


Muscular dystrophies (MDs) are hereditary diseases that cause progressive muscle weakness. Significant variation in disease penetrance and expressivity among the causative genes and pathogenic variants complicates clinical diagnosis. Although muscle biopsy has been the traditional tool used by clinicians to narrow down the subtype of MD in a patient, phenotypic overlap plus nonspecific histological changes results in significant diagnostic difficulties.
Until recently, genetic testing for MDs was only done with Sanger sequencing, considered to be the gold standard for more than 30 years.Reference Mardis 1 Introduced in 2005, NGS is a high-throughput technology that provides a broader and more cost-effective method of genetic testing in comparison to Sanger sequencing’s gene-by-gene analysis.Reference Schuster 2 NGS has the potential to be one of the most powerful diagnostic tools in the workup of patients with histories suspicious for a genetic muscle disease. A handful of studies have already examined the use of NGS in the diagnostic workup of undiagnosed MD. These populations were primarily pediatric and reported detection rates ranging from 15% to 65%.Reference Evilä, Arumilli, Udd and Hackman 3 - Reference Sevy, Cerino and Gorokhova 8
Consideration of an underlying genetic muscle disease does not only occur for patients who present with muscle weakness. Patients with recurrent episodes of rhabdomyolysis or idiopathic hyperCKemia (creatine kinase [CK] >220 U/L) are often referred to neuromuscular specialists for evaluation to rule out neuromuscular disease. Genetic causes for these presentations can include McArdle disease, mitochondrial disease, fatty acid oxidation defects, Duchenne/Becker MD, or malignant hyperthermia susceptibility.Reference Sauret, Marinides and Wang 9 In some cases, rhabdomyolysis and seemingly idiopathic hyperCKemia are later realized to have been the first signs of a latent MD.Reference Klein 10 In other cases, muscle issues such as episodic rhabdomyolysis with or without myoglobinuria have a nongenetic cause such as idiopathic inflammatory myopathies, substance abuse, strenuous exercise, and/or toxins.Reference Huerta-Alardin, Varon and Marik 11
In this study, an NGS panel of 163 to 183 genes associated with MD was used in the workup of patients referred to a Canadian neuromuscular clinic for evaluation of muscle weakness, recurrent episodes of rhabdomyolysis, or asymptomatic hyperCKemia. The population of patients in this study are primarily adults with adult-onset symptoms. This retrospective study adds to the existing literature demonstrating the utility of NGS as a tool in the molecular characterization of MD in both pediatric and adult populations. This study also is one of the first to quantify the utility of genetic testing with a muscle disease focused NGS panel in the workup of patients with recurrent rhabdomyolysis and idiopathic hyperCKemia.
METHODS
Patients
The participants in this study were patients assessed in a tertiary pediatric and adult neuromuscular clinic in Hamilton, Ontario, over a 34-month period. Patients were of differing ethnicities and ages (median age, 43 years; range, 0-80).
The multigene NGS panel was ordered for patients where there were more than two genes on the differential diagnosis. This excluded any patients suspected to have an acquired or inflammatory myopathy (e.g. statin-associated myopathy, sporadic inclusion body myositis) or who reported any additional environmental exposures or risk factors for rhabdomyolysis (e.g. statins). For patients with unexplained hyperCKemia, the NGS panel was ordered if they had any of the following; a recorded CK value that was three times above the upper limit of normal (based on ethnicity), had a family history of similarly elevated CK (nonblack), or was below the age of 12 years of age.Reference Kyriakides, Angelini and Schaefer 12 This study also excluded patients with classic features of a single distinctive MD (e.g., myotonic dystrophy type 1 or Duchenne MD) who underwent genetic testing for only that condition.
The 169 patients who met these criteria were categorized into one of three groups based on the most significant manifestation of their muscle disease at the time of consultation; clinical muscle weakness (n=135), recurrent rhabdomyolysis (n=18), or idiopathic hyperCKemia (n=16).
Sequencing Analysis
Samples were referred to Medical Neurogenetics, LLC, for Clinical Laboratory Improvement Amendments–certified diagnostic NGS testing. The first 38 patients were tested using the 163-gene panel, the next 56 patients were tested using an expanded panel of 176 genes, and the final 76 patients were tested using a further expanded panel of 183 genes. Supplementary Table 4 presents the full list of genes tested in each panel.
Testing was performed using an Illumina HiSeq 1500 (Illumina, San Diego, CA) according to the manufacturer’s guidelines as well as proprietary approaches developed at Medical Neurogenetics, LLC. NGS sequencing was performed and variant classification was compliant with American College of Medical Genetics (ACMG) standards and guidelines.Reference Richards, Aziz and Bale 13 - Reference Richards, Bale and Bellissimo 17 Based on patient clinical information, gene coverage was assessed and sequenced using dideoxy-sequencing (Sanger sequencing) as necessary using an ABI 3730x1 automated sequencer (Life Technologies, Foster City, CA). Annotation was performed using GRCh37.p13 (GCA_000001405.14) assembly.
Variant Classification
Classification of genetic variants for the patient phenotypes were assessed using criteria outlined by the ACMG.Reference Richards, Bale and Bellissimo 17 Variants identified in patients before the publication of the ACMG guidelines underwent reinterpretation in 2017. Both pathogenic and likely pathogenic variants were considered in the calculation of detection rate.
Patient demographics, clinical phenotypes, muscle biopsy pathology, and genetic results are listed in Supplementary Table 1 (muscle weakness), Supplementary Table 2 (recurrent rhabdomyolysis), and Supplementary Table 3 (hyperCKemia). All muscle pathology showed normal immunohistochemistry unless otherwise stated. Mean allele frequency data were obtained from the Exome Aggregation Consortium database.
RESULTS
Pathogenic and likely pathogenic variants were identified in 36.09% of all patients (61/169). No likely causative variants were identified in 55.62% (94/169). Twenty-eight of the pathogenic and likely pathogenic variants reported here are novel and have not previously been associated with disease.
Muscle Weakness Cohort
Pathogenic or likely pathogenic variants were seen in 37.04% (50/135) of patients who presented with muscle weakness (Figure 1A). Variants of uncertain clinical significance possibly related to disease phenotype were identified in 8.14% (11/135). No candidate variants were detected in 54.8% (74/135) of the cases. When examining only the patients with pediatric-onset symptoms, the detection rate was 38.30% (17/47 pathogenic, 1/47 likely pathogenic). Detection rate for patients with adult-onset weakness was 36.36% (26/88 pathogenic, 6/88 likely pathogenic). Testing did not identify any candidate variants in 57.44% and 53.40% of the pediatric and adult cohorts, respectively. ANO5-related myopathies were the most common diagnosis, seen in five patients (Supplementary Table 1).
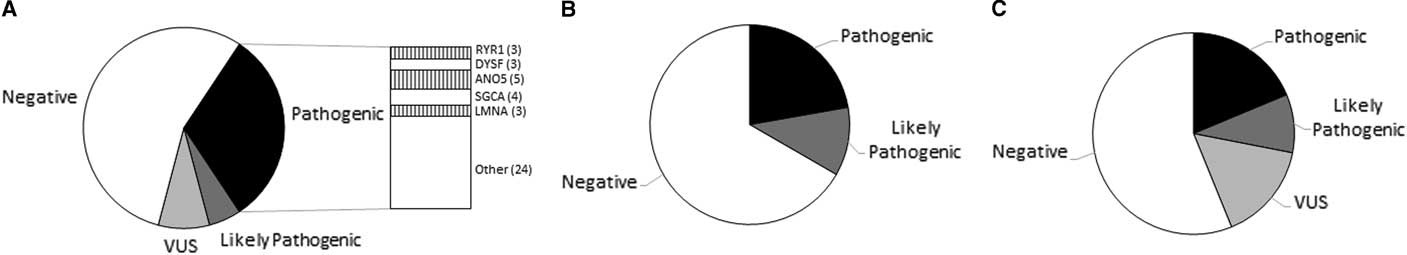
Figure 1 Graphical representation of variant classification of patients referred for evaluation of (A) muscle weakness, (B) recurrent episodes of rhabdomyolysis, and (C) asymptomatic elevation of creatine kinase.
Recurrent Rhabdomyolysis Cohort
Pathogenic or likely pathogenic variants were seen in 33.33% (6/18) of patients with recurring episodes of rhabdomyolysis (Figure 1B). No candidate variants were detected in 66.66% (12/18) of the cases (Supplementary Table 2).
Idiopathic HyperCKemia Cohort
Pathogenic or likely pathogenic variants were seen in 31.25% (5/16) of the idiopathic hyperCKemia cohort (Figure 1C). No candidate variants were detected in 50.0% (8/16) of the cases (Supplementary Table 3).
DISCUSSION
There are now several studies in the literature demonstrating the utility of disease-focused NGS panels in the molecular characterization of undiagnosed muscular dystrophy, reporting detection rates ranging from 15% to 65%.Reference Evilä, Arumilli, Udd and Hackman 3 - Reference Sevy, Cerino and Gorokhova 8 Other studies, such as the MYO-SEQ project, use a different NGS platform genetic test known as whole exome sequencing. Unlike a fixed disease-focused panel, whole exome sequencing is an unbiased transcriptomics technique for sequencing the all the protein-coding regions of the entire human genome. The MYO-SEQ project has reported a detection rate of approximately 49% in patients with limb-girdle weakness.Reference Johnson, Bertoli and Phillips 18 In this present report, the muscle-focused NGS panel used identified pathogenic and likely mutations causative for disease were in 37.04% (50/135) of the muscle weakness cohort. Many of the aforementioned studies had a heavyReference Dai, Wei and Zhao 6 , Reference Seong, Cho and Park 7 or exclusiveReference Chae, Vasta and Cho 5 focus on patients with early-onset/congenital MDs. This is quite different from our cohort, in which 65.19% (88/135) of the patients with muscle weakness did not have symptoms until adulthood; however, the age of onset did not appear to have a significant impact on the detection rates. Pathogenic and likely pathogenic variants were identified in 38.30% of patients with childhood-onset symptoms and in 36.36% with adult-onset symptoms. The similarity in detection rates was unexpected because it is generally accepted that genetic disorders are more likely to be identified in the pediatric population (e.g. early-onset/congenital MDs) because they are generally more severe and is less likely to be an acquired condition.Reference Retterer, Juusola and Cho 19 In contrast, a larger proportion of adult-onset MDs can be sporadic/acquired from external causes such as drugs (e.g. statins), toxins, endocrine abnormalities, aging effects on skeletal muscle, or neuropathies mimicking a myopathy.Reference Chawla 20 This study strictly excluded any patients suspected to have an acquired myopathy (e.g. statin-associated myopathy or sporadic inclusion body myositis), unless there were features on their clinical examination or family history suspicious of a hereditary myopathy. It is possible that the similarly in detection rates between the adult and pediatric populations were due to this strict exclusion of patients with a possible acquired or inflammatory myopathy. There remains the possibility of course that some of the patients included in this study may actually have an acquired condition and that some of the patients excluded may have a hereditary myopathy.
Interestingly, a number of patients found to have the same type of MD showed a high degree of heterogeneity in muscle pathology. In this study, we found that muscle biopsies with central cores (W12 and W68), Trabecular myopathy (W44), neurogenic changes (W93), and dystrophic changes and mitochondrial hyperplasia (W112) were all seen in patients with pathogenic changes in the ANO5 gene. Considering how muscle pathology can often be nonspecific or heterogeneous and the lower cost of NGS, it may be prudent to pursue genetic testing before a muscle biopsy. However, a muscle biopsy can still be an important part of the diagnostic process and may assist in the clarification of variants of uncertain clinical significance found by genetic testing. For example, a muscle biopsy suggestive of a desmin storage myopathy in the muscle biopsy of patient W24, who was found to have a variant of uncertain clinical significance in the DES assisted in the diagnosis when one parent was not available for family studies. Genetic testing, clinical examination, and other diagnostic tools such as electromyography and muscle biopsy are best used in conjunction during the diagnostic process.
This study is one of the first to investigate the utility of using a focused muscle disease NGS panel in the molecular diagnosis of patients referred to a neuromuscular clinic for evaluation of recurrent episodes of rhabdomyolysis or idiopathic hyperCKemia.
The European Federation of Neurological Societies recommends further investigation in individuals with apparently asymptomatic hyperCKemia if the CK level is continually more than three times the upper limit of normal, there are myopathic changes on electromyography, or the patient is younger than age 25 years.Reference Kyriakides, Angelini and Schaefer 12 Most studies to date have used more classic nongenetic diagnostic tools (e.g. muscle biopsy) in the evaluation of patients with asymptomatic elevated CK levels. One review by Moghadam-Kia et al found that combining these diagnostic methods resulted in discovering an etiology of the hyperCKemia in approximately 28% of patients,Reference Moghadam-Kia, Oddis and Aggarwal 21 many of which were found to be related to the use of statins. In excluding statins and other non-neuromuscular causes of hyperCKemia (e.g. alcohol/drug abuse), others found diagnoses of dystrophinopathies and other myopathies in approximately 10%.Reference Prelle, Tancredi and Sciacco 22 However, all but one of the papers in the review by Moghadam-Kia et al were published before 2008, a time when NGS was not available and access to genetic testing was more limited. With NGS testing, we found that 31.25% of our hyperCKemia cohort had pathogenic or likely pathogenic mutations in genes traditionally associated with limb-girdle MD (LGMD) (DYSF, ANO5, SGCA genes) (Supplementary Table 3). At the time of consultation, none of these patients reported any muscle weakness or functional limitations in daily activities. With the identification of mutations in ANO5 and DYSF, all patients were found to have very mild atrophy of the medial calf muscle(s), and some had some slight difficulty standing on their tiptoes. These changes were so mild that neither the patients nor the physicians in the initial examination had appreciated these changes. Interestingly, case CK7 actually had a family history of LGMD in a male sibling with onset in his 20s, but she herself did not show any functional muscle weakness at the age of 50. Our diagnostic rate of 31.25% suggests that genetic testing can be a useful tool in the workup of a patient with apparently idiopathic hyperCKemia. With the lower cost of NGS, it may even be prudent to pursue genetic testing before more invasive testing such as muscle biopsy.
More work has been done on the identification and investigation of genetic defects associated with recurring episodes of rhabdomyolysis. There are currently approximately 31 known inherited neuromuscular disorders that can confer this susceptibility.Reference Scalco, Gardiner and Pitceathly 23 All but one of the six cases of rhabdomyolysis that received a diagnosis in this study were found to have genetic mutations in one of the genes listed in Supplementary Table 1 of the review article by Scalco et al.Reference Scalco, Pitceathly and Gardiner 24 This case, RM11, was found to be homozygous for a pathogenic mutation in SGCA (c.850C>T). This case was published before this study.Reference Tarnopolsky, Hoffman, Giri, Shoffner and Brady 25 Findings such as this suggest that there may be other neuromuscular genes not yet reported to be associated with rhabdomyolysis. Additional studies are needed to assess the detection rates of genetic testing with rhabdomyolysis-focused panels in comparison to MD-focused panels such as the one used in this present study.
In addition to reporting detection rates of genetic testing for various types of muscle disease, this study identified a number of particularly interesting patients representing the complexities of diagnosis with NGS. One of the limitations of NGS, especially with older platforms, is that it may not be able to detect significant copy number variations (CNV). Other technologies such as multiplex ligation-dependent probe amplification (MLPA) and gene-specific array comparative genomic hybridization (aCGH) are considered the gold standard for testing for CNVs. Using one of these methods to test for deletions or duplications in dozens of genes for someone with a nonspecific LGMD can be cost prohibitive, especially with the likely low yield of copy number variants seen in this population. In this study, the identification of one pathogenic or likely pathogenic mutation by the NGS panel allowed for the selection of one or two additional genes that warranted additional testing for copy number variants. This stepwise process allowed for a diagnosis in four patients (W34, W88, W134, and R8). This was especially valuable because all four patients had clinical features of conditions with significant clinical heterogeneity. Case W34 carried a clinical diagnosis of congenital myasthenic syndrome since childhood. NGS testing revealed a de novo single heterozygous variant in AGRN (c.4525C>T) and a 7.9-kb AGRN deletion of exon 1-2 inherited from her mother (paternity and maternity confirmed). Case W88 presented with a nonspecific congenital MD with unremarkable immunohistochemistry. NGS testing revealed a single heterozygous variant in LAMA2 (c.8582T>G), which was found to be in trans with a 218.18-kb duplication of exon 2-4. Case W134 had a generic presentation of adult-onset proximal weakness and scapular winging. The NGS panel identified a pathogenic heterozygous mutation in CAPN3 and MLPA analysis revealed a large deletion of exons 2-8 of CAPN3. Case R8 reported multiple episodes of rhabdomyolysis since childhood and had a family history suspicious of possible autosomal dominant inheritance. NGS testing found the common c.148C>T (p.Arg50*) pathogenic mutation in PYGM, whereas subsequent aCGH showed a 1.1-kb deletion of exon 17. Although CNVs of LAMA2 are relatively common,Reference Oliveira, Santos and Soares-Silva 26 there have only been a few case reports of patients with deletions of CAPN3 Reference Todorova, Georgieva and Tournev 27 or PYGM. Reference Garcia-Consuegra, Rubio and Nogales-Gadea 28 This is the first report of a partial AGRN deletion in a patient with congenital myasthenic syndrome.
Although the combination of NGS followed by gene-specific aCGH or MLPA led to definite diagnoses for these four individuals, there was one case in which no second mutation was detected. Case W60 presented with classic features of hereditary inclusion body myopathies with rimmed vacuoles on muscle biopsy but only one pathogenic mutation in GNE (c.1225G>T). Sanger sequencing and aCGH analysis of GNE was performed but was unable to identify a second mutation or CNV. The NGS panel did not identify any other variants that could explain this patient’s phenotype and muscle pathology, raising the possibility that this patient’s second GNE mutation could occur in an intronic region not covered by testing.
At this time, it is believed that up to 15% of genetic diseases may be caused by intronic mutations.Reference Majewski, Schwartzentruber, Lalonde, Montpetit and Jabado 29 , Reference Botstein and Risch 30 Case reports of patients with deep intronic or other complex pathogenic mutations, proven with messenger RNA analysis and other in vitro studies, are becoming more recognized and have been reported in genes such as LAMA2,Reference Siala, Louhichi, Triki, Morinière, Fakhfakh and Baklouti 31 DOK7, Reference Selcen, Milone and Shen 32 and others.Reference Fanin, Nascimbeni, Tasca and Angelini 33 , Reference Cummings, Marshall and Tukianen 34 RNA sequencing from the muscle tissue of patients with undiagnosed muscular dystrophy is likely to provide a significantly higher diagnostic yield than NGS (panels and whole exome sequencing), though is not yet as widely available.Reference Wrighton 35 One of the most well-described examples of an intronic mutation in muscular dystrophy is the founder mutation in GAA (c.-32-13T>G), which is seen in more than half of all Caucasians with adult-onset Pompe disease.Reference Hule, Chen and Tsujino 36 This mutation was not detected by NGS testing in two cases (W33 and W49), who were later confirmed to have Pompe disease. For case W49, the NGS panel identified a heterozygous mutation (c.2242_2243insG) in GAA but no second mutation. Dried blood spot acid α-glucosidase testing was positive (1.21 pmol/punch/h [Ref>4.49 pmol/punch/h]). Sanger sequencing and MLPA revealed the second mutation as c.-32-13T>G. Both mutations in case W33 (c.-32-13T>G (+) c.525delT) were not detected by the NGS panel (data not shown) and were found after a positive dried blood spot (1.43 pmol/punch/h [Ref> 4.49 pmol/punch/h]) and Sanger sequencing of GAA. Although the proportion of pathogenic mutations found in introns versus exons is gene-dependent, it is important for clinicians to be wary of this restriction in the workup of patients with muscle diseases.
CONCLUSION
The MD-focused NGS panel used in the present study identified pathogenic and likely pathogenic variants in 36.09% (61/169) of the cases assessed in this study.
The detection rate was 37.04% (50/135) in patients with muscle weakness, 33.33% (6/18) with recurrent rhabdomyolysis, and 31.25% (5/16) in the group of patients with idiopathic hyperCKemia. Twenty-eight of the pathogenic and likely pathogenic variants reported here are novel and have not previously been associated with disease.
This study helps to quantify the value of targeted NGS panels in identifying the underlying genetic etiology behind myopathic processes, particularly progressive and nonprogressive muscle weakness, recurrent rhabdomyolysis, and idiopathic hyperCKemia. This study also demonstrates the potential of using large muscular dystrophy focused panels to aid in the investigation of genetic susceptibilities of rhabdomyolysis and/or idiopathic hyperCKemia. However, clinicians should be cognizant of technology and laboratory-specific limitations in detecting a variety of variant types including copy number variants, regulatory sequence variants, trinucleotide repeat expansions, and deep intronic mutations. Clinical examination and other diagnostic tools such as electromyography and muscle biopsy are still an important part of the diagnostic process.
Some studies suggest that an NGS panel may be preferred over whole exome sequencing in this population because panels currently have better exon coverage. However, this may not always be the case as the low-coverage regions continue to be identified and improved in updated whole exome sequencing platforms.Reference Ankala, da Silva and Gualandi 37 The question of ordering an NGS panel or whole exome sequencing for a patient can be evaluated on a case-by-case basis while considering the complexity of phenotype, muscle biopsy findings (if available), and availability of informative family members.
Acknowledgments
We are grateful to all the patients and their family members who participated in this study. This study was partially supported by Jay’s Drive for MD.
DISCLOSURES
LB reports personal fees from Sanofi/Genzyme, outside the submitted work. JS is a stock owner (approximately 12%) of Medical Neurogenetics LLC, and has also received fees from Medical Neurogenetics LLC for diagnostic testing. MAT reports personal fees from Sanofi/Genzyme, outside the submitted work. JS formerly worked for Medical Neurogenetics LLS (exit date: January 2016). LW has nothing to disclose.
SUPPLEMENTARY MATERIALS
To view the supplementary materials for this article, please visit https://doi.org/10.1017/cjn.2017.286



