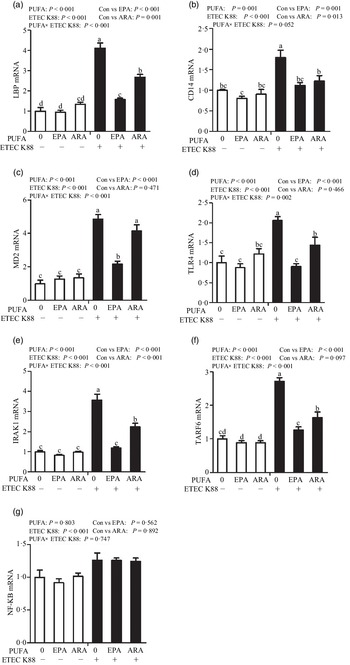
Cells treated with ETEC K88 had higher mRNA abundance of TLR4, LPS-binding protein (LBP), myeloid differentiation factor-2 (MD2), cluster differentiation factor-14 (CD14), IL-1 receptor-associated kinase 1 (IRAK1) and TNF-α receptor-associated factor 6 (TRAF6) (P < 0·001) compared with cells treated with PBS (Fig. 6(a)–(g)). There was a PUFA × ETEC K88 interaction observed for mRNA expression of TLR4, LBP, MD2, IRAK1 and TRAF6 and a trend for a PUFA × ETEC K88 interaction observed for CD14 (P = 0·10) in which the response of these variables to ETEC K88 challenge was lower in those cells receiving the EPA or ARA compared with the ETEC K88 challenged cells treated with PBS, whereas there was no difference for these variables in PBS-treated cells. There was no PUFA × ETEC K88 interaction observed for mRNA abundance of NF-κB. However, the cells treated with the ETEC K88 had higher NF-κB (P < 0·001) mRNA abundance compared with those cells treated with PBS. Neither PUFA nor ETEC K88 had an effect on MyD88 mRNA abundance (data not shown).
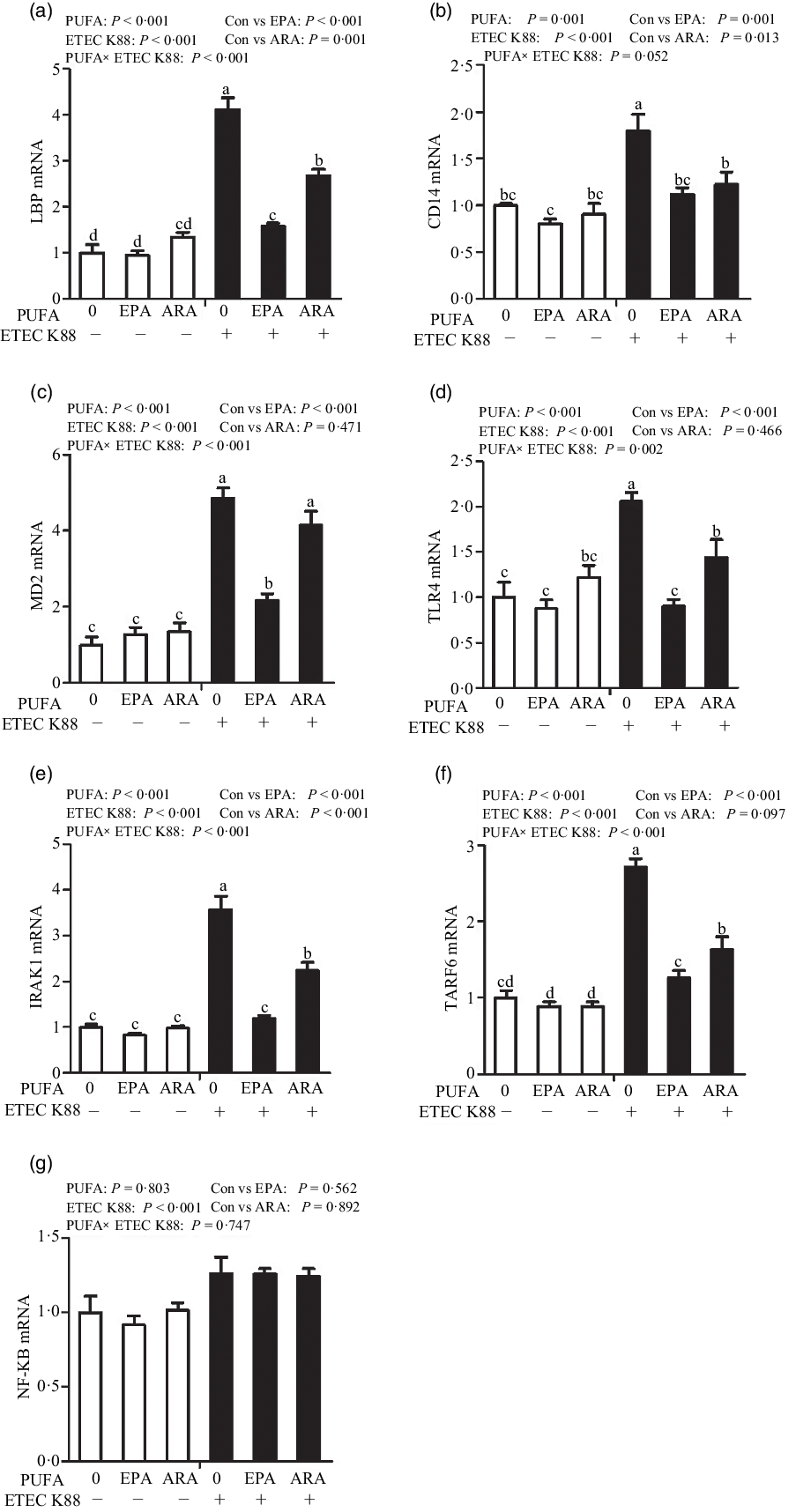
Fig. 6. Effects of EPA and arachidonic acid (ARA) on mRNA expression pf TLR4 signalling pathway after ETEC K88 challenge in IPEC-1 cells. Cells were pre-incubated with 0 or 38 µmol EPA or 10 µmol ARA for 24 h and then treated with PBS or 1 × 108 ETEC K88/ml for 2 h. Values are means ± se, n 6. a,b,cMeans without a common letter differ, P < 0·05. IPEC-1, intestinal porcine epithelial cell 1. CD14, cluster differentiation factor-14; IRAK1, IL-1 receptor-associated kinase 1; LBP, LPS-binding protein; MD2, myeloid differentiation factor-2; TLR4, toll-like receptor; TRAF6, TNF-α receptor-associated factor 6.
Effects of EPA and arachidonic acid on pyroptosis signalling pathway in intestinal porcine epithelial cells 1 cells challenged with ETEC K88
To explore the involvement of the pyroptosis signalling pathway in the beneficial effects of EPA and ARA, we also measured the key signalling molecules of pyroptosis signalling pathway. Cells treated with ETEC K88 had higher mRNA expression of nucleotide-binding oligomerisation domain-like receptor protein (NLRP3, P < 0·001), nod-like receptors family CARD domain-containing protein (NLRC4, P < 0·001), caspase 1 (P < 0·001) and IL-18 (P < 0·001) (Fig. 7(a)–(d)). There was a PUFA × ETEC K88 interaction observed for caspase 1 (P < 0·05) and IL-18 (P < 0·001) mRNA expression in which cells incubated with EPA or ARA had lower mRNA expression of caspase 1 and IL-18 among ETEC K88 groups, whereas caspase 1 and IL-18 mRNA expression did not differ among non-ETEC K88 treated cells. There was an interaction observed for NLRP3 mRNA expression (P < 0·001) in which cells incubated with EPA had lower mRNA expression of NLRP3 among ETEC K88 groups, whereas NLRP3 expression did not differ among non-ETEC K88 treated cells. However, for the cells treated with ARA, ARA alone increased the NLRP3 mRNA expression (P < 0·001) among non-ETEC K88-treated cells and ARA decreased NLRP3 mRNA expression (P < 0·001) among ETEC K88 groups. Totally, ARA had no effect on the expression of NLRP3 mRNA. Cells treated with EPA or ARA had no difference with the control cell in NLRC4 mRNA expression.
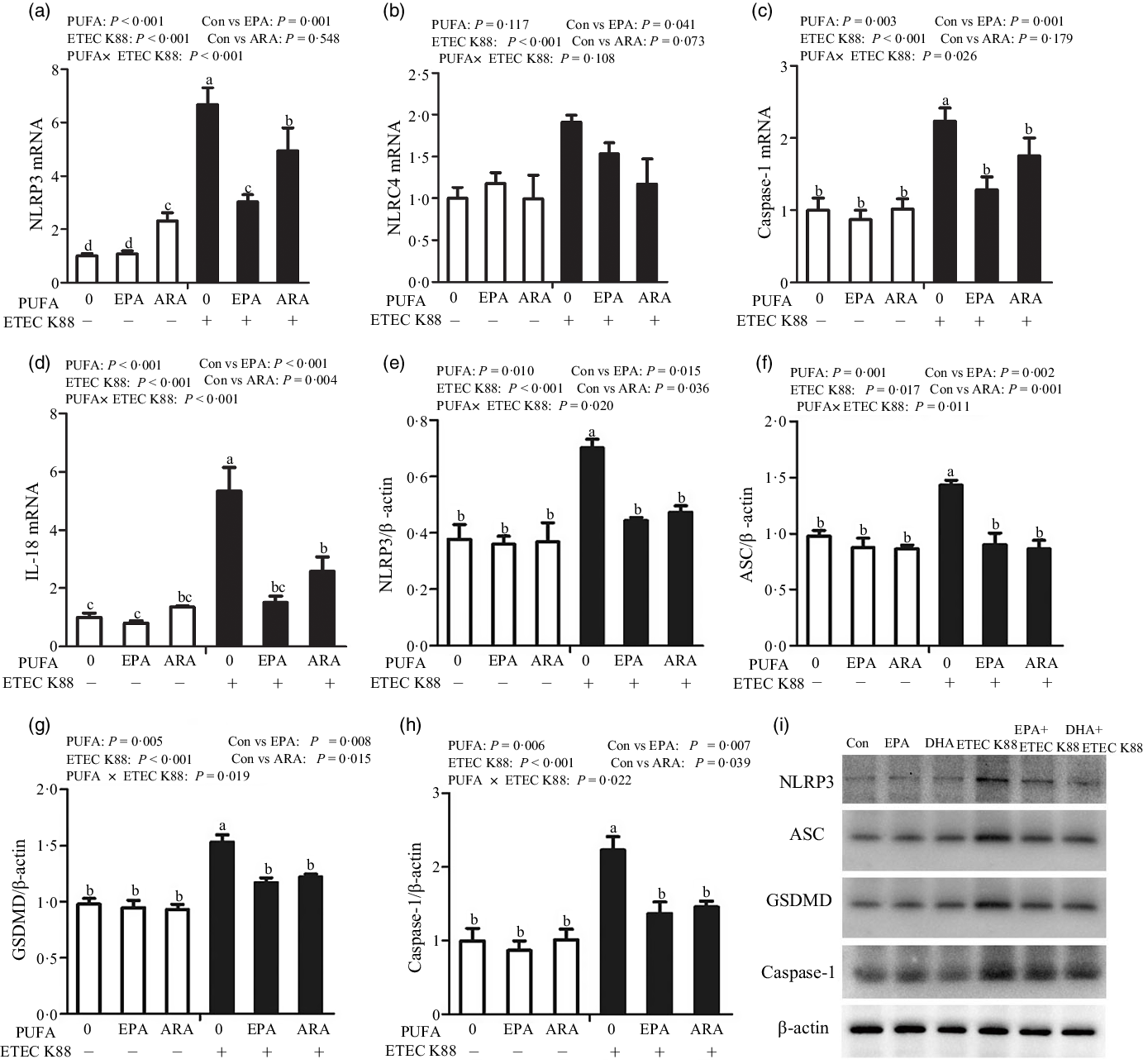
Fig. 7. Effects of EPA and arachidonic acid (ARA) on the mRNA and protein expression of pyroptosis signals after ETEC K88 challenge in IPEC-1 cells. Cells were pre-incubated with 0 or 38 µmol EPA or 10 µmol ARA for 24 h and then treated with PBS or 1 × 108 ETEC K88/ml for 2 h. (a–d) mRNA expressions of pyroptosis signals. (e–i) Protein concentration of pyroptosis signals. Values are means ± se, n 6. a,b,c,dMeans without a common letter differ, P < 0·05. IPEC-1, intestinal porcine epithelial cell 1; NLRP3, nod-like receptor protein 3; NLRC4, nod-like receptors family CARD domain-containing protein; ASC, apoptosis-associated speck-like protein containing a CARD; GSDMD, gasdermin D.
Cells treated with ETEC K88 had higher protein expression of NLRP3 (P < 0·001), apoptosis-associated speck-like protein containing a CARD (ASC, P < 0·05), gasdermin D (GSDMD, P < 0·05) and caspase-1 (P < 0·001) than the control cells (Fig. 7(e)–(i)). There was a PUFA × ETEC K88 interaction observed for NLRP3 (P < 0·05), ASC (P < 0·05), GSDMD (P < 0·05) and caspase-1 (P < 0·05) protein expression in which cells incubated with EPA or ARA had lower protein expression of NLRP3, ASC, GSDMD and caspase-1 among ETEC K88 groups, whereas NLRP3, caspase 1 and IL-18 protein expression did not differ among non-ETEC K88-treated cells.
Effects of EPA and arachidonic acid on necroptosis signalling pathway in intestinal porcine epithelial cells 1 cells challenged with ETEC K88
To examine the involvement of the necroptosis signalling pathway in the protective effects of EPA and ARA, we next measured the mRNA and protein expressions of key signalling molecules in this pathway. Cells treated with ETEC K88 had higher mRNA (P < 0·001) expression of tumour necrosis factor receptor 1 (TNFR1), fas-associated death domain (FADD), caspase 8, RIP1, MLKL, phosphoglycerate mutase 5 (PGAM5), motility-related protein 1 (DRP1) and high mobility protein 1 (Fig. 8(a)–(i)). There was a PUFA × ETEC K88 interaction observed for mRNA expression of FADD (P < 0·001), caspase 8 (P < 0·001), RIP1 (P < 0·001), RIP3 (P < 0·01), MLKL (P < 0·001), PGAM5 (P < 0·05), DRP1 (P < 0·001) and HMGB1 (P < 0·001) in which cells incubated with EPA or ARA had lower mRNA expression of FADD, caspase 8, RIP1, RIP3, MLKL, PGAM5, Drp1 and HMGB1 among ETEC K88 stimulation groups, whereas mRNA expression of above genes did not differ among non-ETEC K88 treated cells. No interaction was observed for TNFR1 expression, and EPA or ARA had no influence on the TNFR1 expression.
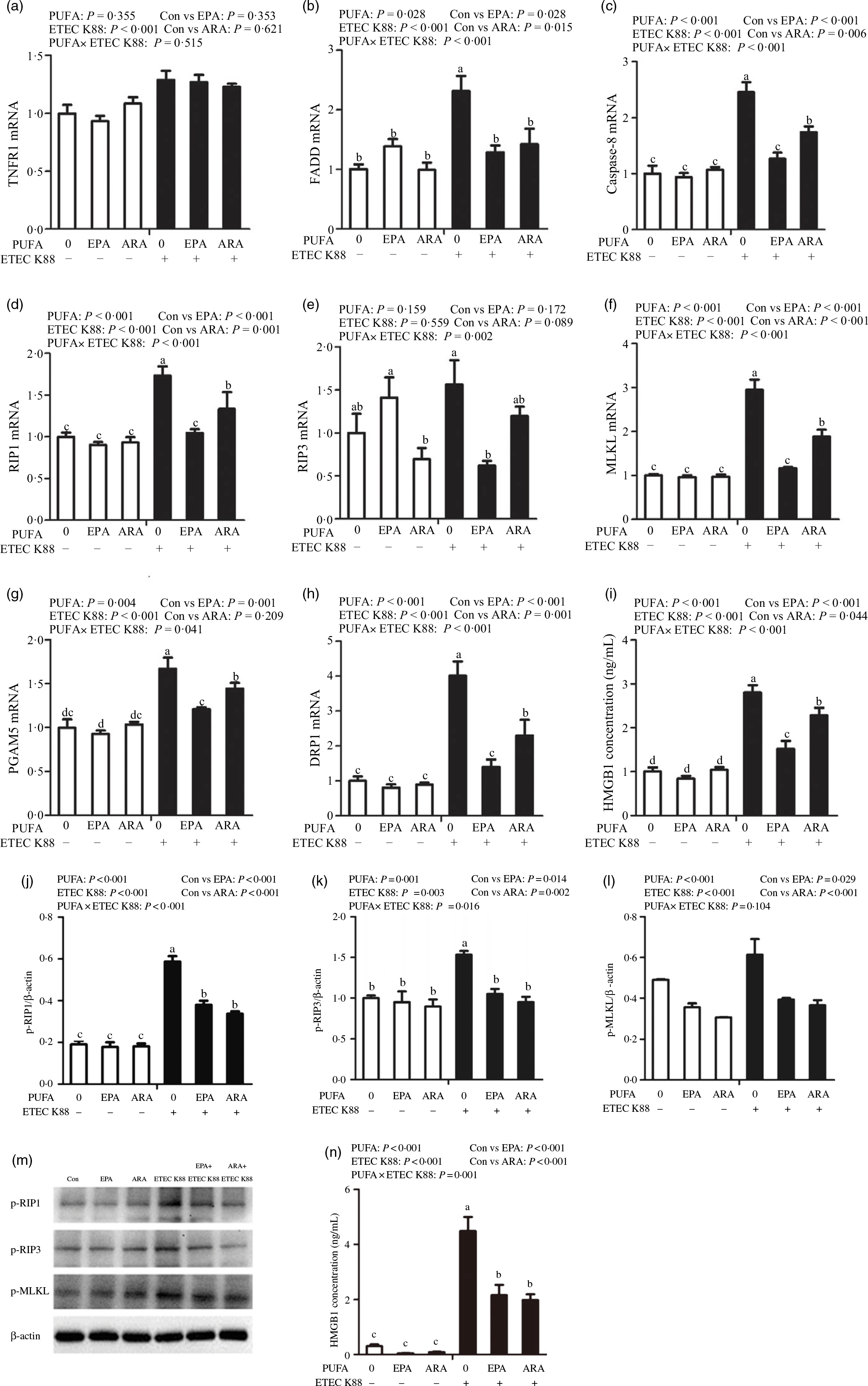
Fig. 8. Effects of EPA and arachidonic acid (ARA) on the mRNA and protein expression of necroptosis signals after ETEC K88 challenge in IPEC-1 cells. Cells were pre-incubated with 0 or 38 µmol EPA or 10 µmol ARA for 24 h and then treated with PBS or 1 × 108 ETEC K88/ml for 2 h. (a–i) mRNA expressions of necroptosis signals. (j–n) Protein concentration of necroptosis signals. Among them (j–l) were the protein expression of p-RIP1, p-RIP3 and p-MLKL, (m) was the representative bands of above three proteins and (n) was the concentration of HMGB1 in supernatant. Values are means ± se, n 6. a,b,c,dMeans without a common letter differ, P < 0·05. IPEC-1, intestinal porcine epithelial cell 1; TNFR1, tumour necrosis factor receptor; RIP1, receptor-interacting protein 1; RIP3, receptor-interacting protein 3; MLKL, mixed-lineage kinase domain like-domain protein; PGAM5, phosphoglycerate mutase family 5; Drp1, dynamin-related protein 1; HMGB1, high mobility group box-1 protein.
Cells incubated with ETEC K88 had higher protein expression of p-RIP3 (P < 0·001), p-RIP3 (P < 0·001), p-MLKL (P < 0·001) and HMGB1 (P < 0·001) than the control cells (Fig. 8(j)–(n)). There was a PUFA × ETEC K88 interaction observed for phosphorylated-RIP1 (p-RIP1) (P < 0·001), p-RIP3 (P < 0·001) and HMGB1 (P = 0·001) in which cells incubated with EPA or ARA had lower protein expression of p-RIP1, p-RIP3 and HMGB1 among ETEC K88 groups, whereas p-RIP1, p-RIP3 and HMGB1 protein did not differ among PBS-treated cells. No PUFA × ETEC K88 interaction was observed for p-MLKL protein expression. Cells treated with EPA or ARA had lower expression of p-MLKL (P < 0·001).
Discussion
It is well known that ETEC infection not only induces severe intestinal inflammation but also impairs cells or tissues in human and animals(Reference Devriendt, Stuyven and Verdonck24,Reference Roselli, Finamore and Britti25) . It can cause many diseases such as severe diarrhoea and sepsis, and even life-threatening diseases under certain conditions. It has been shown that ETEC infection could result in an increased risk of mortality and even morbidity in children and young animals(Reference Dubreuil, Isaacson and Schifferli1,Reference Matsumoto, Miyagawa and Takahashi26) .
Long-chain n-3 PUFA has been reported to play a critical role on intestinal health in animal models and many clinical trials(Reference Calder27–Reference Chen, Li and Tang29). However, relatively little attention has been given to long-chain n-6 PUFA on intestinal health under pathological conditions. EPA and ARA are two representative members of long-chain PUFA. First, we explored the effects of EPA and ARA on bacterial growth, bacterial adhesion to cells and endoxin secretion. In the present study, EPA did not influence the ETEC K88 growth but decreased endotoxin secretion and bacterial adhesion after ETEC K88 challenge. Similarly, dietary n-3 PUFA has been found to decrease serum endotoxin concentration in pigs receiving a porridge meal(Reference Mani, Hollis and Gabler30). Interestingly, in our study, ARA also reduced endotoxin secretion and bacterial adhesion, showing the same effects as EPA. We demonstrated for the first time that n-6 PUFA could inhibit ETEC adherence and endotoxin secretion. It is possible that long-chain PUFA inhibits bacterial adhesion to the epithelial cells therefore suppress endotoxin secretion.
The intestinal epithelial cells serve as a critical barrier separating the internal from the external environment in animals. TEER and mucosal-to-serosal flux of FD4 are important indicators of epithelial cell barrier function, which refers to the permeability of intestinal epithelium. Higher TEER and lower FD4 flux indicate better barrier function. Then we explored the effects of EPA and ARA on epithelial cell barrier function. In our study, expectedly, EPA supplementation improved the TEER and decreased the FD4 flux, suggesting a beneficial role on barrier integrity after ETECK 88 challenge. Similar to EPA, ARA supplementation also improved TEER and reduced FD4 flux. Currently, abundant evidence has reported the protective effects of n-3 PUFA on intestinal barrier function. For example, supplementation of n-3 fatty acids, which included EPA and DHA, prevented the decrease of TEER and the increase of FD4 permeability induced by inflammatory factors and mycotoxins(Reference Xiao, Liu and Qin9,Reference Li, Zhang and Wang31) . Moreover, EPA and DHA also improved epithelial barrier integrity of T84 cell monolayers by improving TEER and reducing IL-4-mediated permeability increase(Reference Willemsen, Koetsier and Balvers23). However, the research on n-6 PUFA regulating cell integrity is very limited. Only research from Jacobi et al. showed that dietary ARA enhanced TER recovery and reduced mucosal-to-serosal flux of 3H-mannitol and 14C-inulin in ischaemia-injured intestine of suckling pigs(Reference Jacobi, Moeser and Corl13). Occludin, claudins and ZOs are crucial components, which joined epithelial cells together. In the present study, EPA and ARA also increased the expression of tight junction proteins and prevented the disruption of tight junction proteins, which further showed the protective role of PUFA on intestinal barrier integrity. Studies have shown that n-3 PUFA could improve occludin and ZO-1 protein expression and prevent the redistribution of tight junction proteins(Reference Xiao, Tang and Yuan32). Fish oil (rich in n-3 PUFA) was found to enhance tight junction proteins occludin and claudin-1 expression in weaned pigs after LPS challenge(Reference Liu, Chen and Odle8). Therefore, EPA, as well as ARA, may partially improve intestinal barrier function via improving the expression of intestinal tight junction proteins.
Cell death contributes to intestinal injury and barrier function impairment. Thus, we further measured cell necrosis using the IncuCyte ZOOM™ Live Cell Imaging System, which can automatically monitor necrotic cells in real time and calculate the living or dead cell density at every time point. In the current study, consistent with improved intestinal cell barrier function, EPA and ARA decreased necrotic cells density after ETEC K88 challenge. Similarly, a report from Kishida et al. showed that DHA enrichment reduced L929 cell necrosis induced by TNF-α (Reference Kishida, Tajiri and Masuzawa33). Research from Xiao et al. showed that EPA and DHA could decrease the percentage of necrotic cells induced by DON stimulation(Reference Xiao, Liu and Qin9). Until now, the research on n-6 PUFA modulating cell necrosis was limited. Therefore, EPA and ARA may improve intestinal integrity partially by inhibiting cell necrosis induced by ETEC K88.
ETEC infection usually activates inflammatory response and leads to the release of pro-inflammatory cytokines(Reference Willemsen, Koetsier and Balvers23). TLR4 is the best-characterised pattern-recognition transmembrane receptors, which can cause inflammation in intestine or other organs(Reference Sabroe, Parker and Dower34). It is well known that TLR4 can recognise many exogenous substances such as endotoxin or LPS from Gram-negative bacteria, and then initiate systemic inflammatory response syndrome. Therefore, we next studied the effects of EPA and ARA on pro-inflammatory cytokines and TLR4 signals. In the current experiment, consistent with improved intestinal integrity, EPA decreased the mRNA and protein expression of TNF-α, IL-8 and IL-6, as well as mRNA abundance of TLR4 and its downstream signals such as LBP, MD2, CD14, IRAK1 and TRAF6, suggesting a beneficial role in suppressing intestinal inflammation via TLR4 signalling pathway. Currently, there is an abundance of research on the modulation of intestinal proinflammatory mediators through supplementation of (n-3) PUFA. Wijendran et al. (Reference Wijendran, Brenna and Wang35) found that EPA and DHA reduced IL-1β-induced proinflammatory cytokines of IL-8 and IL-6 in human fetal intestinal epithelial cells. Liu et al. also found that dietary fish oil (rich in n-3 PUFA) decreased the concentrations of TNF-α and PGE2 via inhibition of the TLR4 and NOD2 signalling pathways in jejunum and ileum after LPS challenge in weaned piglets(Reference Liu, Chen and Odle8). Recently, Zhu et al. also reported that flaxseed oil (rich in n-3 PUFA) attenuated intestinal inflammation by regulating TLR4/NOD signalling pathways following LPS challenge in a piglet model(Reference Zhu, Wang and Wang7). Interestingly, we also found that, similar to EPA, ARA also decreased intestinal inflammation, which was contrary to our traditional thoughts. That is because ARA is usually thought to be the principal substrate for bioactive mediators known as eicosanoids, which was involved in mediating inflammation. In our current study, it is possible the protective effects of EPA and ARA on intestinal inflammation were related to inhibiting the TLR4 signalling pathway.
To elucidate the molecular mechanism(s) by which EPA and ARA might attenuate intestinal inflammation and injury, we examined the role of necroptosis and pyroptosis signalling pathways. Pyroptosis is crucial for controlling microbial infections, which was regulated by inflammasomes and gasdermin D (GSDMD)(Reference Patankar and Becker16). First, pathogen-associated molecular patterns (PAMP) or damage-associated molecular patterns (DAMP) signals was recognised by the NLRP3, linking to the adaptor protein apoptosis-related speckle-like protein (ASC) to activate the caspase-1 and finally the effect protein GSDMD are cleaved by activated caspase 1, forming cell membrane pores and enabling the release of the intracellular contents such as mature pro-inflammatory cytokines(Reference Liu, Zhang and Ruan36,Reference Liu, Zaki and Vogel37) . Additionally, NLRC4 can directly bind and activate caspase-1 independently of ASC(Reference Shi, Zhao and Wang38). Several studies have demonstrated that pyroptosis was involved in dysfunction of intestinal epithelium cells(Reference Zeng, Duan and Hu22,Reference Mandal, Feng and Lyons39) . First, we examined the activation of pyroptosis signalling pathway. In the present study, we observed that EPA or ARA reversed the mRNA or protein expression of NLRP3, ASC, NLRC4, caspase 1 and IL-18 after ETEC K88 challenge, indicating beneficial effects on suppressing pyroptosis signalling pathway. Until now, there is little evidence about the effect of PUFA on pyroptosis signalling pathway. Only Shen et al. reported that PUFA prevented NLRP3 activation and decreased IL-1β protein in human macrophages(Reference Shen, Yang and Ou40). We uncovered for the first time that PUFA could inhibit pyroptosis signalling pathway in epithelial cells. It is possible the protective effects of EPA and ARA on cell injury and inflammation were closely related to inhibiting the pyroptosis signalling pathway, which indicated a novel mechanism for PUFA in maintaining intestinal health.
In the next, we examined the activation of necroptosis signalling pathway. Necroptosis is a newly appreciated pathway of regulated necrosis, which was mainly mediated by the activation of RIP1, RIP3 and p-MLKL. Various factors, such as TNF family members, Fas ligand, LPS, TLRs and endotoxins, can activate necroptosis signalling pathway(Reference Holler, Zaru and Micheau41,Reference Vanlangenakker, Vanden Berghe and Vandenabeele42) . Once initiated by ligands, the receptor TNFR1 was activated to recruit TNFR-associated death domain TRADD forming a complex with RIP1 known as complex I. When RIP1 was deubiquitinated, complex II was formed including RIP1, caspase 8 and FADD. When caspase 8 is inhibited, phosphorylated RIP1 recruits RIP3 to form the RIP1/RIP3/MLKL necrosome with the phosphorylated MLKL, which ultimately leads to cell necroptosis(Reference Hildebrand, Tanzer and Lucet43). The phosphorylated MLKL can translocate to cell membrane to destroy the integrity of cell membrane and release the cell content such as HMGB1 outside the cells(Reference Silke, Rickard and Gerlic44). Furthermore, the RIP1/RIP3/MLKL necrosome can activate the PGAM5 and made it phosphorylated, then the phosphorylated PGAM5 dephosphorylated the mitochondrial fission factor (DRP1), causing the mitochondrial fragmentation and the release of ROS, which finally leads to cell necroptosis(Reference Wang, Jiang and Chen45,Reference Wang, Sun and Su46) . Translocation and secretion of HMGB1 are important steps in late inflammatory responses. Several studies have revealed that necroptosis was involved in inflammation and damage of intestinal epithelium cells(Reference Günther, Neumann and Neurath14,Reference Welz, Wullaert and Vlantis47) . In the present study, similar to pyroptosis signalling pathway, we observed that EPA supplementation decreased mRNA expression of FADD, caspase 8, RIP1, MLKL, PGAM5, DRP1 and HMGB1 and inhibited protein expression of p-RIP1, p-RIP3, p-MLKL and HMGB1 after ETEC K88 challenge. Limited research was conduct about the effects of n-3 PUFA on necroptosis signals. Our previous research found that EPA or DHA alleviated DON- or TNF-α-induced necroptosis in IPEC-1 cells(Reference Xiao, Liu and Qin9). Zhu et al. also reported that dietary flaxseed oil enhanced intestinal integrity via modulating necroptosis signalling pathway(Reference Zhu, Wang and Wang7). Surprisingly, ARA also inhibits the p-RIP1, p-RIP3, p-MLKL and HMGB1 expression, suggesting a beneficial role in regulating cell necroptosis. Until now, there is no research investigating the role of n-6 PUFA on necroptosis. In our current study, we demonstrated for the first time that similar to EPA, ARA also has beneficial effects on inhibiting the necroptosis signalling pathway. So, in our current study, EPA or ARA may ameliorate ETEC-induced intestinal inflammation and cell injury by inhibiting pyroptosis and necroptosis signalling pathways.
In our current study, it is possible that EPA and ARA suppressed ETEC-induced intestinal inflammation and cell injury by improving the fluidity of the cell membrane since PUFA were incorporated in the membrane phospholipid fraction(Reference Willemsen, Koetsier and Balvers23). The cell membrane involved in ion transport, energy conversion, cells recognition and immune regulation. Any alterations of the cell membrane can affect the distribution of enzymes and membrane’s receptors, secretion of lymphokines and antibodies and recognition of antigens. On the other way, EPA and ARA inhibit the signalling pathways of pyroptosis, and necroptosis may be through eicosanoids, such as prostaglandin, thromboxane and leukotriene, which are the derivations of EPA and ARA(Reference Huang, Wang and Peng48). ARA in general is a pro-inflammatory substance. However, some researches have indicated that n-6 PUFA, especially ARA as well as its metabolites, facilitate recovery of damaged intestinal mucosa(Reference Ruthig and Meckling-Gill11–Reference Jacobi, Moeser and Corl13). The mechanism for EPA and ARA on pyroptosis and necroptosis signalling pathways may be direct or indirect. In the future, further studies are needed to explore the mechanisms involving benefits of n-3 and n-6 PUFA on pyroptosis and necroptosis signalling pathways.
In summary, our results demonstrate that long-chain PUFA exerts beneficial role in protecting against ETEC K88-induced inflammation, cell injury and barrier dysfunction. It is possible that the protective role of EPA and ARA on intestinal epithelial cells is closely related to suppressing pyroptosis and necroptosis signalling pathways. Our study also provided a novel principle that long-chain PUFA, especially ARA, has potential therapeutic application in disease of enteric infections.
Acknowledgements
This research was supported by the Innovative Research Groups of the Natural Science Foundation of Hubei Province (2019CFA015), the National Natural Science Foundation of China (No. 31772615) and the projects of Wuhan Science and Technology Bureau (2018020401011304).
Y. L. and K. X. designed the research; Y. Y., Y. Z., K. X., F. H. and Q. L. performed the experiments; Y. L., D. W., J. Z. and K. X. analysed data; Y. L., K. X. and Y. Y. wrote the paper; YL had primary responsibility for final content. All authors read and approved the final manuscript.
The authors declare that there are no conflicts of interest.
Supplementary material
For supplementary material/s referred to in this article, please visit https://doi.org/10.1017/S0007114521005092