Woodhouse–Sakati syndrome (WSS) is a rare autosomal recessive multi-system disorder, included under neurodegeneration with brain iron accumulation (NBIA), characterized by hypogonadism, alopecia, diabetes mellitus, sensorineural deafness, mental retardation, and extrapyramidal symptoms Reference Woodhouse and Sakati1 caused by mutations in the DCAF17 (previously known as C2orf37) gene, which encodes a poorly characterized nucleolar protein. Reference Alazami, Al-Saif and Al-Semari2 We describe a South Indian family with two affected siblings suffering from WSS presenting with childhood-onset writer’s cramp.
A 25-year-old lady (proband), born of third-degree consanguineous parentage, presented with writer’s cramp since 10 years of age which later progressed to affect other activities. By 13 years, she had difficulty in tasks with the left upper limb as well. There was no neck dystonia or bradykinesia. She was a school dropout without any behavioral issues or seizures. She had never attained menarche and had sparse scalp hair noticed since childhood.
Examination showed a high-arched palate, facial dysmorphism with triangular facies, hypertelorism, alopecia over the frontal and temporal regions, and poorly developed secondary sexual characters. She had action dystonia of both upper limbs (left > right) with writer’s cramp and dystonia of both lower limbs (Video segment 1 and 2). There were no features of parkinsonism, pyramidal or cerebellar dysfunction.
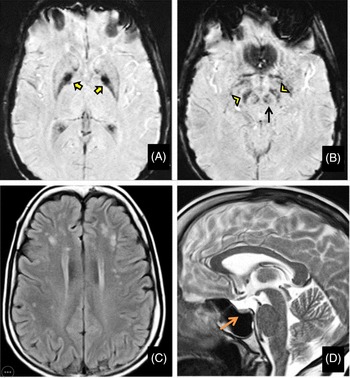
Her routine blood investigations were normal except for hyperglycemia. Electrocardiogram and echocardiography were within normal limits. Pure tone audiometry showed bilateral mild sensorineural hearing loss. Magnetic resonance imaging (MRI) brain showed mineralization in bilateral globus pallidus interna, substantia nigra, and red nucleus on susceptibility-weighted images (SWI) (Figure 1A and B) along with bilateral frontoparietal discrete white matter hyperintensities on T2-weighted images(Figure 1C) and partial empty sella with small-sized hypophysis (Figure 1D). Serum estradiol level of 23.2 pg/mL (normal range in premenopausal females being 30–400 pg/mL) and progesterone levels of 0.04 ng/mL (normal range being 0.1–0.7 ng/mL in the follicular stage and 2–25 ng/mL in the luteal stage) were low, whereas serum follicular stimulating hormone (FSH) level of 33 IU/L(normal range 3–10 ), luteinizing hormone (LH) level of 27.4 IU/L (normal range 2–8 ) were high suggesting a hypergonadotropic hypogonadism secondary to ovarian failure. Ultrasound pelvis showed a small infantile uterus with ill-defined ovaries.
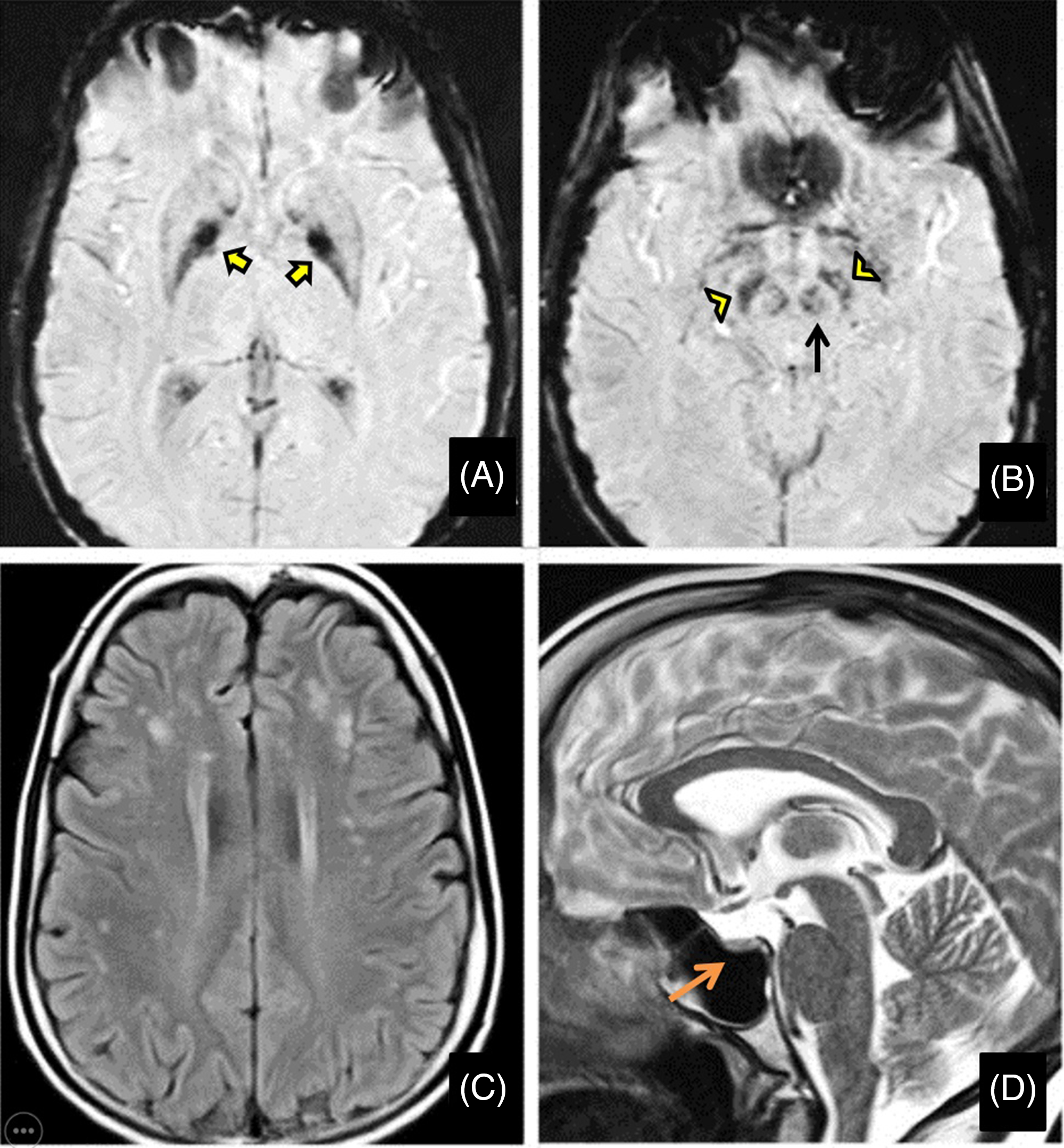
Figure 1: Magnetic resonance imaging (MRI) of the brain shows mineralization in bilateral globus pallidus interna (yellow arrows), substantia nigra (yellow arrowheads), and red nucleus (black arrow) on susceptibility-weighted images (A and B) along with bilateral frontoparietal white matter hyperintensities on T2-weighted images (C) and partial empty sella with small sized hypophysis (D-orange arrow).
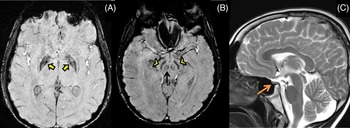
The proband’s 19-year-old brother also had writer’s cramp since 8 years of age which later became non-task specific. He did not have lower limb symptoms, tremors, parkinsonism, or ataxia. He was poor scholastically, had similar sparse scalp hair and impaired secondary sexual characters. Examination revealed action dystonia of both upper extremities (right > left) on writing and other tasks (Video segment 3 and 4). His MRI brain showed iron deposition in bilateral globus pallidus and substantia nigra on SWI images (Figure 2A, B) and a small-sized pituitary gland on T2 weighted images (Figure 2C) with no white matter changes. He had diabetes and a hormonal profile suggesting hypogonadotropic hypogonadism.
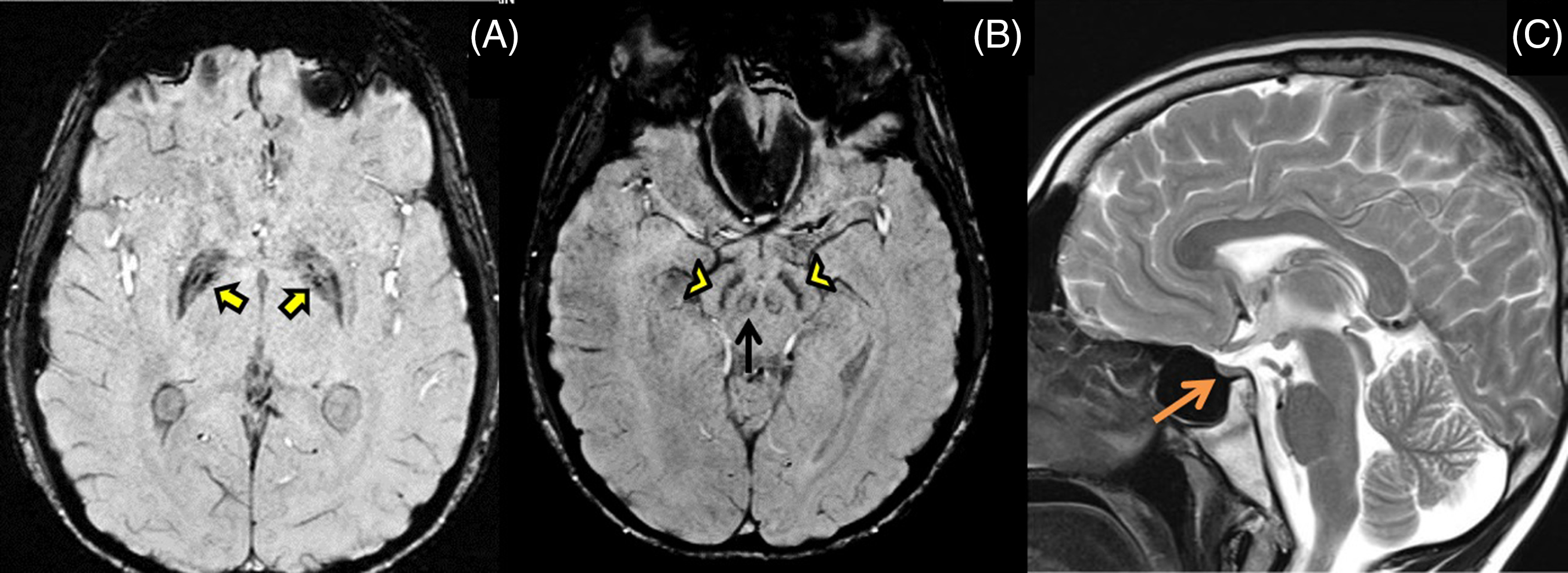
Figure 2: MRI brain shows mineralization in bilateral globus pallidus (yellow arrows), substantia nigra (yellow arrowheads) and red nucleus (black arrow) on SWI images (A,B), and small-sized pituitary gland (orange arrow) on T2 weighted images (C) with no white matter changes.
Targeted gene sequencing (which includes 11 genes implicated in NBIA namely AP4M1, ATP13A2, C19ORF12, COASY, CP, DCAF17, FA2H, FTL, PANK2, PLA2G6, and WDR45) revealed a homozygous pathogenic nonsense mutation c.85C>T (p. Gln29Ter) in exon 1 of DCAF17 gene on chromosome 2q31.1 in the proband and sibling, which causes a premature truncation of the protein, confirming WSS. They were initiated on metformin and low dose insulin for diabetes mellitus, local application of minoxidil for hair growth, a combination pill containing low-dose estrogen and progesterone in monthly cycles for proband and testosterone depo injections once monthly for the brother.
WSS is characterized by facial dysmorphism (long triangular face, hypertelorism, prominent nasal bridge), alopecia, hypogonadism, diabetes mellitus, hypothyroidism, intellectual disability, sensorineural deafness, extrapyramidal features, and ECG abnormalities (ST segment lengthening and flattened or negative T waves). Reference Woodhouse and Sakati1 WSS has been reported from North-East Europe, India, Turkey, Saudi Arabia, Kuwait, Portugal, and Israel and onset is with neurologic, endocrine or skin manifestations during adolescence. Women with WSS have hypergonadotropic hypogonadism, whereas men tend to have hypogonadotropic hypogonadism. This has been ascribed to the greater ovarian damage as compared with testes in this disease and the relative insensitivity of the hypothalomo-pituitary axis (HPA) in males resulting in failure of rise of gonadotropin levels. Reference Bohlega, Abusrair and Al-Ajlan3 The other endocrine manifestations include diabetes mellitus and hypothyroidism which affect 40–50% of individuals, and low serum insulin-like growth factor 1 (IGF-1) level, which is seen in virtually all patients. Reference Agopiantz, Corbonnois and Sorlin4 The lower IGF-1 levels without elevation of growth hormone levels suggest again a HPA dysfunction.
The common neurological features include extrapyramidal signs, mild intellectual disability, dysarthria, dysphagia, spastic paraparesis, sensorineural deafness, seizures, and polyneuropathy. The extrapyramidal features consist of dystonia (focal, segmental, or generalized), chorea, and less commonly tremor and ataxia which progress to cause gait difficulties and immobility later. Reference Schneider and Bhatia5
In a study of 26 genetically proven WSS patients, the most common MRI abnormalities were a small pituitary gland (76.9%), basal ganglia iron deposition (73%), followed by white matter hyperintense lesions which were nonenhancing, frontoparietal and periventricular predominant, sparing subcortical U-fibers helping to distinguish it from other NBIA subtypes. With advancing age, the white matter hyperintense lesions which are initially patchy and discrete (as seen in our case), became more extensive, reflecting a progressive myelin destruction and/or axonal loss. Reference Abusrair, Bohlega and Al-Semari6
The pathogenic mutation is seen in the DCAF17 gene that encodes DDB1- and CUL4-associated factor 17, a nucleolar protein that may function as a substrate receptor for the CUL4-DDB1 E ubiquitin–protein ligase complex. Reference Ali, Shah, Nasir, Steyaert, Coucke and Ahmad7 This protein is highly expressed in the brain, liver, skin, and seminiferous tubules congruent to the neurological, cutaneous and endocrine features. Reference Alazami, Al-Saif and Al-Semari2 Mutated DCAF17 causes diminished lifespan and reduced survival advantages to the endocrine cells. The most common mutation affects exon 4 of DCAF17 gene, whereas our patients had a novel pathogenic homozygous nonsense mutation c.85C>T (p. Gln29Ter) in exon 1 of DCAF17.
The differential diagnosis for WSS includes Perrault’s syndrome, caused by mutations in aminoacyl t-RNA synthetase, characterized by gonadal dysgenesis, primary amenorrhea, cerebellar ataxia, intellectual impairment and sensori-neural hearing loss. A N E syndrome with alopecia, neuropathy, endocrinopathy (hypogonadotropic hypogonadism, adrenal insufficiency, subnormal intelligence) caused by the nucleolar RBM28 dysfunction is another clinical differential of WSS. Given these near-consistent phenotypes and the nucleolar expression of the implicated genes, ribosomal dysfunction seems culpable.
Previously described families from India Reference Koshy, Danda, Thomas, Mathews and Viswanathan8 lacked extrapyramidal features. Ours is the first familial case from India presenting with unique extrapyramidal manifestation, as writer’s cramp, and characteristic MRI/SWI findings and targeted gene sequencing aid in confirming WSS.
Acknowledgements
We acknowledge the help of Ms Anju S (Medical Social Worker) in collecting the patient data.
Conflicts of Interest
None.
Statement of Authorship
1. A. Conception, B. Organization, C. Execution.
2. Manuscript Preparation: A. Writing of the first draft, B. Review and Critique.
MVC: 1A-C, 2A
AC: 1A-C, 2A
DKP: 1A-C, 2B
SK: 1A, 2B
MG: 1B,C, 2B
US: 1A, 2B
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/cjn.2021.201