


I had been working on the endocrine and signalling role of white adipose tissue (WAT) since 1994 following the identification of the ob (Lep) gene(Reference Zhang, Proenca and Maffei1), this after some 15 years investigating the physiological role of brown adipose tissue. The ob gene, a mutation in which it is responsible for the profound obesity of ob/ob (Lep ob/Lep ob) mice, is expressed primarily in white adipocytes and encodes the pleiotropic hormone leptin. The discovery of this adipocyte hormone had wide-ranging implications, including that white fat has multiple functions that far transcend the traditional picture of a simple lipid storage organ.
The recognition that white adipocytes secrete a major hormone, and are key endocrine cells, catalysed an increasingly intensive search for other protein signals and factors that might be released from adipose tissue. Given the rapid developments that were then taking place I felt that an overview of the new perspectives on WAT was timely, and especially for a nutritional sciences audience. There was, of course, considerable interest in adipose tissue as a consequence of the escalating levels of obesity and its associated disorders.
I had a particular motivation in submitting the article to the BJN; I was at the time the Editor-in-Chief and had introduced a new section – ‘Horizons in Nutritional Science’ – which I was concerned to see develop. This section was intended to present fresh perspectives in the form of short reviews of rapidly emerging areas in nutritional science. My colleague Stuart Wood and I had written a paper to launch the Horizons series a year earlier – ‘Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins’ (this article, with > 600 citations, is itself amongst the top 20 most-cited BJN papers)(Reference Wood and Trayhurn2).
What did the 2004 paper say?
Our 2004 article(Reference Trayhurn and Wood3) - the abstract of which is shown in Fig. 1 - had three elements, each of which may have contributed to its success. The first, and major, is that it provided an overview of the emerging roles of white fat, highlighting the extensive secretory properties of the tissue. These include the secretion of leptin and a further key adipocyte hormone, adiponectin, as well as the then rapidly growing list of other protein signals and factors. The secreted proteins encompass a wide range of physiological functions, including insulin sensitivity, haemostasis, lipid metabolism, blood pressure and energy balance – as summarised in a figure from the 2004 paper (Fig. 2). A number are cytokines and chemokines, such as TNFα, IL-1β, IL-6 and MCP1, and other signals linked to inflammation had also been identified. This, together with reports that macrophages are recruited to WAT in the obese(Reference Weisberg, McCann and Desai4,Reference Xu, Barnes and Yang5) , underpinned the view that the tissue becomes ‘inflamed’ as the mass increases, the inflammation leading to the development of obesity-associated disorders such as insulin resistance and the metabolic syndrome.

Fig. 1. Original abstract from 2004 article (Reference Trayhurn and Wood3).
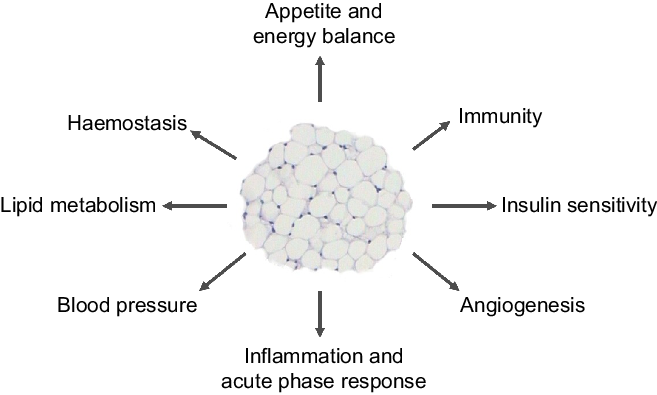
Fig. 2. Figure from the original 2004 paper(Reference Trayhurn and Wood3) illustrating the key metabolic and physiological processes with which white adipose tissue was – and is – considered to be involved through the secretion of various adipokines.
The second, though minor, element of the article was the attempt to systematise the nomenclature associated with the secretions from white adipocytes and encourage a common terminology. The multiple secreted proteins were being referred to as either ‘adipocytokines’ or ‘adipokines’. We argued that the preferred term was adipokine, since adipocytokine can be taken as indicating that these proteins are cytokines, or cytokine-like – and most are not. In practice, adipokine is the name now customarily employed. We also described the totality of protein factors secreted from white adipocytes as the ‘adipokinome’, which together with the various lipid moieties released constitutes the ‘secretome’ of these cells.
The third element of the 2004 paper, which is to me the most significant, was the presentation of a novel hypothesis to explain the development of inflammation in WAT in obesity. For several years, I had been intrigued as to why inflammation should occur in the tissue as it expands, but there was no clear explanation. We proposed that hypoxia – a relative deprivation of oxygen – occurs in WAT in obesity as a consequence of the enlarging adipocytes becoming more and more distant from the vasculature. We further postulated that the key hypoxia-inducible transcription factor, HIF-1, is recruited leading to increased expression of inflammation-related adipokines. Indeed, extensive cellular and metabolic adaptations in response to hypoxia were envisaged (as recognised in several other contexts, such as in tumours), and this has now been firmly established to be the case (Fig. 3).
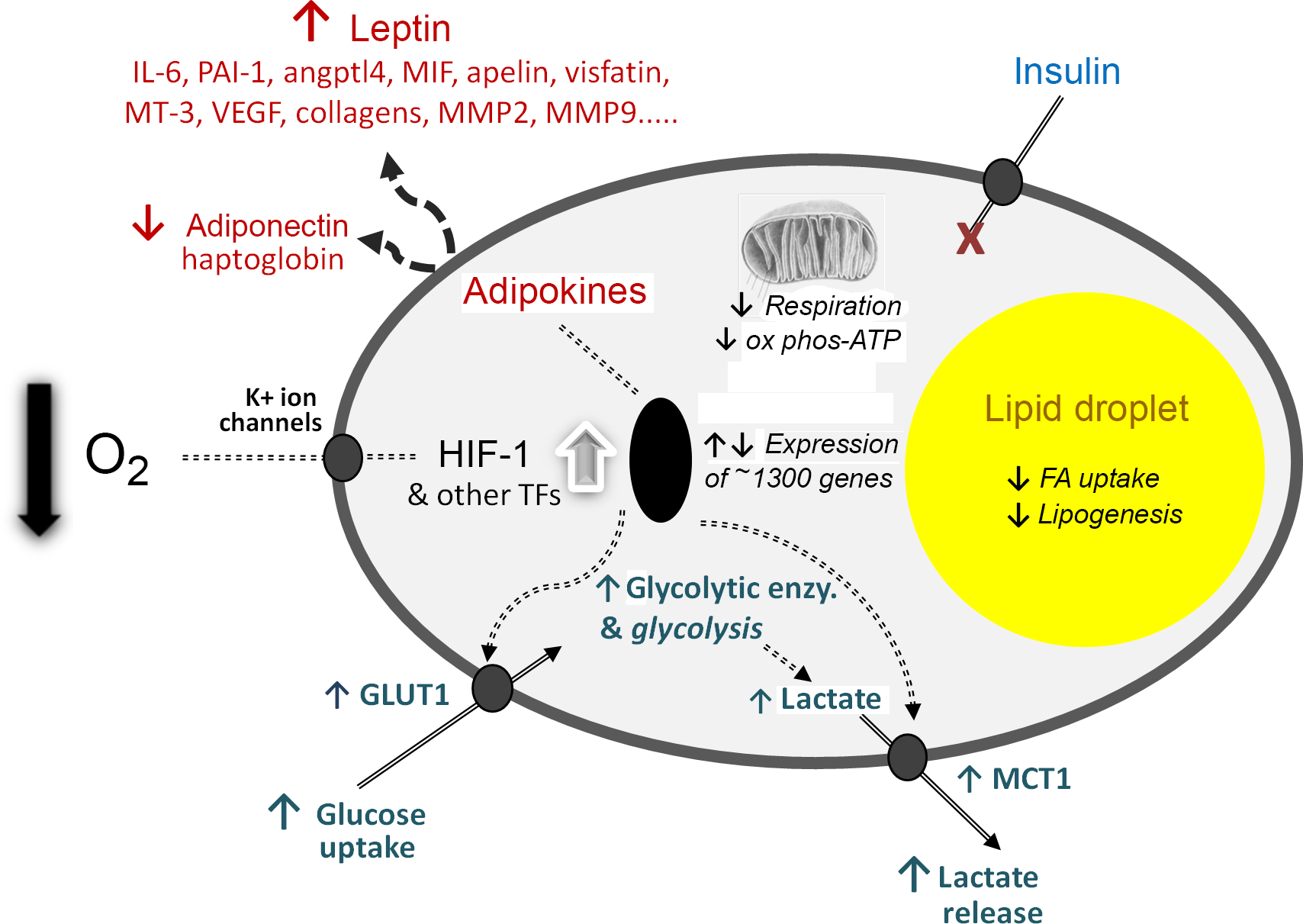
Fig. 3. Schematic representation of some of the central cellular responses to hypoxia (oxygen deficiency) in white adipocytes. The figure illustrates adaptations that are universal to all cell types, particularly the increase in glucose utilisation through anaerobic glycolysis and the reduction in respiration and oxidative phosphorylation (ox phos). Adaptations that are specific to adipocytes are also shown, primarily those relating to lipid utilisation and the production of adipokines as key secretory proteins of these cells; in some of the examples, such as MT-3 (metallothionein-3), only major changes at the gene expression level have been formally documented. angptl4, angiopoietin-like protein-4; enzy, enzyme; FA, fatty acid; GLUT1, facilitative glucose transporter 1; HIF-1, hypoxia-inducible factor-1; MCT1, monocarboxylate transporter-1; MIF, macrophage migration inhibitory factor; MMP, matrix metalloproteinases; PAI-1, plasminogen activator inhibitor-1; TF, transcription factors (additional to HIF-1); VEGF, vascular endothelial growth factor. Reproduced from Trayhurn (2019)(Reference Trayhurn23).
At the time, investigation of the molecular and cellular responses to hypoxia was a limited, though growing, area. It is now a major field with the Nobel Prize for Physiology/Medicine being awarded in 2019 to three of the pivotal figures in hypoxia research.
Subsequent development of my research – what came next?
How did my research evolve following the appearance of the 2004 article? There were in practise two thrusts to the programme. My Unit in Liverpool continued to search for novel adipokines and several were identified; these included nerve growth factor, IL-18 (a member of the IL-1 cytokine gene family), and the lipolytic agent zinc-α 2-glycoprotein(Reference Peeraully, Jenkins and Trayhurn6–Reference Bing, Bao and Jenkins8). Zinc-α 2-glycoprotein was particularly intriguing, expression being substantially increased in WAT of mice bearing tumours with the factor being linked to cachexia and to be abundantly produced by white adipocytes(Reference Bing, Bao and Jenkins8). A considerable number of other adipokines have subsequently been identified(Reference Trayhurn9,Reference Trayhurn10) .
My second, and major, interest was in examining the effects of hypoxia on the function of white adipocytes, work that was undertaken with my colleagues Stuart Wood and Bohan Wang. Importantly, 3 years after the publication of the BJN article, two papers appeared demonstrating reduced O2 tension in WAT depots of both genetic and dietary obese mice(Reference Ye, Gao and Yin11,Reference Hosogai, Fukuhara and Oshima12) , in agreement with the hypoxia hypothesis. However, varying results have been obtained in human studies and it is not yet clear whether hypoxia is a characteristic of WAT in obese humans(Reference Trayhurn9,Reference Trayhurn10) .
Our studies on hypoxia centred on human fat cells differentiated in culture from fibroblastic preadipocytes and began with a candidate gene approach in which the expression of selected adipokine genes associated with inflammation were examined together with the release of their encoded proteins(Reference Wang, Wood and Trayhurn13). Adipocytes incubated in 1 % O2 were compared with those maintained in 20 % O2. Increases in the expression and production of leptin, IL-6 and vascular endothelial growth factor (VEGF), for example, were shown to be increased in response to hypoxia, while adiponectin was decreased(Reference Wang, Wood and Trayhurn13). PCR arrays, followed by full DNA microarrays, were then used to probe the extent of the effects of hypoxia on adipocyte gene expression(Reference Wang, Wood and Trayhurn14,Reference Mazzatti, Lim and O’Hara15) .
The microarray studies demonstrated that the expression of > 1300 genes was modulated in human white adipocytes by hypoxia, approximately half being up-regulated and half down-regulated(Reference Mazzatti, Lim and O’Hara15). Multiple metabolic pathways were found to be altered by low O2 tension, including glucose utilisation, lipid oxidation and cell death(Reference Mazzatti, Lim and O’Hara15). Direct functional studies that we also undertook demonstrated that glucose uptake is substantially increased in human adipocytes under hypoxic conditions, this being facilitated by increased expression and production of the GLUT1 facilitative glucose transporter(Reference Wood, Wang and Lorente-Cebrián16).
Our group was not, of course, alone in examining the effects of hypoxia on adipose tissue function. Important contributions from others included the demonstration of a link between O2 deprivation and the development of tissue fibrosis and cellular dysfunction(Reference Halberg, Khan and Trujillo17), as well as the rapid induction of insulin resistance in adipocytes(Reference Regazzetti, Peraldi and Gremeaux18,Reference Yin, Gao and He19) .
Most of my group’s studies involved comparing adipocytes incubated in 1 % O2 with those under 20 % – so-called ‘normoxia’ – which is the conventional approach to examining the response of cells to hypoxia. However, the O2 tension to which adipocytes (in concert with many other cells) are exposed physiologically is considerably less than the equivalent of 20 % and is actually close to 6–8 %. In a study in which adipocytes were incubated under various levels of O2 between 20 and 1 %, a dose–response was observed in the parameters examined, with the cells titrating small differences in O2 tension including in the expression and secretion of specific adipokines as well as in the uptake and utilisation of glucose(Reference Wood, Stezhka and Trayhurn20). A key observation was that many of the changes occurred between 20 and 10 % O2 such that at physiological levels of O2 there is already a marked ‘hypoxic’ effect. Normoxia, in practise, represents ‘hyperoxia’ and employing this as the reference point both exaggerates the measured response to hypoxia at 1 % O2 and raises an important question of whether using 20 % O2 to culture cells has distorted our view of what is normal cellular metabolism(Reference Trayhurn9,Reference Trayhurn10) .
Although the main focus of our work was on adipocytes, we also conducted studies on human preadipocytes – cells which on differentiation become mature fat cells. Marked differences in the response to hypoxia between preadipocytes and adipocytes were observed, and of particular note was the effect on leptin production(Reference Wang, Wood and Trayhurn21). Expression of the leptin gene is considered as differentiation-dependent, being absent in preadipocytes. However, in preadipocytes exposed to hypoxia, both leptin expression and secretion of the encoded protein were evident. Thus preadipocytes become leptin-secreting endocrine cells under conditions of low pO2. This raises the question of whether at physiological O2 levels in vivo preadipocytes do produce leptin and that the endocrine function of these cells has been misrepresented by culturing them in 20 % O2 – a potent example of normal cellular function being distorted by employing unphysiological, hyperoxic conditions.
One of the outcomes from my interest in hypoxia is the highlighting of O2 as a cellular nutrient; indeed, cell biologists frequently describe it as such. However, nutritionists do not consider O2 as a nutrient, and nutrition textbooks rarely mention it other than in the context of respiration and energy metabolism. I have recently argued that nutritional science should encompass O2 as a macronutrient – it fulfils the key criteria, but it is only the route of entry (nose/lungs rather than mouth/gastrointestinal tract) that accounts for its omission(Reference Trayhurn22,Reference Trayhurn23) .
Coda
I am delighted that the 2004 paper has been so highly cited (> 1500 citations: Web of Science, September 2021) and is the third most cited article in the 75 years of the BJN. I am also pleased, and surprised, that nearly 20 years after its publication it continues to be referenced – despite the number of reviews on WAT that have appeared over the same period. In 2020 alone, it received some 66 citations (Web of Science).
In 2008, my group published a follow-up review specifically on hypoxia and adipose tissue function, again as a Horizons article in the BJN (Reference Trayhurn, Wang and Wood24), and this has received > 300 citations. I was subsequently invited to write articles on hypoxia in WAT in relation to obesity for Physiological Reviews (Reference Trayhurn9) and Annual Reviews of Nutrition (Reference Trayhurn10), and although they have been well cited (> 400 cites for the Physiological Reviews paper), the number of citations do not approach that of the original BJN paper, notwithstanding the considerable prestige of these leading journals.
Acknowledgements
The author is grateful to his colleagues and students at the Obesity Biology Unit, University of Liverpool, for their contributions to the work that led to, and followed, the 2004 paper. The author particularly acknowledges the critical contributions of Drs Chen Bing, Bohan Wang and Stuart Wood.
The author declares that he is not in receipt of any relevant external funding.
P. T. devised and wrote this commentary.
The author declares that there are no conflicts of interest pertaining to this article.



 Open access
Open access