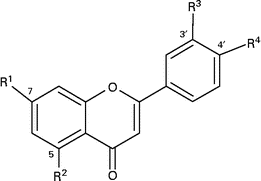
Flavanones, hesperetin and naringenin, are abundant in citrus fruits and constitute a major part of the total flavonoid intake in many European countries(Reference Knekt, Kumpulainen and Jarvinen1, Reference Justesen, Knuthsen and Leth2). Studies on hesperetin and naringenin have shown several health-protective properties such as antioxidant and anti-inflammatory action and prevention of bone loss in vivo (Reference Garg, Garg and Zaneveld3–Reference Kim, Jung and Choi7). Hesperetin and naringenin are naturally present in citrus fruits as their respective 7-O-rutinosides, hesperidin and narirutin(Reference Tomas-Barberen and Clifford8) (Table 1). By enzymatic treatment with α-rhamnosidase, the rutinoside moiety (rhamnose-glucose) can be hydrolysed to yield hesperetin-7-O-glucoside and naringenin-7-O-glucoside, respectively(Reference Nielsen, Chee and Poulsen9). The sugar moiety is a major determinant of the absorption site and bioavailability of the flavonoid since flavonoid monoglucoside bioavailability is several fold higher than flavonoid rutinosides(Reference Nielsen, Chee and Poulsen9–Reference Manach, Morand and Gil-Izquierdo11). Flavonoid monoglucosides are absorbed in the small intestine after hydrolysis by lactase phloridzin hydrolase(Reference Day, Canada and Diaz12, Reference Sesink, Arts and Faassen-Peters13) or cytosolic β-glucosidases(Reference Day, DuPont and Ridley14, Reference Nemeth, Plumb and Berrin15), whereas flavonoid rutinosides must reach the microflora in the colon before they can be hydrolysed and absorbed. Both small intestine and colonic epithelium can metabolise flavonoids via phase 2 conjugation by addition of glucuronic acid or sulphate groups(Reference Brand, van der Wel and Rein16) However, the possibly different roles of these two tissues are not clear for flavonoid conjugation, although it has been recently reported for phenolic acids(Reference Poquet, Clifford and Williamson17). The endogenously produced metabolites are at least in part responsible for the systemic effects of flavonoids, and knowledge about the nature of the metabolites produced is therefore crucial for further studies on putative health effects. The removal of the rhamnose sugar gives an ideal system for studying the effect of small intestine and colon absorption on the metabolic pathways. We demonstrated that the bioavailability of hesperetin was increased when hesperidin in orange juice was enzymatically transformed into hesperetin-7-O-glucoside(Reference Nielsen, Chee and Poulsen9). Here, we further investigate if this enzymatic treatment, as a tool to investigate the absorption mechanism of hesperidin, could also affect the bioavailability of naringenin and identify the major hesperetin and naringenin metabolites excreted in urine after absorption targeted either to the small intestine or colon.
Table 1 Structures of hesperetin and naringenin glycosides and their metabolites

O-Glc-Rha, O-glucose-rhamnose (O-rutinoside); O-GlcU, O-glucuronide.
Materials and methods
Study design
The study was conducted as previously described(Reference Nielsen, Chee and Poulsen9). Sixteen healthy volunteers (eight men and eight women) completed a double blind, placebo controlled, randomised, three-treatment crossover study. After an overnight fast, the subjects were randomly assigned to one of the following three treatments of orange juice: (1) orange juice naturally containing 2 and 0·83 mg/kg body weight hesperidin and narirutin, respectively; (2) α-rhamnosidase-treated orange juice providing 1·52 and 0·52 mg/kg body weight hesperetin-7-O-glucoside and naringenin-7-O-glucoside, respectively; (3) hesperidin-fortified orange juice providing 6 mg/kg body weight total hesperidin, for metabolite identification (all values are based on the dose of aglycone equivalents). Samples of blood (4·5 ml) were collected at 0·5, 1, 1·5, 2, 3, 4, 5, 6, 7, 8, 9 and 10 h after consumption. Urine was collected just before treatment and in three different fractions: 0–5; 5–10; 10–24 h. Treatments were separated by no less than a 3-d washout period. The present study was conducted according to the guidelines laid down in the Declaration of Helsinki and all procedures involving human subjects/patients were approved by the Ethical Committee of Nestlé (Lausanne, Switzerland; protocol 03.15MET). Written consent was obtained from all subjects.
Chemicals
Orange juice was obtained from Nestlé (Bangkok, Thailand). Hesperidinase (67 000 units/g) was from Amano Enzyme Co., Nagoya, Japan. The isotopically labelled internal standards, 13C3 daidzein and 13C3O-desmethylangolensin, were purchased from the School of Chemistry, University of St Andrews, St Andrews, UK. The naringenin-7-O-glucoside and narirutin standards were both purchased from Extrasynthese, Genay, France. Purity of the compounds was determined to be 88–90 % using weight and spectrophotometry. The flavanone standards, hesperetin and naringenin, were obtained from Sigma Chemicals Co., St Louis, MO, USA. The enzymes used for enzymatic hydrolysis of metabolites for calibration curves were β-glucuronidase expressed in Escherichia coli (0·14 mmol/min per mg; Roche Diagnostics, Mannheim, Germany) and Aerobacter aerogenes sulphatase (0·0025 mmol/min per mg protein; Sigma-Aldrich). All other reagents and solvents were HPLC grade.
Naringenin analysis in orange juice, plasma and urine
Naringenin-7-O-glucoside and narirutin were extracted from orange juice essentially as described previously(Reference Breinholt, Svendsen and Dragsted18) but without hydrolysis. Aliquots of 5 ml juice were centrifuged (3000 rpm, 10 min) and applied to Bond Elute columns (2 g; Varian, Middelburg, The Netherlands). The pellet was subsequently extracted twice with 5 ml 1 % aqueous formic acid and three times with 5 ml methanol. The total volume after elution with methanol was 30 ml. Aliquots of 200 μl were evaporated, reconstituted (0·5 % aqueous acetic acid, 10 % acetonitrile) and analysed on the HPLC–UV–MS system with diode array detector (DAD) as described previously(Reference Nielsen and Sandstrom19). The method of analysis was as reported(Reference Nielsen, Chee and Poulsen9) with minor modifications. Eluents: A, 0·5 % aqueous acetic acid, 10 mm-ammonium acetate (v/v); B, acetonitrile. Negative electrospray ionisation mass spectra were obtained in selected ion monitoring mode. Conditions for electrospray ionisation: gas temperature, 350°C; drying gas, 12 litres/min; nebuliser pressure, 414 kpa; capillary voltage (negative), 3500 V. Calibration curves for naringenin-7-O-glucoside and narirutin were obtained by spiking orange juice (1) (natural content of hesperetin and naringenin rutinosides) with 3·5, 7, 14 and 21 μm-naringenin-7-O-glucoside standard and (2) α-rhamnosidase-treated orange juice with 10, 20, 30 and 40 μm-narirutin as standard, respectively. The spiked juice samples were extracted and analysed exactly the same way as the samples. Analysis of hesperetin in orange juice and the concentration of naringenin aglycone in urine and plasma were determined as previously described for hesperetin(Reference Nielsen, Chee and Poulsen9).
Identification of hesperetin and naringenin metabolites in urine
Initially, the identity of metabolites was established on the basis of their MS spectra. Where possible, the exact sites of conjugation were subsequently determined by 1H NMR data after isolation (by collecting fractions directly from the HPLC), differential spectrophotometry and by comparison of their HPLC retention times with authentic synthetic standards. The chemical synthesis of hesperetin conjugates will be published elsewhere.
NMR spectroscopy
NMR spectra were acquired on a Bruker AV 400 WB spectrometer (Bruker, Rheinstetten, Germany) operating at 400·13 MHz for 1H. A 1 mm triple-resonance inverse probe was used (1H observe, 13C, 77Se decouple). Samples were dissolved min 10 μl d 6-dimethyl sulphoxide and transferred to 1 mm NMR tubes.
Typical parameters: 32 768 points were acquired in the time domain with a sweep width of 6000 Hz. The 90° pulse was 5·7 μs. For suppression of the signal from residual water from the freeze-drying process and from d 6-dimethyl sulphoxide, a 1D-noesypresat (Bruker pulse programme library) pulse sequence was used with a pre-saturation time of 1 s, mixing time of 0·3 s and decoupler power level of 70 dB. Receiver gain and number of acquired transients were adjusted according to sample amount and amount of residual formate (from the HPLC mobile phase). Typically, thirty-two transients were acquired for standards and between 2048 and 32 900 for samples.
Spectra were zero-filled to 131 072 complex data points and processed with an exponential multiplication corresponding to an increase in line width of 1 Hz. Spectra were interpreted by comparison with authentic non-metabolised standards.
Identification of hesperetin-3′-O-sulphate
The hesperetin-sulphate in urine samples did not respond well to enzymatic hydrolysis with sulphatase and it was not possible to get a clear spectrum with NMR analysis. Instead, its identity as a hesperetin monosulphate was confirmed by high-resolution LC–quadrupole time-of-flight (Bruker Daltonics, Bremen, Germany) analysis. The monoisotopic mass of hesperetin-sulphate (382·0359 Da) was detected with a mean error of 1·4 parts per million, and information on the position of conjugation was determined indirectly by differential pH spectrophotometry(Reference Xie, Xu and Wang20). Urine samples were analysed on HPLC–DAD as described earlier. However, to obtain pH 6 and 8, a constant delivery of 0·055 and 0·1 ml/min 1 m-Tris base, respectively, was applied to the gradient post-column. UV detection was carried out at 280 and 330 nm, with peak scanning between 210 and 600 nm (2 nm steps).
Analysis and quantification of hesperetin and naringenin metabolites in urine
Urine samples were filtered through a 0·2 μm filter and portioned into aliquots of 200 μl. An internal standard mix (10 μl, 13C3 daidzein and 13C3O-desmethylangolensin, 25 ng/μl dimethyl sulphoxide) was added before adjusting to approximately pH 3 with 10 % aqueous acetic acid. A total volume of 220 μl was analysed on HPLC–DAD–MS. UV detection was carried out at 280 nm with peak scanning between 210 and 600 nm (2 nm steps). UV quantification was preferred due to tendencies to ion suppression in MS, and metabolites were quantified on the basis of their UV absorption unless otherwise stated.
Hesperetin-7-O-glucuronide and hesperetin-3′-O-glucuronide were quantified with calibration curves of their respective authentic synthetic standards (chemical synthesis to be reported elsewhere). For hesperetin-sulphate-glucuronide estimation, urine samples were evaporated and reconstituted in solvent (pure water adjusted to pH 5 with acetic acid). Calibration curves were made by diluting 1:1 in solvent four times in duplicate, resulting in two calibration curves with four concentrations (100, 50, 25 and 12·5 %). To the samples in one calibration curve were added 3 μl β-glucuronidase, 20 μl sulphatase and 5 μl internal standard (20 ng 13C daidzein/μl dimethyl sulphoxide), and to the samples in the other calibration curve were added 23 μl solvent and 5 μl internal standard. All samples were left at 37°C for 1 h. Before analyses on HPLC–DAD–MS, all samples were added to 25 μl 0·5 % acetic acid and 25 μl methanol and centrifuged (10 000 rpm, 5 min). Hesperetin aglycone calibration curves were analysed along with the hydrolysed and unhydrolysed calibration curves for estimation of hesperetin-sulphate-glucuronide.
Calibration curves for naringenin-7- and -4′-O-glucuronide were obtained similar to hesperetin-sulphate-glucuronide as described earlier, except no sulphatase was added, and naringenin aglycone calibration curves were analysed along with the hydrolysed and unhydrolysed calibration curves. Due to co-eluting compounds, naringenin-7- and -4′-O-glucuronide could not be quantified on the basis of UV data but were quantified from MS data.
For analysis of hesperetin-3′-O-sulphate, urine was evaporated and reconstituted in 0·1 m-HCl. Calibration curves were then made by diluting 1:1 five times in duplicate. For one calibration curve, 80 μl of sodium acetate (2 m) was added resulting in a pH of about 5 and a total volume of 130 μl and left at room temperature for 1 h. For the other calibration curve (total volume 50 μl), samples were heated to 95°C for 1 h. Before HPLC–DAD–MS analysis, the heated samples were added to 10 μl of acetonitrile and 40 μl of pure water, while the samples left at room temperature were added to 20 μl of acetonitrile.
Hesperetin aglycone calibration curves were analysed along with the hydrolysed and unhydrolysed calibration curves. Correlation coefficients of all calibration curves were >0·99.
Statistics
The incremental area under the curve (AUC) was determined by a mixed log-linear trapezoidal model, where the linear rule (trapezoidal) is applied to the range where concentration is ascending and the log-linear rule is applied to the range where concentration is descending. Combination of the two trapezoidal methods enhances the estimation of the AUC in reducing errors inherent to both methods by applying them under the conditions of best estimate. Values below the limit of quantification were set to limit of quantification (plasma naringenin limit of quantification = 0·0037 μmol/l). To compare groups 1 and 2, no adjustment for multiplicity was used and all tests were performed at α level 5 %. AUC, C max and T max were calculated using Kinetica (Innaphase, Philadelphia, PA, USA); all other statistical analyses were done with SAS software (version 8.2; SAS Institute, Inc., Cary, NC, USA). From 1 week before the intake of the first treatment, subjects were randomly allocated to one of the three different sequences from the Latin square: [A]–[B]–[C]; [B]–[C]–[A]; [C]–[A]–[B]. The randomisation was done on age and with BMI as the balancing factor.
Results
Plasma bioavailability of total naringenin as aglycone (area under the curve)
The AUC for total plasma naringenin (Fig. 1) after subjects consumed the α-rhamnosidase-treated orange juice (2) was about 4-fold higher compared with consumption of the untreated orange juice (1) and C max was 5·4-fold higher (Table 2).

Fig. 1 Plasma concentration v. time curve of total naringenin in healthy human subjects after consumption of two orange juice treatments. ▲, Natural juice (1); ■, α-rhamnosidase-treated juice (2). Values are means with their standard errors, n 16 (1), n 15 (2).
Table 2 Pharmacokinetic measurements for total naringenin in healthy human subjects after the consumption of two orange juice treatments (untreated (1) and α-rhamnosidase-treated (2))
(Mean values and standard deviations)

AUC, area under the curve; C max, peak plasma concentration; T max, time to maximum plasma concentration.
Intake and urinary excretion of naringenin (as aglycone equivalents)
The total naringenin excretion over 24 h was calculated by pooling the three fractions of urine collected, and the data were expressed as a percentage of naringenin intake. The relative urinary excretion of total naringenin of the subjects was significantly higher (6·7-fold) after consuming α-rhamnosidase-treated orange juice (2) than after consumption of untreated orange juice (1) (Table 2).
Hesperetin and naringenin metabolites excreted in urine
The consumption of orange juice containing the natural untreated dose of hesperidin and narirutin (1), hesperetin-7-O-glucoside and naringenin-7-O-glucoside (2) or high dose of hesperidin (3) resulted in the detection of a total of six urinary metabolites of hesperetin and two metabolites of naringenin by HPLC–MS (Fig. 2). NMR analysis allowed the structural elucidation of four of the six hesperetin and two naringenin metabolites in human urine (Table 3). The identified hesperetin metabolites were: hesperetin-3′-O-glucuronide; hesperetin-7-O-glucuronide; hesperetin-5,7-O-diglucuronide; hesperetin-3′,7-O-diglucuronide. The substitution position of hesperetin-3′-O-sulphate was identified by differential pH spectrophotometry. In addition, a hesperetin-sulphate-glucuronide was detected, but it was not possible to specify the exact substitution positions. Hesperetin-sulphate-glucuronide was identified by MS fragmentation patterns and by enzymatic hydrolysis with sulphatase and glucuronidase which required both enzymes to generate the aglycone. Hesperetin-7- and -3′-O-glucuronides were confirmed by comparison with synthetic standards. Hesperetin-5,7-O-diglucuronide and hesperetin-7,3′-O-diglucuronide were not quantified due to insufficient amounts. The limit of quantification of these diglucuronides could not be accurately determined, since no standards are available. The naringenin metabolites were naringenin-7-O- and -4′-O-glucuronide. Hesperetin and naringenin metabolites were quantified in urine collected at the peak excretion time, which was 0–5 h for the group consuming α-rhamnosidase-treated juice (2) and 5–10 h for (1) and (3) (Fig. 3).

Fig. 2 Representative chromatogram of hesperetin and naringenin metabolites in human urine 0–5 h after ingestion of α-rhamnosidase-treated orange juice (2). (X) Hesperetin-3′,7-O-diglucuronide, (IX) hesperetin-5,7-O-diglucuronide, (XIV) hesperetin-sulphate-glucuronide, (XIII) naringenin-7-O-glucuronide, (XII) naringenin 4′-O-glucuronide, (VIII) hesperetin 7-O-glucuronide, (VII) hesperetin 3′-O-glucuronide, (XI) hesperetin 3′-O-sulphate, (IS1) internal standard 13C3 daidzein, (IS2) internal standard 13C3O-desmethylangolensin.
Table 3 1H NMR, LC–MS and UV data of hesperetin and naringenin metabolites excreted in human urine after consumption of three different kinds of orange juice (see Table 1 and Fig. 2)

d, Doublet; NO, not observed; dd, double doublet; ad, apparent doublet; #, multiplet; s, singlet; FI, fragment ion(s); ND, not detected; UV abs, UV absorption.

Fig. 3 Metabolite excretion in urine as percentage of intake. (a) Hesperetin metabolites. ![]() , Hesperetin-7-O-glucuronide;
, Hesperetin-7-O-glucuronide; ![]() , hesperetin-3′-O-glucuronide; □, hesperetin-sulphate-glucuronide;
, hesperetin-3′-O-glucuronide; □, hesperetin-sulphate-glucuronide; ![]() , hesperetin-3′-O-sulphate. (b) Naringenin metabolites. □, Naringenin-4′-O-glucuronide;
, hesperetin-3′-O-sulphate. (b) Naringenin metabolites. □, Naringenin-4′-O-glucuronide; ![]() , naringenin-7-O-glucuronide. Metabolites were measured in urine collected at the peak excretion time, which was 0–5 h for the α-rhamnosidase-treated juice (2) and 5–10 h for the natural (1) and the fortified juice (3). Means with their standard errors, n 13 (2 and 3), n 15 (1).
, naringenin-7-O-glucuronide. Metabolites were measured in urine collected at the peak excretion time, which was 0–5 h for the α-rhamnosidase-treated juice (2) and 5–10 h for the natural (1) and the fortified juice (3). Means with their standard errors, n 13 (2 and 3), n 15 (1).
Discussion
We show for the first time that naringenin-7-O-glucoside is more bioavailable than narirutin (naringenin-7-O-rutinoside) in human subjects. This is as judged by the total naringenin AUC, C max and urinary excretion data that were significantly higher for naringenin-7-O-glucoside compared with the natural untreated orange juice containing narirutin. This is in accordance with hesperetin(Reference Nielsen, Chee and Poulsen9) and quercetin(Reference Hollman, Bijsman and van Gameren10). Others have studied the bioavailability of naringenin in rats(Reference Felgines, Texier and Morand21) where the urinary naringenin recovery as percentage of dose was 28, 31, and 14 % after aglycone, glucoside or rhamnoglucoside (naringin) intake, respectively. The urinary naringenin recovery as percentage of dose in the present study was 7 and 47 % after narirutin and naringenin-7-O-glucoside intake, respectively. When naringenin was given as an aglycone to human volunteers, the urinary recoveries for naringenin were approximately 6 % of the administered dose, i.e. there was no increase in bioavailability (as observed in rats)(Reference Kanaze, Bounartzi and Georgarakis22). It seems therefore that in human subjects naringenin bioavailability is highest when consumed as a monoglucoside. Higher bioavailability of monoglucosides might provide health protective effects in lower doses since increased bioavailability of hesperetin-7-O-glucoside resulted in the same level of protection against bone loss as compared to higher levels of hesperidin (hesperetin-7-O-rutinoside) in rats (0·25 and 0·5 % of the diet, respectively)(Reference Habauzit, Nielsen and Gil-Izquierdo23).
Other studies on bioavailability of naringenin from orange juice have reported values for relative urinary excretion from 1 to 18 % of narirutin intake(Reference Manach, Morand and Gil-Izquierdo11, Reference Erlund, Meririnne and Alfthan24, Reference Mullen, Archeveque and Edwards25); the present study on untreated orange juice for naringenin is in the middle of this range. The biochemical pathway accounting for the increased bioavailability of naringenin-7-O-glucoside has been described in several papers. The absorption of flavonoid glucosides in the small intestine is facilitated by the presence of lactase phloridzin hydrolase in the gut lumen brush border and β-glucosidase in the intestinal cells(Reference Day, Canada and Diaz12, Reference Sesink, Arts and Faassen-Peters13, Reference Day, Gee and DuPont26). Both enzymes are capable of hydrolysing flavonoid glucosides but not rutinosides, which are only absorbed after hydrolysis by the colonic microflora.
We have found six hesperetin and two naringenin metabolites in human urine after consumption of (1) natural untreated orange juice; (2) α-rhamnosidase-treated orange juice; (3) orange juice fortified with three times the original content of hesperetin-7-O-rutinoside. We have identified the exact site of conjugation and quantified several hesperetin and both naringenin metabolites. Two recent studies have identified hesperetin and naringenin metabolites in human urine after consumption of orange juice(Reference Mullen, Archeveque and Edwards25, Reference Brett, Hollands and Needs27). Both have found hesperetin-7-O-glucuronide, naringenin-7- and -4′-O-glucuronide. Brett and co-workers have identified hesperetin-3′-O-glucuronide and hesperetin-3′-O-sulphate as well. In addition to these metabolites, we have further identified hesperetin-5,7-O-diglucuronide and hesperetin-3′,7-O-diglucuronide. All quantification of metabolites was based on the corresponding metabolite either as synthetic standard or collected directly from urine with high metabolite concentration. This excludes a potential source of error from quantitative estimates based on non-identical metabolites.
In the present study, glucuronides of hesperetin were the most abundant metabolites recovered. This result is in agreement with several other studies(Reference Manach, Morand and Gil-Izquierdo11, Reference Mullen, Archeveque and Edwards25, Reference Matsumoto, Ikoma and Sugiura28, Reference Yamada, Tanabe and Arai29). We have also found a substantial amount of hesperetin-3′-O-sulphate excretion after all treatments. It has been suggested that flavonol sulphate-glucuronides are glucuronidated before sulphation, and the sulphation then occurs in the liver(Reference Mullen, Edwards and Crozier30, Reference O'Leary, Day and Needs31). In contrast, Jaganath et al. (Reference Jaganath, Mullen and Edwards32) did not find evidence of sulphation of quercetin after intervention with tomato juice containing rutin (quercetin-3-O-rutinoside which is absorbed in the colon), whereas quercetin-3′-O-sulphate is one of the dominating metabolites in plasma from human volunteers fed fried onions (containing quercetin-4′-O-glucoside and quercetin-3,4′-O-diglucoside which are absorbed in the small intestine)(Reference Mullen, Edwards and Crozier30, Reference Day, Mellon and Barron33). In the present study, the amount of hesperetin-3′-O-sulphate in the urine was not affected by the site of absorption indicating that either hesperetin can be sulphated equally by both small intestinal and colonic sulphotransferases or hesperetin sulphation predominantly occurs in the liver after absorption. It has already been shown that liver cells can deconjugate certain quercetin glucuronides and then subsequently sulphate them(Reference O'Leary, Day and Needs31).
The phase 2 enzymes, UDP glucuronosyl transferase and sulphotransferase, catalyse the conjugation of polyphenols with glucuronic acid and sulphate groups, respectively(Reference Brand, van der Wel and Rein16, Reference Silberberg, Morand and Mathevon34, Reference Scalbert and Williamson35). Both enzymes exist as numerous isoforms and are present in several tissues including the small intestine, colon and liver(Reference Strassburg, Nguyen and Manns36, Reference Glatt, Engelke and Pabel37). Expression of both enzyme classes shows common characteristics in the human alimentary tract; however, the pattern of the forms expressed differs between various segments and opens up the possibility that different metabolites can be formed according to where in the alimentary tract the aglycone is absorbed(Reference Strassburg, Nguyen and Manns36, Reference Glatt, Engelke and Pabel37). The present study provides no information on the mechanisms involved in conjugation of hesperetin and naringenin; however, it is evident that following the release of aglycone, hesperetin and naringenin are subjected to glucuronidation and/or sulphation, and the pattern of excreted metabolites is not affected by the site (small intestine or colon) of absorption. Furthermore, increasing the dose of hesperidin 3-fold only affected the total concentration but did not have any impact on the profile of the conjugates formed after colonic absorption. Colonic microflora produce lower molecular weight phenolics that can be absorbed, such as dihydrocinnamic, phenylacetic and benzoic acids. However, these have not been characterised in the present study.
It is generally agreed that conjugate position is important in relation to the biological activity of flavonoids in human subjects, but only a few studies have looked at the biological activities of the conjugates(Reference Williamson, Barron and Shimoi38). Hesperetin glucuronides, but not the hesperetin aglycone, protected against UV-A-induced necrotic cell death(Reference Proteggente, Basu-Modak and Kuhnle39). Quercetin glucuronides inhibited Cu2+-induced oxidation of human LDL(Reference Manach, Morand and Crespy40). Xanthine oxidase was inhibited by quercetin conjugates, and the inhibition varied 50–800-fold depending on the site of conjugation(Reference Day, Bao and Morgan41). Hesperetin aglycone protected against oxidative injury to primary cortical neurons whereas hesperetin-7-O-glucuronide did not(Reference Vauzour, Vafeiadou and Rice-Evans42), probably related to the inability of glucuronide metabolites to passively traverse membranes, unless a transporter is present(Reference O'Leary, Day and Needs31). In general, the responses to flavonoid conjugates are weaker than those to the aglycones(Reference Williamson, Barron and Shimoi38).
In conclusion, we have shown that the bioavailability of naringenin-7-O-glucoside is increased about 4-fold based on AUC compared to narirutin in healthy human subjects consuming orange juice. There was no significant difference between the pattern of metabolites excreted in urine after consumption of orange juice containing either glucosides or rutinosides, demonstrating that metabolism of hesperetin and naringenin after absorption in the small intestine is not significantly different from the metabolism after absorption in colon. In addition, it was shown for hesperidin that a 3-fold increase in dose does not affect the pattern of excreted metabolites. The eight identified metabolites were all phase 2 conjugates, of which seven were structurally elucidated.
Acknowledgements
We would like to thank all the volunteers of the study. We also wish to thank laboratory technician Anni Schou for skilful technical assistance, senior scientist Henrik Frandsen for valuable scientific assistance and Nestlé Research Center, Lausanne, Switzerland, for funding the present study. L. B., S. E. R. and C. C. have no conflicts of interest. I. L. F. N., D. B., F. B., E. O.-C. and G. W. are, or were, full- or part-time employees of the Nestlé Research Center, Lausanne, Switzerland. I. L. F. N., E. O.-C. and G. W. planned the present study. I. L. F. N. carried out the present study. D. B. provided and characterised hesperetin standards used in the present study. L. B. and S. E. R. analysed the samples, interpreted the data and wrote the first draft of the present paper. F. B. carried out the statistical analysis. C. C. carried out and interpreted the NMR data. G. W., L. B. and I. L. F. N. wrote the manuscript.