


Acute inflammatory demyelinating polyradiculoneuropathy (AIDP) is most frequently associated with rapidly progressive flaccid paralysis and areflexia. The estimated childhood incidence of AIDP (0–15 years of age) is 0.34 to 1.34 per 100,000 per year and the diagnosis of AIDP is often delayed in up to 2/3 of pre-school age children due to nonspecific clinical symptoms and intrinsic difficulties in performing neurological examinations in this age group.Reference Roodbol, de Wit, Walgaard, de Hoog, Catsman-Berrevoets and Jacobs 1 Further contributing to diagnostic inaccuracy is the presence of AIDP variants such as Miller-Fisher syndrome and the pharyngeal-cervical-brachial variant. The major concern with such phenotypic variations is that misdiagnosis or delay in treatment can have substantial effects on acute mortality and chronic morbidity.Reference Roodbol, de Wit, Walgaard, de Hoog, Catsman-Berrevoets and Jacobs 1 We report a case of a three year-old previously healthy girl with a purely autonomic variant of an AIDP-like syndrome consisting only of urinary and stool retention with nerve root enhancement with normal strength, (CSF) and nerve conduction studies responsive to intravenous immunoglobulin (IVIG) treatment.
Case Report
A previously healthy, developmentally and neurologically normal three year-old girl presented with a two-week history of acute onset urinary retention. Her symptoms began with abrupt onset painful swelling of the left knee with normal knee x-rays. The following day, she presented to her local emergency department with abdominal pain and no urinary voiding for over 16 hours. She was treated with in-and-out catheterization, prescribed an antibiotic and discharged. Her urine culture from that day later grew 1×106 coagulase-negative staphylococcus. She returned the following day to the emergency department with repeated urinary retention over 12 hours. She was transferred to a larger community hospital and managed with repeat in-and-out catherization. During her two week admission, she also began retaining stool with loss of spontaneous bowel movements. Bladder catheter removal resulted in significant abdominal distension and urinary retention requiring re-catheterization. The intermittent right knee pain continued with no identifiable cause. She was later transferred to our tertiary facility two weeks after onset of her initial symptoms.
The past medical history of this patient was unremarkable. She was a previously healthy child, born at term after an uneventful pregnancy. Her immunizations were up-to-date with no recent immunizations prior to her symptoms. She was on no medications and a comprehensive urine toxicology screen was negative. She lived with her parents on a farm and they reported keeping all herbicides and pesticides in a locked cabinet. These materials were sprayed annually on the farm but not immediately prior to the development of her symptoms.
The neurology team discovered a history of a viral respiratory illness preceding the patient’s symptoms by two to three weeks. Her initial neurological exam was remarkable for absent right patellar and right ankle reflexes with all other deep tendon reflexes being normal. She had a normal level of consciousness and verbal interaction, normal cranial nerves, full 5/5 muscle strength in both the proximal and distal muscles of the upper and lower extremities, with normal tone and gait. Although she was often not cooperative with serial examinations, she appeared to have no sensory deficits and showed reaction to light touch/pinprick/temperature and vibration testing. She did not complain of pain and her spine was normal with no sacral dimple. See the Table 1 for a summary of pertinent investigations and results. Notably, cerebrospinal fluid (CSF) studies were normal including white blood cell count (WBC), protein and negative oligoclonal bands. Cerebrospinal fluid cultures were negative.
Table 1 Summary of Investigations
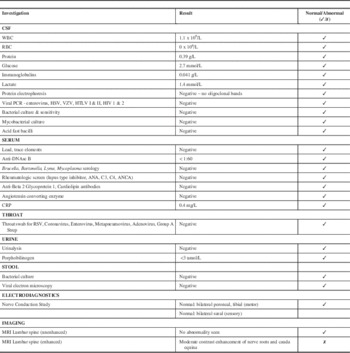
Notably, nerve conduction studies bilaterally of the peroneal and tibial motor nerves as well as sural nerve sensory studies were normal.
The initial non-enhanced 1.5T magnetic resonance imaging (MRI) of the brain and spinal cord was normal, with no evidence of cord tethering or myelopathy. Despite the normal neuroimaging and CSF studies, the working diagnosis was a possible transverse myelitis and was treated with methylprednisolone at 20 mg/kg over five days, nearly three weeks after symptom onset. There was no improvement in her neurologic exam or her symptoms of urine or stool retention.
A gadolinium-enhanced MRI brain and spine performed 30 days after symptoms onset revealed enhancement of the L3 and L4 lumbar nerve roots and the cauda equina, consistent with an acute demyelinating polyradiculoneuropathy (Figure 1). The patient was started on a five-day course of IVIG at 400 mg/kg/day. Over the IVIG treatment and thereafter, the patient steadily regained both bladder and bowel function. At neurological followup one month post-discharge she had six to eight spontaneous bladder voids daily and normal bowel movements. Her strength and gait continued to be normal with continuing absence of her right patellar and Achilles tendon reflexes. Her recovery was complicated by a relapse in bladder dysfunction requiring recurrent catheterizations and frequent urinary tract infections six months later. She continued to have a depressed (1+) right patellar deep tendon reflex. A repeat enhanced cervical and lumbar spine MRI 14 months after the initial MRI was normal and showed resolution of the previously noted root enhancement.
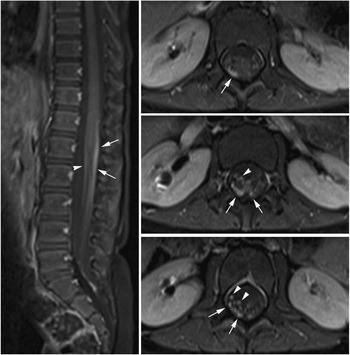
Figure 1 MR of the spine with contrast. Fat-suppressed post-gadolinium T1-weighted images at sagittal and axial planes of the thoracolumbar spine show thickening and moderate contrast enhancement of the nerve roots of the cauda equina. Both the anterior (arrowheads) and posterior (arrows) nerve roots are involved, but more prominent changes are seen in the posterior groups of the nerve roots. This posterior predominance is unusual and was not seen in the previously reported large cohort of children with Guillain–Barre syndrome.Reference Yikilmaz, Doganay, Gumus, Per, Kumandas and Coskun 2
Discussion
Autonomic dysfunction is well described in AIDP, occurring in up to two-thirds of cases. Problems include blood pressure variability, orthostatic hypotension, cardiac dysrhythmias and bowel and bladder dysfunction. The degree of dysautonomia is variable and is most commonly seen later in the course of severe cases of AIDP. Despite this, there are several reports of autonomic dysfunction occurring as the presenting feature in AIDP, predominantly in the adult literature. The pediatric literature is limited to three cases of dysautonomia in older children as a presenting feature of AIDP, all in the context of severe dysesthesia. Our case is the first to describe a toddler with a purely autonomic, painless presentation of an AIDP-like syndrome. While the patient did not meet strict clinical or electrodiagnostic criteria for AIDP, there was evidence of nerve root enhancement and response to IVIG that suggests this case falls in the spectrum of an AIDP-like syndrome, and highlights the need for consideration of this diagnosis in this young population.
The most recent pediatric report of an autonomic, dysesthetic variant of AIDP/Guillain-Barre syndrome (GBS) was described by Dixon et al. in 2001.Reference Dixon, Appleton, Davidson, Hughes and Tedman 3 Two children, aged 14 and 10 years, presented with burning distal dysesthesias after a presumed viral infection. They were hypertensive (patient 1 also had postural hypotension) and had a hypertensive encephalopathy with headache and symptomatic seizures. Their bowel and bladder function were normal. As in our case, their neurologic examinations were significant for retained deep tendon reflexes and normal motor and sensory function (aside from the dysesthesia). Cerebrospinal fluid analyses were normal. Nerve conduction studies were also normal but quantitative somatosensory testing was abnormal and consistent with a small fiber neuropathy affecting both sensory and autonomic fibers. An MRI brain showed cortical changes consistent with a hypertensive encephalopathy. Intravenous immunoglobulin and steroids did not result in any noticeable symptom improvement. Both patients’ painful dysesthesias and autonomic instability gradually resolved over approximately three months.
Nass and ChutorianReference Nass and Chutorian 4 described three children aged 11 years, 10 years and 8 years with an acute, self-limited syndrome of painful distal dysesthesias, sustained hypertension and autonomic dysfunction in the context of normal motor function and deep tendon reflexes. All had normal CSF studies and two patients had features of mild sensory neuropathy on nerve conduction studies. Two of the patients described mild constipation but all had normal bladder function. The outcomes of these patients were not reported.
Singer and BackReference Singer and Back 5 reported a seven year old child with persistent extremity and back pain over five days and new hypertension with one episode of ventricular tachycardia. Bowel function was normal as were muscle strength and deep tendon reflexes. On day three of admission, deep tendon reflexes were absent and later nerve conduction velocities were reduced with elevated CSF protein. The patient quickly deteriorated and required mechanical ventilation for 19 days. After discharge the hypertension resolved and the patient’s neurologic recovery was complete.
In our case, we suspect the nerve conduction studies (NCS) were normal due to small fiber involvement and sparing of the large nerve fibers tested by NCS. The focal MRI nerve root enhancement and thickening was thought to be a sign of pathology rather than post-lumbar puncture enhancement due its focal location, posterior predominance and persistence at one month post lumbar puncture with correlation to areas clinically affected in the patient. There was no sensory disturbance, in particular no saddle anesthesia, to suggest conus medullaris syndrome.
Currently our patient does not meet clinical, electrodiagnostic or CSF criteria for chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) however close clinical and neuroimaging followup will be required in light of the nerve root thickening and enhancement that can be seen in a significant proportion of children with CIDP. In the majority of cases, both children and adults describe significantly painful dysesthesias that prompted patients to rapidly seek attention and likely served as a useful diagnostic clue to physicians. Our case is unique in that it is the first report of a purely autonomic, painless AIDP-like syndrome occurring in a toddler. Despite the rarity of a purely autonomic presentation of AIDP in children, this case serves as a reminder for clinicians to consider atypical presentations of AIDP in children. A delay in proper diagnosis can result in worsening respiratory insuffiency and autonomic instability and delays in treatment can adversely affect long-term outcomes.
Disclosures
Claire Hinnell, Xing-Chang Wei, Jean Mah and Thilinie Rajapakse do not have anything to disclose.
Jong Rho has the following disclosures: UCB Canada, Consultant/Speaker, Honoraria; Eisai Canada, Consultant/Speaker, Honoraria.


