



The liver has a major role in regulating metabolic homeostasis; it is a central cross-road for fatty acid and glucose metabolism. It serves as an intermediary organ between dietary (exogenous) and endogenous energy sources and other extra-hepatic organs/tissues that consume energy; perturbations in its metabolism have the potential to impact widely on metabolic disease risk. In health, the liver rapidly adapts to alterations in nutritional state and the nutrient fluxes that occur from a fasted (postabsorptive) to fed (postprandial) state. However, the deposition of fat (ectopic fat) in non-adipose tissues such as the liver has been suggested to be an important factor in the development of obesity related metabolic abnormalities(Reference Lewis, Carpentier and Adeli1, Reference Yki-Jarvinen2). The net retention of intra-hepatic fat is a prerequisite for the development of non-alcoholic fatty liver disease (NAFLD)(Reference Angulo3), which is now recognised as the hepatic manifestation of the metabolic syndrome(Reference Yki-Jarvinen2). A well-recognised risk factor for NAFLD is obesity, and both are risk factors for more severe metabolic diseases such as type-2 diabetes and CVD; the prevalence of NAFLD parallels that of type-2 diabetes(Reference Vernon, Baranova and Younossi4). Why the liver starts to accumulate fat is not well understood but is likely to occur when fatty acid input and synthesis exceed the liver's capacity for removal (e.g. for secretion or oxidation)(Reference Diraison and Beylot5–Reference Hodson and Frayn7). The intra-hepatic fatty acid pool represents fatty acids from multiple sources, including NEFA that are derived from hydrolysis of peripheral or visceral adipose tissue, hepatic fatty acids that are produced by de novo lipogenesis (DNL) or stored TAG in the cytosol of hepatocytes, and in the postprandial period, this will also include dietary fatty acids (Fig. 1a). The delivery of fatty acids to the liver and synthesis and partitioning (into esterification or oxidation pathways) within the liver appear to be dependent on a number of metabolic factors; not least ambient and postprandial insulin concentrations. The aim of the present paper is to summarise the findings from in vivo human metabolic studies performed by ourselves and others that have helped to understand the way in which fatty acids are synthesised and partitioned within the human liver.
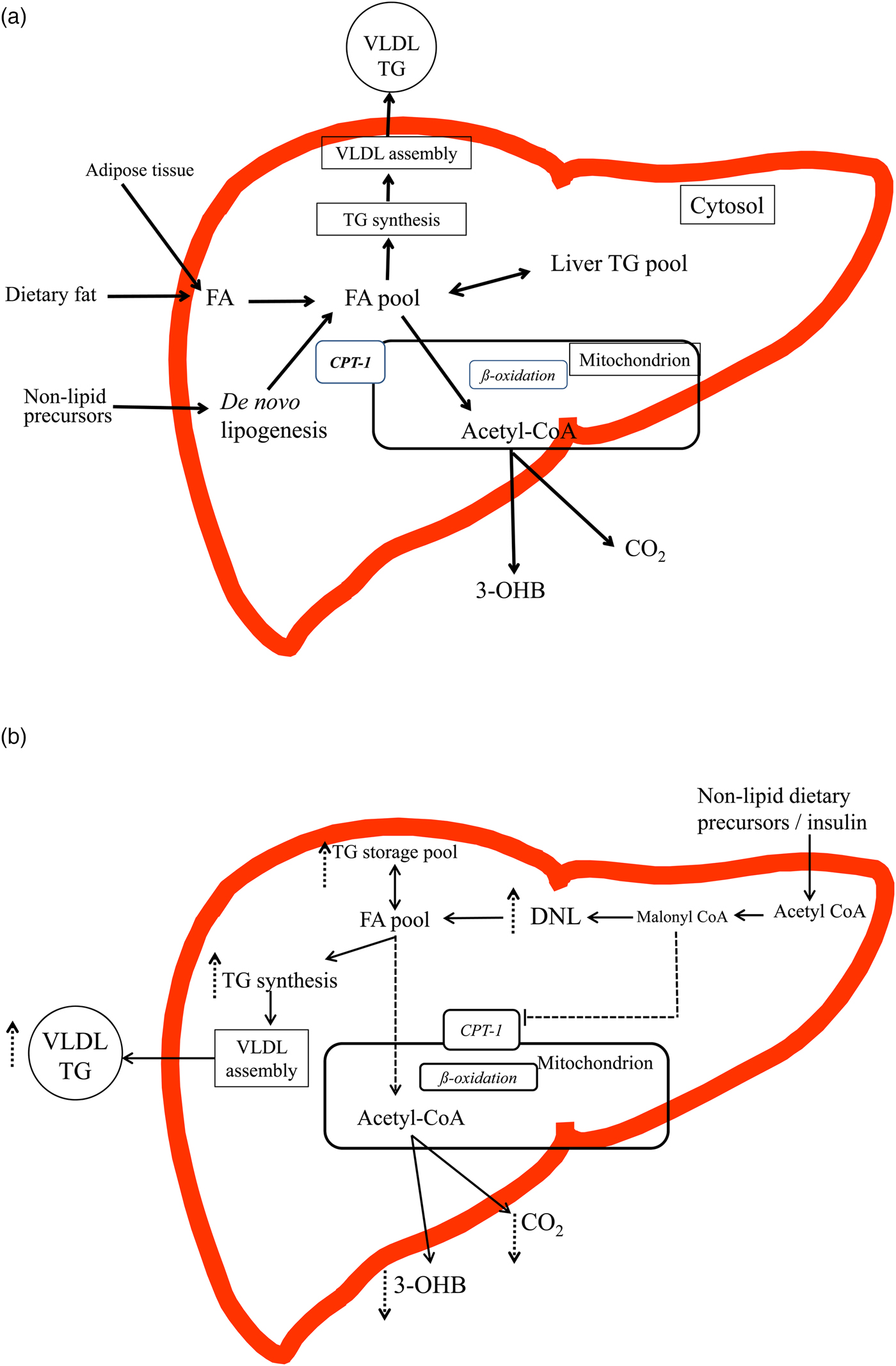
Fig. 1. (Colour online) (a) and (b) An overview of the intra-hepatic pathways of hepatic fatty acid (FA) metabolism (figures are based on previous schematics overviews in Hodson and Frayn(Reference Hodson and Frayn7) and Pramfalk et al.(Reference Pramfalk, Pavlides and Banerjee10)). (a) In the fasting state FA from the lipolysis of subcutaneous and visceral adipose tissue enter the liver and mix with FA from the cytosolic TAG (TG) pool and those from de novo lipogenesis (DNL). FA are then preferentially partitioned towards the oxidative pathway where the acetyl-CoA produced can enter the tricarboxylic acid cycle or ketogenic pathway to produce CO2 or 3-hydroxybutyrate (3-OHB). FA are also esterified to TAG and enter a cytosolic storage pool that is constantly turning over. TAG is then hydrolysed to release FA which are then re-esterified to TAG and utilised in the production of VLDL particles. In the transition to the postprandial state, FA from the diet also enter the liver and mix with endogenous sources. The postprandial increase in plasma insulin concentrations suppresses adipose tissue lipolysis and up-regulates the DNL, which would shift the cellular metabolism of FA away from oxidative pathways towards esterification (TAG synthesis). (b) In individuals with an ‘unhealthy’ phenotype (e.g. insulin resistance, hepatic fat accumulation) in both the fasting and the postprandial state it is proposed that the DNL pathway will be up-regulated and FA from the intra-hepatic FA pool will be partitioned towards esterification (TAG production) pathways and utilised in the production of VLDL particles or stored in the cytosolic TAG pool rather than entering oxidative pathways. The increase in DNL, will increase malonyl-CoA, which will inhibit carnitine-palmitoyl-transferase 1, located on the outer mitochondrial membrane, therefore decreasing the flux of FA into the mitochondria for oxidation, leading to decreases in hepatic CO2 production and plasma 3-OHB concentrations.
Studying in vivo human hepatic metabolism
The liver by virtue of its anatomical position makes studying hepatic processes directly in vivo in human subjects challenging. Direct assessment of the liver can only be achieved by arterio-venous difference measurements, which is impractical in human subjects due to the inaccessibility of the portal vein. However, surrogate markers, such as VLDL-TAG and the ketone body, 3-hydroxybutyrate (3-OHB) which are produced and secreted by the liver offer the opportunity to gain insight into hepatic processes such as hepatic fatty acid esterification and oxidation pathways(Reference Hodson and Frayn7). It has been suggested that the TAG composition in VLDL reflects that of liver TAG(Reference Donnelly, Smith and Schwarzenberg8, Reference Peter, Cegan and Wagner9) although differentiating the contribution of different fatty acid sources (dietary, adipose tissue derived and DNL derived) cannot be determined. The use of metabolic tracers, such as stable isotopes offers the opportunity to trace the fate of specific fatty acid sources through hepatic esterification and/or oxidation pathways (Fig. 1a (based on Hodson and Frayn(Reference Hodson and Frayn7) and Pramfalk et al.(Reference Pramfalk, Pavlides and Banerjee10))). Moreover, as human subjects spend a large proportion of a 24 h period in the postprandial state(Reference Ruge, Hodson and Cheeseman11, Reference McQuaid, Hodson and Neville12) studies in the postprandial state, in combination with stable-isotope methodologies offer the best insight into human hepatic fatty acid metabolism.
Fatty acid sources: adipose tissue
In the fasting state, plasma NEFA arise predominantly from the hydrolysis of adipose tissue TAG and are a major contributor to the intra-hepatic fatty acid pool(Reference Donnelly, Smith and Schwarzenberg8). NEFA release (per unit mass) from subcutaneous abdominal (upper-body) adipose tissue has been inversely associated with fasting plasma insulin concentrations(Reference Karpe, Dickmann and Frayn13). Studies assessing whole-body adipose tissue lipolysis have reported insulin-resistant subjects with NAFLD have an elevated lipolytic rate when compared with insulin-sensitive subjects without NAFLD(Reference Bugianesi, Gastaldelli and Vanni14–Reference Korenblat, Fabbrini and Mohammed16). Whether the higher lipolytic rate observed in insulin-resistant individuals with NAFLD is a cause or a consequence of NAFLD remains unclear. Although often suggested to be the major contributor to liver TAG, Donnelly et al.(Reference Donnelly, Smith and Schwarzenberg8) using stable-isotope tracer methodology, reported that plasma NEFA contributed between 45 and 74 % to liver TAG (after a 4 d stable-isotope labelling procedure); thus it is plausible that some, but not all, individuals develop NAFLD due to an increased fatty acid flux from adipose tissue to the liver. Positron emission tomography/computed tomography in combination with labelled palmitate (11C) or a palmitate analogue fluoro-6-thia-heptadecanoic acid have been used to assess hepatic fatty acid uptake(Reference Iozzo, Bucci and Roivainen17, Reference Immonen, Hannukainen and Kudomi18). It was found that hepatic fatty acid uptake was higher, although not significantly different, in obese compared with overweight control subjects(Reference Iozzo, Bucci and Roivainen17) whilst in morbidly obese individuals hepatic fatty acid uptake is significantly higher before and 6 months after bariatric surgery than in lean controls, despite liver fat content being normalised(Reference Immonen, Hannukainen and Kudomi18). The authors suggested that the persistence of high hepatic fatty acid uptake compared with the lean control subjects, in the combination with the reduction in liver fat content indicates fatty acids may be preferentially directed into oxidation rather than esterification pathways(Reference Immonen, Hannukainen and Kudomi18).
The relative contribution of systemic NEFA (that is fatty acids predominantly from subcutaneous adipose tissue) to VLDL-TAG in lean/insulin-sensitive individuals has been reported to be between 75 and 84 %(Reference Fabbrini, Mohammed and Magkos15, Reference Barrows and Parks19, Reference Vedala, Wang and Neese20–Reference Hodson, McQuaid and Humphreys22) whilst for obese/insulin-resistant individuals and those with NAFLD, the contribution is between 42 and 72 %(Reference Donnelly, Smith and Schwarzenberg8, Reference Fabbrini, Mohammed and Magkos15, Reference Vedala, Wang and Neese20–Reference Hodson, McQuaid and Humphreys22). In the fasting state, the remaining fatty acids come from non-systemic NEFA, some of which would include fatty acids derived from the lipolysis of visceral adipose tissue. As liver fat content is strongly associated with visceral adiposity(Reference Kotronen, Westerbacka and Bergholm23), it is plausible that increasing amounts of visceral fat would contribute to a greater extent to the fatty acid supply being released into the portal vein(Reference Nielsen, Guo and Johnson24). Indeed, Meek et al.(Reference Meek, Nair and Jensen25) have previously demonstrated that moderate hyperinsulinaemia appeared to increase the proportions of fatty acids reaching the liver from visceral fat compared with systemic sources.
In the transition to the postprandial state, and after consumption of a mixed meal, the antilipolytic action of insulin will suppress the hydrolysis of adipose tissue TAG, resulting in decreased plasma NEFA concentrations(Reference Frayn, Arner and Yki-Jarvinen26). This is reflected in the relative contribution of plasma NEFA to VLDL-TAG in the postprandial state where it is between 28 and 57 % for lean/insulin-sensitive individuals(Reference Barrows and Parks19, Reference Hodson, McQuaid and Humphreys22, Reference Hodson, Bickerton and McQuaid27) and 28–30 % for obese/insulin-resistant individuals and those with NAFLD(Reference Donnelly, Smith and Schwarzenberg8, Reference Hodson, McQuaid and Humphreys22). The degree to which adipose tissue hydrolysis is suppressed may be influenced by insulin resistance as some(Reference McQuaid, Hodson and Neville12, Reference Coppack, Evans and Fisher28) but not all(Reference Bickerton, Roberts and Fielding29) studies have reported postprandial NEFA suppression occurs to a lesser degree in insulin-resistant compared with insulin-sensitive individuals. One plausible explanation for the apparent lower degree of NEFA suppression reported in insulin-resistant individuals is that spillover fatty acids, derived from the peripheral lipoprotein lipase mediated lipolysis of chylomicron-TAG may somewhat reduce this effect. We have recently demonstrated that the contribution of spillover fatty acids to the systemic NEFA pool is lower in obese/insulin-resistant compared with lean insulin-sensitive individuals(Reference Piche, Parry and Karpe30). In the situation where suppression of adipose tissue hydrolysis is attenuated, such as insulin resistance, it is plausible there is a greater delivery of adipose tissue derived fatty acids to the liver in the postprandial state compared with more insulin-sensitive individuals. We have previously reported the relative contribution of endogenous systemic NEFA to VLDL-TAG was lower in the insulin-resistant when compared with the insulin-sensitive subjects(Reference Hodson, Bickerton and McQuaid27). This finding suggests other fatty acid sources were contributing to a greater extent to VLDL-TAG production in the insulin-resistant group; however when expressed as a concentration, the contribution from endogenous systemic NEFA was similar in the groups(Reference Hodson, Bickerton and McQuaid27). We did not observe a difference in either the relative or absolute contribution of systemic NEFA to VLDL-TAG in abdominally obese insulin-resistant males, compared with lean insulin-sensitive controls when fed three sequential meals, 5 h apart over a 24 h period(Reference Hodson, McQuaid and Humphreys22).
Fatty acid sources: de novo synthesised
DNL is the process where excess non-lipid precursors (e.g. sugars and protein) are synthesised to fat; in human subjects this primarily occurs in the liver(Reference Hellerstein, Schwarz and Neese31). DNL is a complex process: it takes place in the cellular cytoplasm, and acetyl-CoA is required as the precursor and the principal building blocks, with the major quantitative product being palmitoyl-CoA(Reference Hellerstein, Schwarz and Neese31, Reference Wilke, French and Goh32). The synthesis of one molecule of palmitoyl-CoA from acetyl-CoA is an energy inefficient process costing seven molecules of ATP and requiring the conversion of fourteen high-energy NADPH molecules to NADP+(Reference Solinas, Boren and Dulloo33). Despite being energetically inefficient and likely a minor route for storing excess energy, the contribution of de novo fatty acids to VLDL-TAG has been reported to be up to about 10 % in healthy, insulin-sensitive adults(Reference Pramfalk, Pavlides and Banerjee10, Reference Barrows and Parks19, Reference Diraison, Moulin and Beylot34–Reference Pramfalk, Pavlides and Banerjee38) and 14–22 % in individuals with insulin resistance or NAFLD(Reference Donnelly, Smith and Schwarzenberg8, Reference Diraison, Moulin and Beylot34, Reference Lambert, Ramos-Roman and Browning36–Reference Pramfalk, Pavlides and Banerjee38). As individuals with insulin resistance/NAFLD have notably higher hepatic DNL compared with those without, it is often speculated, although unsubstantiated, that enhanced DNL is a causal factor in the development of insulin resistance/NAFLD; thus it remains unclear if insulin resistance/NAFLD causes enhanced hepatic DNL or vice versa.
The consumption of a mixed meal exposes hepatic lipogenic enzymes to higher concentrations of insulin and this has been shown to stimulate enzyme activity by increasing the transcription of the genes for fatty acid synthase and acetyl-CoA carboxylase(Reference Lewis, Carpentier and Adeli1). In the postprandial state the contribution of DNL fatty acids to VLDL-TAG has been reported to be between 8 and 13 % in insulin-sensitive individuals(Reference Barrows and Parks19, Reference Timlin and Parks35, Reference Pramfalk, Pavlides and Banerjee38) and higher, between 12 and 26 %, in individuals who are insulin resistant or with NAFLD(Reference Donnelly, Smith and Schwarzenberg8, Reference Pramfalk, Pavlides and Banerjee38).
The amount of dietary carbohydrate and/or fat consumed may influence the contribution of DNL fatty acids to VLDL-TAG. Wilke et al.(Reference Wilke, French and Goh32) compared the effects of a short-term (3 d) high-fat (about 37 % total energy (TE), carbohydrate 48 % TE) diet to a lower-fat (about 23 % TE, carbohydrate 59 % TE) diet on hepatic DNL in obese individuals with and without type-2 diabetes. In both groups hepatic DNL was notably up-regulated on the low-fat diet with palmitate accounting for >70 % of the newly synthesised fatty acids in VLDL-TAG; whilst on the higher-fat diet the synthesis of fatty acids were notably reduced(Reference Wilke, French and Goh32). Thus, when investigating the effect of phenotype on the contribution of DNL derived fatty acids to VLDL-TAG, it would be important that dietary intakes are standardised prior to measuring hepatic DNL.
Fatty acid sources: dietary
The contribution of dietary fat to liver fat accumulation and the appearance in VLDL-TAG is dependent on the amount and frequency of fat intake. Dietary fat can enter the liver either as chylomicron remnants or chylomicron-derived spillover NEFA(Reference Hodson and Fielding39). By feeding a single high-fat meal, labelled with 13C fatty acids and tracing the trafficking of the labelled fatty acids with 13C-MRI/spectroscopy, it was demonstrated that there was a transient increase in liver fat content over the postprandial period(Reference Ravikumar, Carey and Snaar40, Reference Lindeboom, Nabuurs and Hesselink41) which is in line with the liver being a dynamic organ that responds rapidly to changes in nutritional status. Dietary fatty acids have been reported to contribute 15 % of liver-TAG, in individuals with NAFLD(Reference Donnelly, Smith and Schwarzenberg8); as individuals consumed stable-isotope tracers for 4 d prior to the biopsy it is unclear how reflective this is as there would have been ‘recycling’ of the tracer from previous meals, which may suggest a higher contribution.
The relative contribution of dietary fatty acids to VLDL-TAG has been reported to be 19–39 % for lean/insulin-sensitive individuals(Reference Barrows and Parks19, Reference Hodson, McQuaid and Humphreys22, Reference Hodson, Bickerton and McQuaid27), and between 12 and 28 % in obese/insulin-resistant individuals and those with NAFLD(Reference Donnelly, Smith and Schwarzenberg8, Reference Hodson, McQuaid and Humphreys22, Reference Hodson, Bickerton and McQuaid27). Few studies have compared differences in contribution between individuals with varying degrees of insulin resistance. We have previously compared the contribution of dietary fatty acids to VLDL-TAG between insulin-sensitive and insulin-resistant individuals and found no difference in the relative or absolute contribution of dietary fatty acids to VLDL-TAG at the end of a 6 h postprandial period, after consumption of a mixed-test meal(Reference Hodson, Bickerton and McQuaid27). After feeding three sequential test meals, 5 h apart, we found the relative contribution of dietary fatty acids to VLDL-TAG was lower in abdominally obese insulin-resistant males compared with lean insulin-sensitive males; however there was no difference in the absolute contribution(Reference Hodson, McQuaid and Humphreys22). Differences in the reported contributions of dietary fatty acids to VLDL-TAG (and potentially liver TAG) can in part be explained by differences in the amount and type of fat consumed in the test meal, the frequency of meals, the amount of time after meal consumption the measurement was made, and the phenotype of the individual; all of which differ/vary across studies. The contribution of dietary fat to VLDL-TAG or liver TAG is not often measured and is rarely reported in intervention studies where the amount and composition of dietary fat has been altered. This would be of interest to do as the type of fat consumed appears to influence liver fat content as previously reviewed(Reference Green and Hodson42–Reference Parry and Hodson44). Taken together it has been found that diets overfeeding SFA increase liver fat content to a greater extent than diets overfeeding unsaturated (primarily monounsaturated and n-6 PUFA) fatty acids(Reference Bjermo, Iggman and Kullberg45–Reference Luukkonen, Sadevirta and Zhou47). Supplementing the diet with n-3 fatty acids (namely EPA and DHA) at 4 g/d (as ethyl esters) for periods between 8 weeks and 16 months has also been reported to decrease liver fat(Reference Cussons, Watts and Mori48–Reference Hodson, Bhatia and Scorletti50).
Although these studies have investigated the effect of dietary fat quality on liver fat content, there is limited work investigating the effect on intra-hepatic fatty acid synthesis and partitioning. In the study by Luukkonen et al.(Reference Luukkonen, Sadevirta and Zhou47) it was found that fasting hepatic DNL was unchanged after 3 weeks overconsumption of SFA, whilst fasting hepatic DNL significantly increased after overconsumption of the carbohydrate-enriched diet. Rosqvist et al.(Reference Rosqvist, Iggman and Kullberg46) over fed participants with SFA or n-6 PUFA and found no change in fasting plasma 3-OHB concentrations.
We have previously found no difference in the contribution of systemic NEFA and dietary fatty acids to VLDL-TAG, when participants consumed a mixed-test meal after short-term (3 d) consumption of a high-fat (40 % TE) or high-carbohydrate (75 % TE) diet(Reference Chong, Hodson and Bickerton51); however there was a higher contribution of splanchnic fatty acids (which would include fatty acids from DNL, liver TAG and visceral fat) after the high-carbohydrate diet. Indeed, the lipogenic index in VLDL-TAG (a surrogate marker of hepatic DNL) was significantly increased after the high-carbohydrate diet(Reference Chong, Hodson and Bickerton51). As this was a short-term intervention, it would be of interest to investigate how the composition of dietary intake may influence the contribution of fatty acid sources to VLDL-TAG (and presumably liver TAG) over a longer period of time. We have undertaken a pilot study to investigate the effect of EPA and DHA supplementation on hepatic fatty acid partitioning(Reference Hodson, Bhatia and Scorletti50). We found that long-term (16–18 months) supplementation with EPA and DHA (total 4 g/d as ethyl esters) significantly decreased the contribution of DNL fatty acids to VLDL-TAG in the postprandial state, in individuals who showed an increase of >2 % in erythrocyte DHA compared with those who did not(Reference Hodson, Bhatia and Scorletti50). In the individuals with an increased erythrocyte DHA, we found a significantly higher incorporation of 13C (which was given in a test meal) appear in plasma 3-OHB over the postprandial period, suggesting that a greater proportion of recently ingested fatty acids were undergoing oxidation to produce 3-OHB(Reference Hodson, Bhatia and Scorletti50). Given the effect that different fatty acids are reported to have on liver fat content, it would be of interest to study the effects they have on hepatic fatty acid synthesis and partitioning to understand their role in liver fat metabolism.
Intra-hepatic partitioning of fatty acids: esterification
Fatty acids within the hepatocyte are broadly partitioned between two pathways: (i) esterification to form mainly TAG that will enter the hepatic TAG pool that may be secreted as VLDL-TAG or stored as cytosolic TAG and (ii) oxidation (Fig. 1a). Typically the rate of VLDL-TAG secretion has been investigated in the fasting state(Reference Fabbrini, Magkos and Mohammed21, Reference Adiels, Taskinen and Packard52–Reference Hodson, Banerjee and Rial55) and has not often been reported in the postprandial period(Reference Barrows and Parks19). In the fasting state, secretion of TAG has been reported to be higher in individuals with NAFLD, who are insulin resistant compared with those without NAFLD, who are by comparison insulin sensitive(Reference Adiels, Taskinen and Packard52). In the postprandial state, Barrows and Parks(Reference Barrows and Parks19) reported a decrease in rate of appearance of VLDL-TAG upon consumption of a meal; a comparison of the rate of VLDL-TAG secretion in the postprandial state has not been undertaken between insulin-sensitive and insulin-resistant individuals.
Increases in insulin, either induced experimentally using a euglycaemic/hyperinsulaemic clamp protocol or during the transition to the postprandial state, suppress VLDL production in insulin-sensitive individuals(Reference Malmstrom, Packard and Caslake56). However, this suppressive effect of insulin on VLDL production is blunted in individuals with NAFLD(Reference Adiels, Westerbacka and Soro-Paavonen53). During the postprandial period, there are approximately ten times more VLDL than chylomicron particles(Reference Schneeman, Kotite and Todd57); thus for individuals who do not suppress VLDL production during the postprandial period this would result in a more exaggerated postprandial lipaemia. It is suggested that as ApoB-100 secretion is not increased in NAFLD patients(Reference Fabbrini, Mohammed and Magkos15) the particles secreted are more TAG-rich and thus they have an increased particle size(Reference Adiels, Olofsson and Taskinen58). Evidence of the effect of diet on VLDL-TAG secretion is lacking. Given the pronounced effect that not only the nutrient, but fatty acid composition of the diet has on liver fat accumulation and potentially intra-hepatic fatty acid synthesis and partitioning it is important to determine the effect of dietary components on VLDL-TAG secretion; VLDL-TAG secretion is a pathway of disposal for intra-hepatic TAG (Fig. 1a).
Intra-hepatic partitioning of fatty acids: oxidation
Fatty acids entering the β-oxidation pathway generate intra-mitochondrial acetyl-CoA and this may then enter either the tricarboxylic acid cycle (Krebs cycle) for oxidation to CO2, or undergo ketogenesis to form ketone bodies (Fig. 1a). In the ketogenic pathway, two molecules of acetyl-CoA are combined within the mitochondrion to form acetoacetic acid, from which the other major ketone body, 3-OHB is produced. Systemic concentrations of 3-OHB are often used as a surrogate marker of hepatic fatty acid oxidation whilst direct assessment of hepatic CO2 production, in human subjects in vivo is challenging. A major regulator of hepatic fatty acid oxidation, particularly ketogenesis is undoubtedly the rate of supply of fatty acids from adipose tissue.
A number of studies have investigated hepatic fatty acid partitioning and its hormonal regulation in human subjects. By using a radio-tracer, in individuals who had undergone an 18 h fast it was demonstrated that of the fatty acids entering the liver, approximately 2-fold more fatty acids were converted to ketone bodies than to VLDL-TAG and there are twice as many fatty acids converted to VLDL-TAG than CO2, demonstrating that ketogenesis is considerably greater than complete oxidation(Reference Havel, Kane and Balasse59). In hyperlipidaemic subjects fatty acids were converted in similar proportions to ketone bodies, VLDL-TAG and CO2(Reference Havel, Kane and Balasse59).
By using a combination of stable-isotope labelled tracers (2H and 13C) Sunny et al.(Reference Sunny, Parks and Browning60) found mitochondrial oxidation, which represented the tricarboxylic acid cycle to be twice as high in subjects with NAFLD (17 % liver fat content) compared with subjects without NAFLD (3 % liver fat content); with a strong direct association with flux through the cycle and liver fat content. They found no difference in ketone body production between the groups(Reference Sunny, Parks and Browning60). By using 13C-acetate infusion in combination with a 13C-MRS methodology Petersen et al.(Reference Petersen, Befroy and Dufour61) reported similar rates of hepatic mitochondrial oxidation (based on a mathematical model using the appearance of 13C in the tricarboxylic acid cycle intermediate glutamate) in subjects with high (about 9 %) and low (about 2 %) liver fat content. Aside from differences in methodologies used, the difference in findings between the studies of Sunny et al.(Reference Sunny, Parks and Browning60) and Petersen et al.(Reference Petersen, Befroy and Dufour61) may in part be explained by differences in participant age, BMI (the participants in the study by Petersen et al.(Reference Petersen, Befroy and Dufour61) were notably younger and leaner), sex (Petersen et al.(Reference Petersen, Befroy and Dufour61) studied only men), ethnicity (Sunny et al.(Reference Sunny, Parks and Browning60) studied Hispanic and African American) and the degree of liver fat content; the high liver fat group in the study of Sunny et al.(Reference Sunny, Parks and Browning60) has twice as much liver fat as the high liver fat group of Petersen et al.(Reference Petersen, Befroy and Dufour61). As it was not known how long the high liver fat individuals had their liver fat accumulation, it is also plausible that the difference in findings were due to metabolic adaptation; we have previously suggested that greater fatty acid oxidation may be an adaptive mechanism to prevent liver fat accumulation(Reference Hodson, McQuaid and Humphreys22).
Studies measuring plasma 3-OHB concentrations in the fasting state as a marker of hepatic fatty acid oxidation have reported mixed findings, with concentrations being decreased(Reference Croci, Byrne and Choquette62), similar(Reference Hodson, McQuaid and Humphreys22, Reference Hodson, Bickerton and McQuaid27, Reference Pramfalk, Pavlides and Banerjee38, Reference Kotronen, Seppala-Lindroos and Vehkavaara63) or increased(Reference Bugianesi, Gastaldelli and Vanni14, Reference Sanyal, Campbell-Sargent and Mirshahi64) in subjects with insulin resistance and/or NAFLD compared with those without insulin resistance and/or NAFLD.
There is a limited amount of evidence for differences in postprandial hepatic fatty acid β-oxidation in human subjects. No direct assessment of hepatic CO2 production has been undertaken in human subjects over the postprandial period; however plasma concentrations of 3-OHB have been measured. We have previously found mixed results with some of our studies showing a significantly lower(Reference Hodson, Bickerton and McQuaid27), similar(Reference Pramfalk, Pavlides and Banerjee38) or higher(Reference Hodson, McQuaid and Humphreys22) postprandial response of blood 3-OHB in insulin-resistant compared with insulin-sensitive individuals. We have also found that men have a notably lower postprandial 3-OHB concentration when compared with women(Reference Pramfalk, Pavlides and Banerjee10). This is in agreement with the work of Halkes et al.(Reference Halkes, van Dijk and Verseyden65) who also found higher postprandial blood 3-OHB concentrations in females compared with males. The reported sexual dimorphism in postprandial plasma 3-OHB concentrations may be due to males having a more prolonged pattern of hepatic DNL than females(Reference Pramfalk, Pavlides and Banerjee10).
In the postprandial period ketogenesis is suppressed, compared with the fasting state, as adipose tissue lipolysis is suppressed(Reference Pramfalk, Pavlides and Banerjee10, Reference Hodson, McQuaid and Humphreys22, Reference Hodson, Bickerton and McQuaid27, Reference Pramfalk, Pavlides and Banerjee38). Using stable-isotope tracers we have demonstrated that dietary fatty acids in the form of chylomicron derived ‘spillover’ fatty acids and chylomicron remnant fatty acids are still utilised for ketogenesis(Reference Pramfalk, Pavlides and Banerjee10, Reference Hodson, McQuaid and Humphreys22). Using this methodology we have demonstrated that incorporation of 13C from recently ingested dietary fat into plasma 3-OHB was higher in insulin-resistant compared with insulin-sensitive individuals over 24 h(Reference Hodson, McQuaid and Humphreys22). More recently and in contrast to our previous finding, we found greater incorporation of 13C from dietary fat, in insulin-sensitive compared with insulin-resistant individuals(Reference Pramfalk, Pavlides and Banerjee38) and in women compared with men(Reference Pramfalk, Pavlides and Banerjee10). These findings demonstrate that recently ingested dietary fatty acids enter the ketogenic pathway; however the regulation of this process still remains to be elucidated.
It remains unclear if insulin regulates intra-hepatic fatty acid partitioning into the ketogenic pathway. The transport of fatty acyl-CoA into the mitochondrion for oxidation is achieved via the ‘carnitine shuttle’, and the key regulated enzyme is carnitine-palmitoyl-transferase 1, located on the outer mitochondrial membrane. Malonyl-CoA, an intermediate in the pathway of DNL is a potent inhibitor of carnitine-palmitoyl-transferase 1(Reference McGarry and Foster66), thus when DNL is stimulated under conditions of carbohydrate excess both directly via the carbohydrate response element binding protein, and via insulin, fat oxidation is consequently suppressed via inhibition of carnitine-palmitoyl-transferase 1 (Fig. 1b (based on Hodson and Frayn(Reference Hodson and Frayn7) and Pramfalk et al.(Reference Pramfalk, Pavlides and Banerjee10)). This will complement the suppression of fatty acid release from adipose tissue which is under the influence of insulin. Indeed, we have previously found strong inverse associations between hepatic DNL and plasma 3-OHB concentrations in the postprandial period in some, but not all, of our participants(Reference Pramfalk, Pavlides and Banerjee10, Reference Pramfalk, Pavlides and Banerjee38) suggesting a dissociation between the pathways. It could be speculated that some individuals have hepatic insulin resistance (which is not obvious at a systemic level) and this leads to a lack of suppression in ketone body production in the postprandial period. Alternatively, the difference in results may be explained by the fact that postprandial studies typically feed a standard test meal and it is possible that there may be variations from true energy balance of the individual and this could result in either suppression or enhancement of fatty acid oxidation, along with perturbations of other plasma metabolites, such as insulin.
Discrepancy in findings regarding fatty acid oxidation between studies is most likely explained by differences in phenotypes, methodology to assess fatty acid oxidation and duration/severity of disease. It is also plausible that the higher oxidation reported in insulin resistance, compared with insulin-sensitive individuals is a compensatory response to try and normalise liver (ectopic) fat content in insulin-resistant compared with insulin-sensitive males(Reference Hodson, McQuaid and Humphreys22). However, this was an observation study and it would be of interest to undertake studies where dietary fatty acid oxidation was measured regularly over time, in individuals who were on a trajectory to become insulin resistant or get NAFLD (e.g. increasing body fat, particularly abdominal fat) to see if fatty acid oxidation changed.
Conclusions
When we consume dietary fat, a series of complex metabolic processes ensures that fatty acids are absorbed, transported around the body and used/stored appropriately. The liver is a central metabolic organ within the human body and has a major role in regulating fat and carbohydrate metabolism; it serves as an intermediary organ between dietary (exogenous) and endogenous energy sources and other extra-hepatic organs/tissues that consume energy. Hepatic fat accumulation represents an imbalance between pathways of fatty acid delivery and fatty acid removal. Why hepatic fat starts to accumulate is, surprisingly, not well understand but must involve alterations in hepatic fatty acid input, synthesis and removal. Whether fatty acids are partitioned towards oxidation or esterification pathways appears to be dependent on a number of metabolic factors; not least ambient insulin concentrations. Moreover, along with the phenotype of an individual (i.e. BMI, plasma insulin concentrations), it is apparent that lifestyle factors (e.g. habitual diet) of an individual may also play a role (and explain some of the variation and discrepancies between studies). It is becoming increasingly clearer that dietary composition (macronutrient and fatty acid composition) may play pivotal roles in determining if intra-hepatic fat accumulates, although what remains to be elucidated is the influence these nutrients have on intra-hepatic fatty acid synthesis and partitioning.
Acknowledgements
I am indebted to all my work colleagues and collaborators, past and present for their help, support, kindness and friendship. I would like to pay particular thanks to my mentors, Professors Murray Skeaff, Keith Frayn and Fredrik Karpe and my former colleague Barbara Fielding.
Financial Support
Financial support for our studies where the work is acknowledged here was from The Wellcome Trust, the Hepatic and Adipose Tissue and Functions in the Metabolic Syndrome’ (available at http://www.hepadip.org/) which was supported by the European Commission as an integrated project under the Sixth Framework Programme, and the British Heart Foundation. Over the course of the studies I was Girdlers’ Health Research Council New Zealand Fellow, a British Heart Foundation Intermediate Research Fellow in Basic Science and am currently a British Heart Foundation Senior Research Fellow in Basic Science.
Conflict of Interest
None.
Authorship
The author had sole responsibility for all aspects of preparation of this paper.

