


Chronic inflammatory demyelinating polyneuropathy (CIDP) is an acquired neuropathy resulting from an autoimmune response toward peripheral nerve axo-glial antigens predominately presenting with symmetric weakness of proximal and distal muscles.Reference Köller, Kieseier, Jander and Hartung1 The immunologic cause of most forms of CIDP remains unclear; however, immunoglobulin G4 (IgG4)-subclass antibodies against cell adhesion molecules at the nodes of Ranvier have been detected in some patients. Contactin 1 (CNTN1) is a paranodal axonal adhesion molecule which interacts with its glial trans-binding partner, neurofascin-155, forming an axo-glial junction. Autoantibodies against CNTN1 are believed to disrupt the tripartite junction, resulting in delayed nerve conduction. Patients with seropositive CNTN1-associated CIDP often have a severe phenotype associated with motor involvement, sensory ataxia, and paresthesia.Reference Querol, Nogales-Gadea and Rojas-Garcia2
Several case reports have described the concurrence of CIDP and membranous nephropathy (MN), an autoimmune disease which accounts for up to one-third of biopsied cases of nephrotic syndrome, but a disease mechanism linking the conditions has not yet been elucidated. Only a handful of these cases have been found positive for anti-CNTN1 antibodies,Reference Taieb, Le Quintrec and Pialot3,Reference Hashimoto, Ogata and Yamasaki4 but their role in the association of CIDP and MN remains unknown. Here we present a case of CIDP and MN in a patient that was anti-CNTN1 positive and propose potential mechanisms linking the diseases.
A 43-year-old male was referred due to an aggressive generalized polyneuropathy. For the first two months, symptoms were exclusively sensory, with persisting numbness and bilateral paresthesia of the lower extremities, which began to ascend. His past medical history revealed treated hypothyroidism and corrected B12 deficiency. His condition progressively worsened over greater than 2 months resulting in wheelchair-dependency from sensorimotor deficits, with absent bulbar and respiratory involvement. Cerebrospinal fluid level was elevated at 4.0 g/L. Comprehensive blood work including serum protein electrophoresis, anti-myelin-associated glycoprotein, and anti-GM1 was unrevealing. Nerve conduction studies showed prominent demyelinating patterns including prolonged distal latencies, low-amplitude compound muscle action potentials with conduction block [i.e., 63% median block: (wrist) 4.3 mV, (elbow) 1.6 mV], reduced sensory and motor conduction velocities, and prolonged F-wave latencies (Figure 1). Findings met the European Federation of Neurological Societies/Peripheral Nerve Society electrophysical criteria for definitive CIDP. Enzyme-linked immunosorbent assay (ELISA) for paranodal antibodies demonstrated anti-CNTN1 IgG4 antibodies. A treatment course of intravenous immunoglobulin (IVIG) and tapering prednisone was initiated with good clinical and electrophysiologic response (Figure 1).

Figure 1: (A) Strength increased significantly over the first 28 weeks of intravenous immunoglobulin (IVIG) therapy and prednisone. DF=dorsiflexors; EF=elbow flexors; KE=knee extension. Note: grip strength was measured in kg, whereas EF, KE, and DF were measured in Nm. (B) Median, ulnar, and fibular motor conduction parameters uniformly improved over 50 months of therapy. TML=terminal motor latency; ms=milliseconds; CV=conduction velocity; m/s=meters per second.
Unlike other reported cases, nephrotic syndrome was not concurrent with the initial neuropathy presentation but developed over 12 months after, suggesting a temporal gap which has not been identified previously. It also reveals that IVIG alone is not sufficient in preventing nephrotic syndrome. Testing revealed a 24-hour urine protein of 22 g/day, serum albumin 15 g/L, and total cholesterol 8.84 mmol/L, meeting diagnostic criteria for nephrotic syndrome. Renal biopsy and electron microscopy revealed glomerular basement membrane and mesangial immune complex deposits, indicative for stage 2 MN. Immunofluorescence was positive for IgG, but the patient was negative for serum anti-phospholipase A2 receptor (PLA2R) antibodies. Furthermore, the patient's biopsy showed extensive CNTN1 staining within the glomerulus, which was minimal in control samples (Figure 2). The patient began cyclosporin with continuation of prednisone, and protein levels fell to 0.6 g/day within 18 months. Unlike some previously reported concurrent cases in which there was rapidly worsening neuropathy and axonal involvement despite usual therapies, we saw positive recovery of both nephrotic and CIDP symptoms. This suggests a variable disease progression which remains poorly understood and may be related to delayed MN onset.
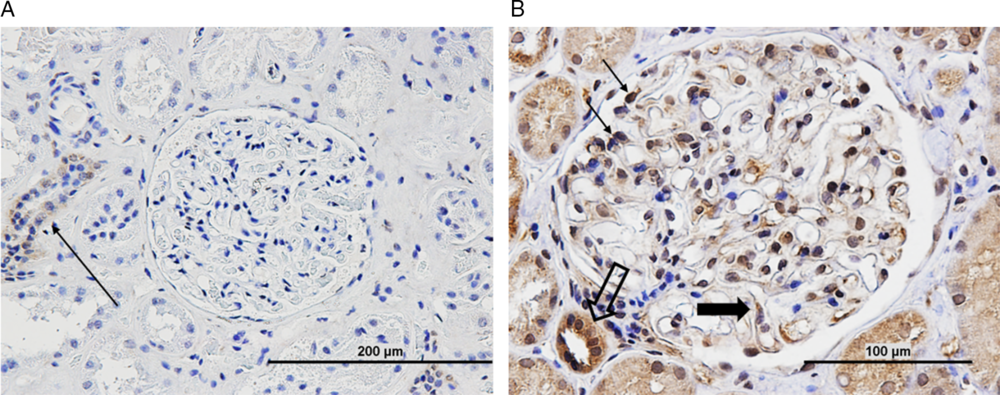
Figure 2: (A) Normal control renal tissue (nephrectomy sample) shows minimal or nonspecific contactin-1 staining in tubules (arrow). (B) Patient biopsy sample shows extensive contactin-1 antibody staining in glomerulus (thin arrow – podocytes, thick arrow – endothelial cell, open arrow – distal tubule). Scale as indicated.
CIDP is a rare disease, and patients with IgG4 anti-CNTN1 antibodies account for less than 10% of all CIDP patients.Reference Querol, Nogales-Gadea and Rojas-Garcia2 Thus, the number of reported patients with anti-CNTN1 antibody-positive CIDP and concurrent MN is minimal, limiting insight into the association between the two autoimmune diseases. The role of anti-CNTN1 antibodies in CIDP has been well-established, with evidence that these antibodies disrupt axo-glial interactions at the paranodes; however, their role in MN remains unclear. Interestingly, the patient in this case was negative for anti-PLA2R antibodies, which are found in 50–80% of MN cases.Reference Francis, Beck and Salant5 Despite this, there was evidence of IgG4 deposition on the glomerular membrane, which still suggests immune-mediated podocyte damage as the underlying cause. What remains unclear is the mechanism of immune damage. Previous reports have speculated that the dense deposits visualized on electron microscopy may represent the deposition of CNTN1-containing immune complexes which in turn are responsible for podocyte dysfunction. To evaluate this further, future studies may look to isolate the proposed immune complexes and transfer them to suitable models to assess for subsequent MN.
A majority of patients reported with concurrent CIDP and MN have presented simultaneously with both conditions or had the two diagnoses within 2 months, which may suggest a common antigenic target.Reference Hashimoto, Ogata and Yamasaki4 Here, the patient's biopsy revealed extensive CNTN1 staining throughout the glomerulus. The widespread immunoreactivity of anti-CNTN1 antibody suggests the possibility of binding to de novo membrane expressed CNTN1 antigen. This implies direct immune attack of the kidney as the underlying cause as opposed to secondary immunocomplex deposition. However, due to the prolonged temporal gap of the CIDP and nephrotic syndrome presentations in this patient, a common antigenic target may be unlikely. Despite this, prior studies have detected low levels of CNTN1 messenger RNA (mRNA) expression in healthy kidney cells.Reference Reid, Bronson, Young and Hemperly6 It may be beneficial to conduct mRNA sequencing studies to clarify expression of CNTN1 in healthy and MN kidney samples to determine if it may possibly be a novel antigen underlying MN. Additionally, passive IgG transfer studies may contribute to our understanding of the pathogenic effect of these antibodies.
Despite uncertainty as to the role of anti-CNTN1 autoantibodies in the concurrence of CIDP and MN, the presence of CNTN1 antibodies may serve as a novel biomarker and warrant renal investigation in patients presenting with aggressive CNTN1-antibody-positive CIDP. This includes testing those that are already being treated with IVIG, as this case has shown that it may not be sufficient to prevent the variable onset of MN. Therefore, using CNTN1 as a biomarker and an indication for renal assessment may allow for detection of early kidney impairment, and as a direct result, allow for targeted management and prevention of additional renal damage.
Disclosures
SN, DT, and PJM have nothing to disclose. EKM reports grants from GBS/CIDP Foundation International and grants from National Health and Medical Research Council, during the conduct of the study. SKB reports personal fees from AKCEA Therapeutics Canada, personal fees from Alnylam Pharmaceuticals, personal fees from Octapharam, other from Allergan Pharmaceutical Company, and other from Grifols, outside the submitted work.
Statement of Authorship
SN organized and wrote the manuscript. EKM ran the ELISA studies of the research project and reviewed and critiqued the manuscript. DT and PJM were responsible for the kidney biopsy/staining and reviewed and critiqued the manuscript. SKB conceived, organized, and executed the research project and reviewed and critiqued the manuscript.


