


Relapse, namely the re-emergence of depressive symptoms following successful acute treatment of major depressive disorder (MDD), is a significant clinical concern (Reference Segal, Pearson and ThaseSegal et al, 2003). The current guidelines suggest that antidepressant therapy should be maintained for between 9 months and 1 year (World Health Organization, 1989). Failure to adhere to these guidelines may increase the risk of relapse by 70% (Reference Melfi, Chawla and CroghanMelfi et al, 1998). Preclinical studies indicate that duloxetine is a potent dual reuptake inhibitor with similar affinities for serotonin and noradrenaline transporters (Reference Bymaster, Dreshfield-Ahmad and ThrelkeldBymaster et al, 2001), and it has been shown to be efficacious in the acute treatment of MDD (Reference Detke, Lu and GoldsteinDetke et al, 2002). To determine the efficacy of duloxetine in preventing relapse of MDD, and to assess its long-term safety and tolerability, a randomised relapse prevention study was conducted. We postulated that MDD patients who showed clinically significant symptom improvement in response to duloxetine (60 mg daily) during 12 weeks of open-label treatment would have a significantly longer time to relapse and lower relapse rates when maintained on duloxetine (60 mg daily) compared with patients who were randomised to placebo for 6 months.
METHOD
Selection of participants
All of the study participants (both men and women) were at least 18 years of age. Ethics review boards at each site approved the study protocol before the study began at that site. Written informed consent was provided by all participants before any study procedures were initiated. All of the participants met the DSM–IV (American Psychiatric Association, 1994) criteria for MDD without psychotic features as assessed using the Mini International Neuropsychiatric Interview (MINI; Reference Sheehan, Lecrubier and SheehanSheehan et al, 1998). Baseline disease severity was assessed using the 17-item Hamilton Rating Scale for Depression (HRSD; Reference HamiltonHamilton, 1960) and the Clinical Global Impression–Severity (CGI–S) scale (Reference GuyGuy, 1976). At both the screening and second study visits, all study participants were required to meet the entry criteria of HRSD17 score ≥18 and CGI–S score ≥4, indicating at least moderate depression. In addition, participants must have had at least one other major depressive episode before the episode that was being experienced at the time of entry to the study.
Reasons for exclusion from the study included the following: having a current and primary Axis I disorder other than MDD; anxiety disorder as a primary diagnosis within 1 year of entry to the study; treatment-resistant depression; serious suicidal risk; and serious medical illness.
Study design
The design was a randomised active-treatment lead-in double-blind placebo-controlled multicentre parallel-group study of out-patients who met the DSM–IV criteria for MDD. The study was conducted in France, Italy, Spain and the USA. The study design (Fig. 1) was similar to that of previous relapse-prevention studies (Reference Fava, Schmidt and ZhangFava et al, 2002; Reference Schmidt, Fava and ZhangSchmidt et al, 2002). During the initial screening phase and before the second visit (week 0), all participants underwent screening tests in addition to both psychiatric and physical examinations.

Fig. 1 Patient disposition flow chart. Two of the 280 participants who met the entry criteria for randomisation chose not to continue in the study, and are therefore accounted for in acute-phase discontinuation, even though they completed the acute phase. Of the 200 participants who entered the follow-up phase (continuation phase: duloxetine (60 mg daily) (n=76), placebo (n=54); rescue phase: duloxetine (60 mg daily) (n=47), duloxetine (60 mg twice daily) (n=23)), 193 individuals completed the phase.
Acute phase
All of the participants who met the enrolment criteria received open-label duloxetine (60 mg daily) for 12 weeks. If it was necessary to address problems with regard to tolerability, investigators could reduce the patient's daily duloxetine dose to 30 mg until week 4.
Continuation phase
Participants were eligible to enter the continuation phase if they met the following criteria: HRSD17 score ≤9; CGI–S score 42; and no longer meeting the DSM–IV criteria for MDD in weeks 10 and 12 of the acute phase. Eligible patients were randomised at week 12 to either duloxetine (60 mg daily) or placebo for 26 weeks. Patients who were randomised to the placebo arm were tapered with duloxetine (30 mg daily) for 1 week in a double-blind manner. All continuing participants were assessed during at least nine scheduled visits from week 13 to week 38. Re-emergence of depressive symptoms at any time was defined as an HRSD17 score of ≥12, prompting weekly visits until an HRSD17 score of <12 was obtained or the patient met the criteria for relapse. Relapse was defined in the protocol as an increased CGI–S score of ≥2 points compared with that obtained in week 12, as well as meeting the MINI depression module criteria for MDD at two consecutive visits at least 2 weeks apart. Participants who experienced a relapse were offered the option of entry into the rescue phase.
Rescue phase
Participants who experienced relapse during the continuation phase were offered the option of entry into the double-blind rescue phase at the investigator's discretion. Relapsed patients who had been randomised to placebo were re-initiated on duloxetine (60 mg daily). Relapsed patients who had been randomised to duloxetine (60 mg daily) had their dose of duloxetine increased (to 60 mg twice daily). The rescue phase will be discussed in detail in a later paper.
Follow-up phase
All of the participants entered the follow-up phase after either completing or discontinuing either the continuation phase or the rescue phase. If patients were receiving duloxetine, their duloxetine dose was reduced by 50% for 3 days. Placebo-treated patients continued to receive placebo. In both cases, participants received no study drug after the third day of the follow-up phase. After approximately 1 week, efficacy and safety data were collected.
Assessments
Efficacy
The primary efficacy measure was time from randomisation to relapse for participants during the continuation phase. Secondary efficacy measures included HRSD17 total and sub-scale scores, CGI–S scale score, Patient Global Impression–Improvement (PGI–I; Reference GuyGuy, 1976) scale score, painful physical symptoms as assessed by the Symptom Questionnaire – Somatic Sub-scale (SQ–SS; Reference KellnerKellner, 1987) and visual analogue scales (Reference DeLoach, Higgins and CaplanDeLoach et al, 1998), the Quality of Life in Depression Scale (QLDS; Reference McKenna and HuntMcKenna & Hunt, 1992) score and the Sheehan Disability Scale (SDS) score to measure disruption of the patient's work, social and home life by their symptoms.
Safety and tolerability
During all of the treatment phases, safety and tolerability were evaluated by collecting and reporting the incidence, severity and seriousness of adverse events, the reasons for discontinuation, vital signs, body weight and laboratory investigation results.
Statistical methods
The primary efficacy analysis compared the time from randomisation to relapse for patients who were receiving duloxetine (60 mg daily) and patients who were on placebo, using the log-rank test. Time to relapse was defined as the number of days from the randomisation date to the relapse date. The analysis included as patients experiencing relapse only those participants who satisfied the protocol definition of relapse described above.
During the continuation phase, other continuous measures were compared for duloxetine v. placebo by analysis of covariance (ANCOVA), analysis of variance (ANOVA) and/or mixed-model repeated-measures analysis (Reference Mallinckrodt, Clark and DavidMallinckrodt et al, 2001). Unless otherwise specified, mean changes from baseline to end-point were compared, the baseline and end-point being defined as the initial and last observations respectively during each study phase. The ANCOVA model included baseline, treatment and investigator, the ANOVA model included treatment and investigator, and the mixed-model repeated-measures analysis included baseline, visit, treatment, investigator, treatment × visit interaction and baseline × visit interaction. For categorical measures, treatments were compared by Fisher's exact test.
During the acute phase, baseline and end-point values were compared within groups using the paired t-test. The mixed-model repeated-measures analysis during the acute phase included only visit and investigator. Statistical significance was defined as P≤0.05.
RESULTS
Study flow
The study design is summarised in the flow chart shown in Fig. 1. A total of 533 participants met the enrolment criteria during the screening phase, entered the acute phase of the study, and received open-label duloxetine (60 mg daily) for up to 12 weeks. Of these, 280 participants (52.5%) met the response criteria for entering the continuation phase. All but 2 participants (n=278) agreed to enter the double-blind continuation phase. In total, 142 patients were randomised to placebo and 136 patients were randomised to duloxetine (60 mg daily).
Baseline characteristics of patients
There were no significant differences in baseline characteristics (ethnic origin, age or gender) between patients who received duloxetine (60 mg daily) and those who received placebo during the continuation phase (Table 1). At the time of randomisation in week 12 (and therefore at baseline for the continuation phase of the study) there were no significant differences between participants who were randomised to the two treatment groups with regard to HRSD17 total score, CGI–S score, PGI–I score and visual analogue scale overall score.
Table 1 Patient baseline characteristics
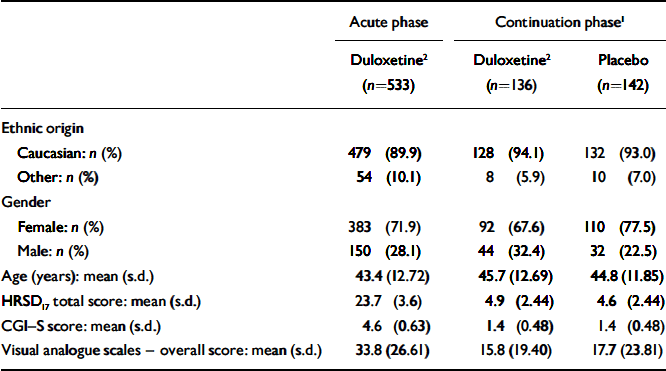
| Acute phase | Continuation phase1 | ||
|---|---|---|---|
| Duloxetine2 (n=533) | Duloxetine2 (n=136) | Placebo (n=142) | |
| Ethnic origin | |||
| Caucasian: n (%) | 479 (89.9) | 128 (94.1) | 132 (93.0) |
| Other: n (%) | 54 (10.1) | 8 (5.9) | 10 (7.0) |
| Gender | |||
| Female: n (%) | 383 (71.9) | 92 (67.6) | 110 (77.5) |
| Male: n (%) | 150 (28.1) | 44 (32.4) | 32 (22.5) |
| Age (years): mean (s.d.) | 43.4 (12.72) | 45.7 (12.69) | 44.8 (11.85) |
| HRSD17 total score: mean (s.d.) | 23.7 (3.6) | 4.9 (2.44) | 4.6 (2.44) |
| CGI—S score: mean (s.d.) | 4.6 (0.63) | 1.4 (0.48) | 1.4 (0.48) |
| Visual analogue scales — overall score: mean (s.d.) | 33.8 (26.61) | 15.8 (19.40) | 17.7 (23.81) |
Efficacy
Patients who received duloxetine (60 mg daily) during the continuation phase had a significantly longer time to relapse than patients who were on placebo (P=0.004). The Kaplan–Meier plot of time to relapse showed that patients on duloxetine separated from those on placebo as early as 1 month after randomisation (Fig. 2), and that this separation increased over time. At the end of the study, the estimated probabilities of relapse were 38.3% and 19.7% for patients who received placebo and duloxetine (60 mg daily) respectively. Significantly fewer patients who received duloxetine (60 mg daily) relapsed compared with patients who received placebo (P<0.05) (Table 2).
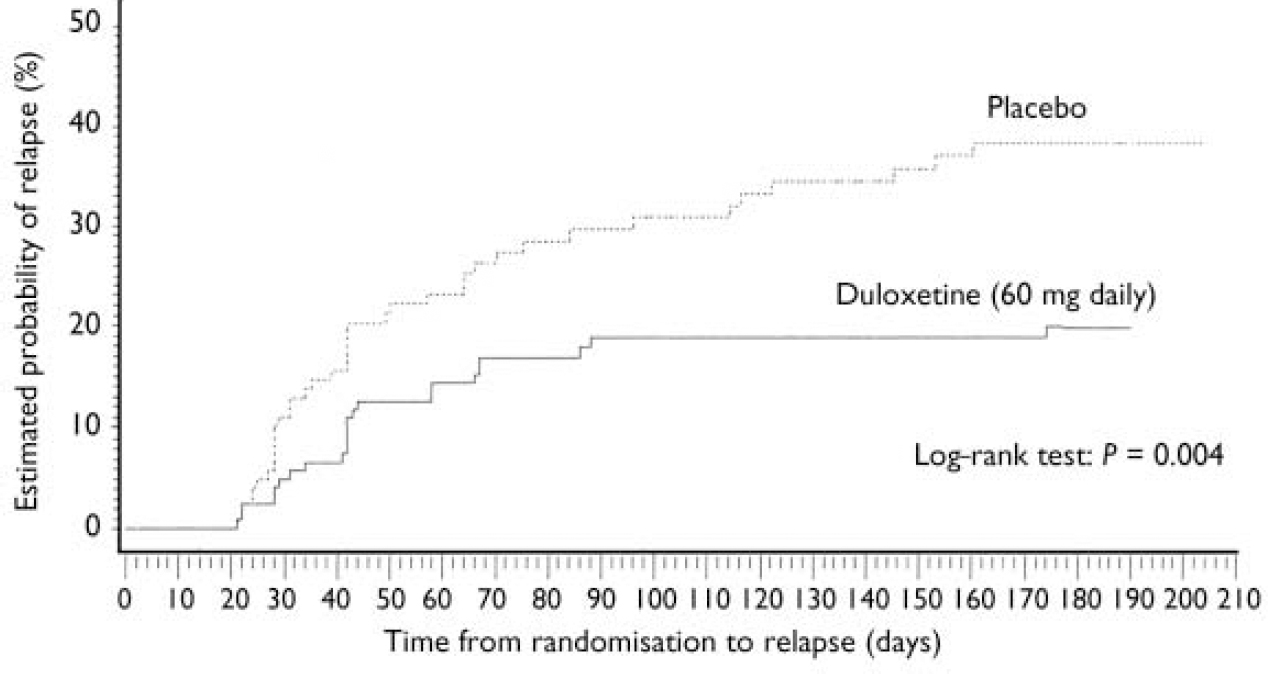
Fig. 2 Kaplan–Meier plot of time from randomisation to relapse. The estimated probability of relapse following randomisation was compared for participants who received either duloxetine (60 mg daily) or placebo during the continuation phase using the log–rank test. The treatment groups separated as early as 1 month after randomisation. Duloxetine-treated patients had a longer time to relapse than patients who received placebo (P=0.004).
Table 2 Relapse rates for patients who received duloxetine (60 mg daily) v. placebo during the continuation phase
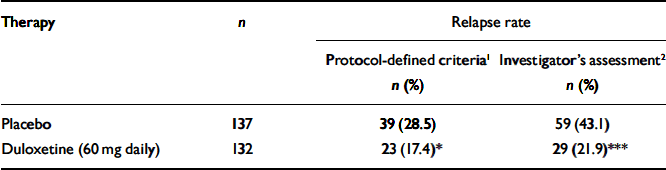
| Therapy | n | Relapse rate | |
|---|---|---|---|
| Protocol-defined criteria1 n (%) | Investigator's assessment2 n (%) | ||
| Placebo | 137 | 39 (28.5) | 59 (43.1) |
| Duloxetine (60 mg daily) | 132 | 23 (17.4)* | 29 (21.9)*** |
In total, 26 participants (6 patients from the duloxetine (60 mg daily) treatment group and 20 patients from the placebo treatment group) entered the rescue phase without completely satisfying the protocol's criteria for the definition of relapse. They did have two consecutive visits which met the requirements for relapse, but the second visit occurred less than 2 weeks after the first one. They were entered into the rescue phase because the investigator judged them to require additional therapy, independent of the protocol criteria. However, their average HRSD17 total score before entry into the rescue phase was 18.8. These patients were not considered to be relapsed in the primary efficacy analysis. This is a statistically conservative approach. If these 26 patients had been included as relapsed in the log-rank test, the relative advantage of duloxetine in preventing relapse compared with placebo would have been even greater (Table 2).
Secondary efficacy measures
At the end-point of the acute phase, patients who were receiving duloxetine (60 mg daily) had a response rate of 68% (defined as a reduction of ≥50% in HRSD17 total score) and a remission rate of 53% (defined as an HRSD17 total score of ≤7) (Table 3). Compared with baseline (week 0), patients’ HRSD17 total scores had decreased after 1 week of duloxetine (60 mg daily), and were reduced at all subsequent visits (P<0.005) throughout the acute phase for patients who remained in the study (Fig. 3).

Fig. 3 HRSD17 total scores during the acute and continuation phases. In total, 533 patients who were diagnosed with major depressive disorder received open-label duloxetine (60 mg daily) during the acute phase. In the continuation phase, patients who met the protocol criteria in week 12 were randomised to either continue duloxetine (60 mg daily) (n=136) or to receive placebo (n=142) in a double-blind manner. Acute phase: HRSD17 total score (least square mean) decreased from week 1 to week 12 (P≤0.001). Continuation phase: HRSD17 total score (least square mean) decreased for patients who received duloxetine (60 mg daily) compared with patients who received placebo at all visits starting in week 14 (P≤0.001). HRSD17, 17=item, 17=item Hamilton Rating Scale for Depression.
Table 3 Response and remission rates during treatment with duloxetine

| Study phase | Treatment | n | Response rate1 n (%) | Remission rate2 n (%) |
|---|---|---|---|---|
| Acute phase | Duloxetine (60 mg daily) | 511 | 347 (68.0) | 270 (52.8) |
| Rescue phase | Duloxetine (60 mg daily) | 56 | 43 (76.8) | 32 (57.1) |
| Duloxetine (60 mg twice daily) | 29 | 18 (62.1) | 11 (37.9) |
During the continuation phase, patients who received duloxetine (60 mg daily) experienced less worsening of depressive symptoms compared with patients who received placebo (P<0.01), as assessed by their change in HRSD17 total scores at all time points from week 14 to week 38 (Fig. 3). The 29 patients who received duloxetine (60 mg daily) and the 58 patients who received placebo who satisfied the protocol's criteria for relapse (see Method) or who were judged by the investigator to have relapsed (as described earlier) were then offered the option of entry into the study's rescue phase (Fig. 1). Response and remission rates at the end-point of the rescue phase are shown in Table 3.
The mean change from baseline to end-point for multiple secondary efficacy measures was also examined during the continuation phase. Patients who received duloxetine (60 mg daily) achieved or maintained significantly greater improvement in physical and emotional symptoms (on all measures except visual analogue scales for overall pain) compared with patients who received placebo (Table 4). In addition, for all visits from week 14 to week 38, duloxetine (60 mg daily) treatment was superior to placebo treatment in terms of patients’ PGI–I scores.
Table 4 Secondary efficacy measures during continuation phase
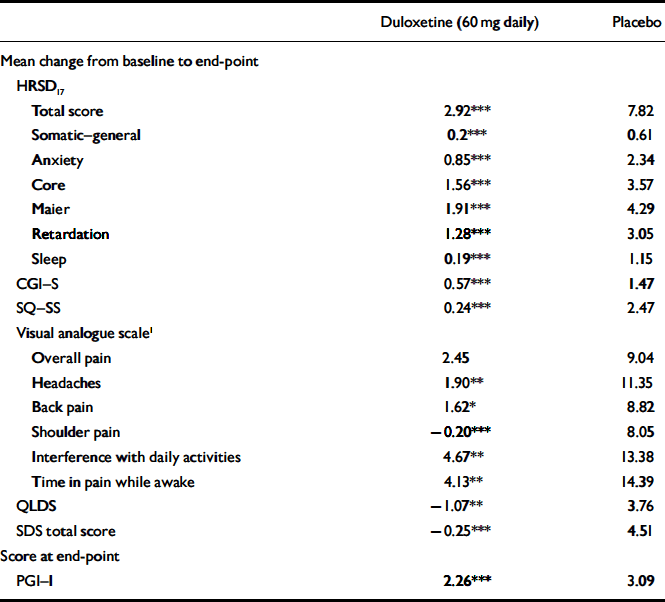
| Duloxetine (60 mg daily) | Placebo | |
|---|---|---|
| Mean change from baseline to end-point | ||
| HRSD17 | ||
| Total score | 2.92*** | 7.82 |
| Somatic—general | 0.2*** | 0.61 |
| Anxiety | 0.85*** | 2.34 |
| Core | 1.56*** | 3.57 |
| Maier | 1.91*** | 4.29 |
| Retardation | 1.28*** | 3.05 |
| Sleep | 0.19*** | 1.15 |
| CGI—S | 0.57*** | 1.47 |
| SQ—SS | 0.24*** | 2.47 |
| Visual analogue scale1 | ||
| Overall pain | 2.45 | 9.04 |
| Headaches | 1.90** | 11.35 |
| Back pain | 1.62* | 8.82 |
| Shoulder pain | -0.20*** | 8.05 |
| Interference with daily activities | 4.67** | 13.38 |
| Time in pain while awake | 4.13** | 14.39 |
| QLDS | -1.07** | 3.76 |
| SDS total score | -0.25*** | 4.51 |
| Score at end-point | ||
| PGI—I | 2.26*** | 3.09 |
Tolerability and adverse events
Acute phase
Investigators rated the severity of adverse events as mild, moderate or severe based on the patient's discomfort, the health risk and/or interference with the patient's activity. The five most frequently reported treatment-linked adverse events were nausea (n=191 (36%); 90% mild to moderate severity), headache (n=108 (20%); 86% mild to moderate severity); dry mouth (n=96 (18%); 96% mild to moderate severity); somnolence (n=72 (14%); 85% mild to moderate severity) and insomnia (n=56; (11%); 79% mild to moderate severity). Adverse events that were reported as reasons for discontinuation of treatment during the acute phase (expressed as number of patients and percentage for whom ‘adverse event’ was given as the reason for discontinuation) included nausea (n=11; 2.1%), somnolence (n=4; 0.8%), suicide attempts (n=3; 0.6%) and vomiting (n=3; 0.6%). In addition, one patient died by suicide on day 16 during the acute phase of the study. The investigator judged the suicide to be unrelated to the study drug.
Continuation phase
There were no statistically significant differences in the rate of adverse event reports between patients who received duloxetine and those who received placebo. During the continuation phase, anxiety, cholelithiasis, spastic colon, ejaculation failure and gastroesophageal reflux disease were reported by 1 patient each as reasons for discontinuation of duloxetine (60 mg daily) (n=5; 3.7%). Back pain, hypomania, insomnia, abnormal liver function tests and paraesthesia were each reported by 1 patient who was taking placebo (n=5; 3.5%) as reasons for study discontinuation.
Follow-up phase
No adverse events were reported as reasons for discontinuation during the follow-up phase. For patients who entered the follow-up phase from the continuation phase, adverse event rates of more than 2% were reported by duloxetine-treated patients for the following: dizziness (13.2%); insomnia (3.9%); headache, abnormal dreams, asthenia, tinnitus and fatigue (2.6% each). Abdominal pain (upper) and pruritis were each reported by one placebo-treated patient.
Cardiovascular safety
There were no clinically significant mean changes in blood pressure or heart rate between baseline and the end-point in any of the groups in any of the study phases.
Acute phase
There was no significant mean change in patients’ diastolic blood pressure between baseline and the end-point during this phase. Average heart rate increased by approximately 1.7 beats/minute (P<0.005) and average systolic blood pressure increased by approximately 1.3 mmHg (P<0.05).
Continuation phase
During this phase there were no significant differences in mean changes in systolic blood pressure or heart rate between patients who received duloxetine (60 mg daily) and those who received placebo. Patients who received duloxetine (60 mg daily) showed increased mean changes in diastolic blood pressure compared with patients who received placebo (2.0 mmHg v. 71.0 mmHg; P=0.005).
Laboratory values
There were no clinically significant or persistent changes in laboratory results for patients who received duloxetine during any of the study phases. Mild to moderate increases in serum aspartate aminotransferase or alanine aminotransferase levels were reported for 37 patients during the acute phase. During the continuation phase, 4 placebo-treated patients and 7 duloxetine-treated patients had elevated alanine aminotransferase levels, and 3 placebo-treated patients and 4 duloxetine-treated patients had elevated aspartate aminotransferase levels. In all cases, the elevated hepatic enzyme levels resolved spontaneously and the patients remained in the study. During the continuation phase, one patient who was treated with placebo for 14 weeks experienced concomitant elevated transaminase and bilirubin levels, with no evidence of haemolysis or hyperbilirubinaemia. The patient was withdrawn from the study and the elevated transaminase and bilirubin levels resolved spontaneously within 1 month.
One female patient had an increased serum prolactin level (36.5 mg/l; normal range 1.39–54.2 mg/l) 2 days before starting the study drug in the acute phase, and was prescribed nomegestrol acetate for 173 days. At the time of randomisation to duloxetine (60 mg daily) her prolactin level was still elevated (35.6 mcg/l). This patient completed the continuation and follow-up phases of the study.
DISCUSSION
In addition to the undoubted impact of depression on individuals and their social networks, the effective treatment of depression, including the prevention of subsequent depressive relapses, has important economic benefits. In 2000, the estimated total cost of adult depression in England was over £9 billion (in excess of US$ 15.5 billion), with nearly 110 million working days lost and 2615 deaths (Reference Thomas and MorrisThomas & Morris, 2003). In the USA, during the same year the economic burden of depression was estimated to be over US$ 83 billion (£47 billion) (Reference Greenberg, Leong and BirnbaumGreenberg et al, 2003).
Continuing treatment
Once an antidepressant has proved to be effective for and well tolerated by an individual patient during the acute treatment of a depressive episode, treatment guidelines (e.g. World Health Organization, 1989) recommend continuing treatment with that antidepressant for at least a further 6 months to minimise the risk of depressive relapse. This recommendation is based on data which clearly show the effectiveness of a number of antidepressants in reducing the risk of depressive relapse. A recent systematic review of 31 antidepressant trials found that continuation treatment with antidepressant reduced the odds of depressive relapse by 70% compared with placebo (Reference Geddes, Carney and DaviesGeddes et al, 2003). Another recent literature review estimated relapse rates of approximately 33–50% in patients for whom antidepressant treatment is not maintained, compared with only 10–15% in patients who receive continued treatment (Reference HirschfeldHirschfeld, 2001). It must therefore be possible to demonstrate that an antidepressant has the ability to prevent relapses of depression as well as having efficacy in the acute treatment of a depressive episode. In the present study, after 12 weeks of acute treatment with duloxetine (60 mg daily), 82.6% of patients who were randomised to continue duloxetine treatment at the same dose for a further 26 weeks remained relapse-free, compared with 71.5% of patients who were randomised to placebo. This is a statistically and clinically significant difference which demonstrates the efficacy of duloxetine in the prevention of depressive relapse. These results are consistent with the findings of similar studies of other antidepressants, including mirtazapine (Reference Thase, Entsuah and RudolphThase et al, 2001), citalopram (Reference Montgomery, Rasmussen and TanghojMontgomery et al, 1993) and escitalopram (Reference Rapaport, Bose and ZhengRapaport et al, 2004).
Symptom resolution
The patient's expectation of antidepressant therapy is resolution of the full spectrum of depressive symptoms, and resolution of both the emotional and somatic symptoms associated with depression is increasingly being recognised as an important treatment goal (Reference FavaFava, 2002). During the continuation phase of this study, when compared with patients who received placebo treatment, participants who received duloxetine (60 mg daily) experienced better maintenance of efficacy as assessed by multiple clinician-rated and patient self-reported measures of both emotional symptoms (including the HRSD17 total score, multiple sub-scales of the HRSD17, PGI–I and CGI–S) and somatic symptoms (including the HRSD17 item 13 (somatic–general), the SQ–SS, and 4 out of 5 measures of the visual analogue scales for pain, namely headaches, back pain, shoulder pain and pain while awake). In addition, duloxetine-treated patients reported increased interest in daily activities, improved quality of life (as assessed by the QLDS) and decreased disability (as assessed by the SDS total score), compared with patients who received placebo.
Transient worsening and adverse events
Although patients who were treated with duloxetine had better outcomes on the primary outcome measure and on 17 out of 18 secondary outcome measures compared with placebo-treated patients, an unexpected finding was the apparent transient worsening (as indicated by an increase in mean HRSD17 total score) of the condition of patients in both the duloxetine- and placebo-treated groups for approximately 10 weeks after randomisation, before there was a subsequent improvement (Fig. 3). The cause of this might be related to a possible state of uncertainty and anxiety induced in patients who were aware that after the period of open-label treatment they had a 50% chance of being randomised to continuation treatment with placebo. Although investigators were also blinded to the patients’ treatments after randomisation, investigator bias during the evaluation of patients might also be implicated in this observation, and it should be noted that the success of blinding for patients and assessors was not evaluated. A further possible explanation for the transient worsening of the condition of study participants after randomisation is the occurrence of adverse events linked to discontinuation in patients who were randomised to placebo. Such adverse events may mimic the symptoms of a depressive relapse if they are sufficiently severe and/or long-lived, and were not explicitly elicited in this study by a specific scale such as the Discontinuation, Emergent Signs and Symptoms scale (Reference Rosenbaum, Fava and HoogRosenbaum et al, 1998). Arguing against this possible explanation is the observation that transient worsening after randomisation occurred in patients who were randomised to both placebo and duloxetine, where in the latter group there was no interruption of duloxetine treatment to permit the appearance of discontinuation-linked adverse events. The tolerability of duloxetine for patients with MDD has been well characterised in 6 double-blind placebo-controlled and active comparator-controlled clinical trials (Reference Nemeroff, Schatzberg and GoldsteinNemeroff et al, 2002). In this study, nausea was the most frequently reported treatment-linked adverse event during the acute phase. It was most often mild or moderate in severity, and it was reported as a reason for discontinuation for 2.1% of patients. These findings are consistent with the results of previous studies of duloxetine in the acute treatment of MDD. For those patients who met the entry criteria and proceeded to the continuation phase, there were no significant differences in the frequencies of treatment-linked adverse event reporting between patients who received duloxetine and those who received placebo. The number of adverse events was low overall, with only 5 patients in each treatment group (duloxetine (60 mg daily), n=136; placebo, n=142) reporting adverse events as reasons for discontinuation. Among patients in whom duloxetine (60 mg daily) was well tolerated (as evidenced by completion of the 12-week acute phase) and efficacious (as evidenced by meeting the criteria for entry into the continuation phase), only 3.6% reported any treatment-linked adverse events as reasons for discontinuation over the next 26 weeks of the study. During both the acute phase and the continuation phase, duloxetine treatment of patients with MDD was not associated with clinically important changes in cardiovascular function or laboratory results between baseline and the end-point. Although in some cases there were statistically significant changes in some parameters, these changes were of small magnitude and were not clinically significant, which suggests that overall duloxetine is well tolerated during long-term therapy, and that its treatment-linked adverse event profile is similar to that reported previously for duloxetine in acute trials and for marketed selective serotonin reuptake inhibitors (SSRIs).
Strengths and limitations
Particular strengths of this study include the following: the relatively large sample size compared with that of similar studies; the similarity of the acute phase to clinical practice by virtue of the use of open-label treatment; the reduction of bias during the continuation phase because of the subsequent double-blinding of both patients and investigators to active treatment v. placebo and the taper of duloxetine for patients who received placebo; the multiple and independent measures of both the emotional and somatic symptoms associated with MDD; and the evaluation of the efficacy of dose-doubling as a strategy for patients who are experiencing relapse (to be discussed elsewhere).
The limitations of the study include the fact that the design did not address the comparison of duloxetine with currently available antidepressants. In addition, caution should be exercised when extrapolating the results reported here (and in most clinical trials) to clinical practice, as the patients in this study may not be entirely representative of the general clinical population for a number of reasons, including the use of inclusion and exclusion criteria during the selection of study participants, and the number and frequency of study visits, neither of which are representative of typical clinical practice.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
▪ Prevention of relapse by continuation of therapy beyond12 weeks is an important aspect of the treatment of depressive illness.
-
▪ Duloxetine, an inhibitor of serotonin and noradrenaline reuptake, is effective in preventing depressive relapse.
-
▪ Continuation treatment with duloxetine (60 mg daily) was not associated with the occurrence of adverse events at a greater frequency than that seen with placebo treatment.
LIMITATIONS
-
▪ Caution should be exercised when extrapolating these results to general clinical practice, because of the highly selected nature of the population that was studied.
-
▪ The study did not employ an established antidepressant drug as an active comparator.
-
▪ Discontinuation-linked adverse events may mimic some signs of depressive relapse, and were not specifically elicited in this study.
Acknowledgement
The authors gratefully acknowledge the editorial and graphical assistance of Sharie Sipowicz, Eli Lilly and Company.








eLetters
No eLetters have been published for this article.