



Introduction
Background
Beryl as the most common beryllium mineral is of important geochemical significance in different geological contexts. Ideally Be3Al2Si6O18, beryl is a cyclosilicate mineral of hexagonal crystal structure. Six-membered rings of Si tetrahedra are linked by tetrahedrally coordinated Be2+ (T2 site) and octahedrally coordinated Al3+ (A site) in planes parallel to (0001) (Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988, Reference Aurisicchio, Grubessi and Zecchini1994; Černý, Reference Černý and Grew2002; Groat et al., Reference Groat, Giuliani, Marshall and Turner2008). The rings form channels parallel to the crystallographic c axis, which in natural beryl typically contain variable amounts of primarily alkali cations and neutral molecules of water, CO2 and CH4 (Ginzburg, Reference Ginzburg1955; Wood and Nassau, Reference Wood and Nassau1968; Hawthorne and Černý, Reference Hawthorne and Černý1977; Aines and Rossman, Reference Aines and Rossman1984; Artioli et al., Reference Artioli, Rinaldi, Stahl and Zanazzi1993; Charoy et al., Reference Charoy, De Donato, Barres and Pinto-Coelho1996; Pankrath and Langer, Reference Pankrath and Langer2002; Gatta et al., Reference Gatta, Nestola, Bromiley and Mattauch2006; Groat et al., Reference Groat, Giuliani, Marshall and Turner2008; Fridrichová et al., Reference Fridrichová, Bačík, Bizovská, Libowitzky, Škoda, Uher, Ozdín and Števko2016). The accommodation of monovalent alkali cations on the two channel vacancy sites charge balances the substitution of divalent or trivalent cations (e.g. Mg2+, Fe2+/3+ or Mn2+) for Al3+ at the A site, or the substitution of primarily Li+ for Be2+ at the T2 site (Folinsbee, Reference Folinsbee1941; Wood and Nassau, Reference Wood and Nassau1968; Bakakin et al., Reference Bakakin, Rylov and Belov1969; Hawthorne and Černý, 1977; Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988). The smaller alkali Na+ occupies the narrower channel site (C2/2b), while the larger K+, Cs+ or Rb+ as well as H2O and CO2 prefer the wider channel site (C1/2a) (Aurisicchio et al., Reference Aurisicchio, Grubessi and Zecchini1994; Łodziński et al., Reference Łodziński, Sitarz, Stec, Kozanecki, Fojud and Jurga2005; Fukuda and Shinoda, Reference Fukuda and Shinoda2008). Pure beryl is colourless (var. goshenite), whereas different substitutions give rise to distinct colours of blue (var. aquamarine, Fe), green (var. emerald, Cr/V), yellow (var. heliodor, Fe) or pink (var. morganite, Mn) (Lum et al., Reference Lum, Viljoen, Cairncross and Frei2016; Tempesta et al., Reference Tempesta, Bosi and Agrosì2020).
Following early ultraviolet (UV), visible, and IR spectroscopy (Wood and Nassau, Reference Wood and Nassau1967, Reference Wood and Nassau1968), the lattice position, molecular orientation, bonding state and dynamic behaviour of water in natural and synthetic beryl, and at variable temperatures, have been characterised spectroscopically (Kim et al., Reference Kim, Bell and McKeown1995; Charoy et al., Reference Charoy, De Donato, Barres and Pinto-Coelho1996; Kolesov and Geiger, Reference Kolesov and Geiger2000; Łodziński et al., Reference Łodziński, Sitarz, Stec, Kozanecki, Fojud and Jurga2005; Fukuda and Shinoda, Reference Fukuda and Shinoda2008, Reference Fukuda and Shinoda2011). Based on these studies, water on the C1/2a sites has been defined as type I or type II with a molecular dipole moment orientated perpendicular or parallel with respect to the c axis, respectively. In addition the preference for each type is controlled by the channel alkali content.
Xuebaoding beryl
One notable occurrence of beryl is the Xuebaoding greisen-type W–Sn–Be deposit in the Songpan–Ganzi (or Songpan–Garzê) orogenic belt in Sichuan Province, western China (Zhang et al., Reference Zhang, Peng, Coulson, Hou and Li2014). Explored since the 1950s, the occurrence is known for scheelite, cassiterite and beryl of exceptional size and gem quality, with quartz, fluorite and minor apatite formed in a post-magmatic hydrothermal process (Lin et al., Reference Lin, Cao, Liu, Li, Zhang and Ying2000; Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002; Liu et al., Reference Liu, Deng, Cai, Zhou, Wang, Zhou, Gao, De xiu, Fu yu and Zhu2005). The abundant beryl is typically transparent and colourless to pale blue. In contrast to the common columnar prismatic habit of beryl parallel to the c axis, Xuebaoding beryl is primarily of tabular habit parallel to the basal pinacoid.
Even though a range of studies have addressed the gemmology (Qi et al., Reference Qi, Ye, Xiang, Pei and Luo2001a, Reference Qi, Pei, Zhou, Shi and Luob, Reference Qi, Xiang, Liu, Pei and Luoc, Reference Qi, Ye, Xiang, Pei and Shid) and mineralogy of beryl from Xuebaoding over more than two decades (Guo et al., Reference Guo, Wang, Xu, Fontan, Luo and Cao2000a, Reference Guo, Wang and Xub; Lin et al., Reference Lin, Cao, Liu, Li, Zhang and Ying2000; Ye et al., Reference Ye, Qi, Luo, Zhou and Pei2001; Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002; Liu et al., Reference Liu, Deng, Li and Shi2007, Reference Liu, Deng, Shi and Sun2012), its crystal chemistry has not yet been well established with several analytical results either inconclusive or in part contradictory, leading to a confusing picture even about elementary questions of composition and structure. Further, contrasting hypotheses were proposed, with crystal growth controlled by defects and dislocations (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b) or by high alkali content at the Be and channel sites (Liu et al., Reference Liu, Deng, Shi and Sun2012) being responsible for the tabular habit.
This work
The goal of this work is a systematic chemical, including trace element, and structural classification of beryl from Xuebaoding including comparison with beryl from other localities, notably of comparable tabular habit. Further, water in its different structural states and its vibrational response are quantified by correlating single-crystal X-ray diffraction with Raman and IR micro-spectroscopy and imaging.
Geographic and geological setting
The sample localities are generally referred to as Xuebaoding (Sichuan Province, China) situated northwest of the Longmen Shan range, at the eastern margin of the Tibetan plateau, at elevations of 3900–4200 m. The locality is named after the mountain Xuebaoding (‘Snow Treasure Crown’, Tibetan name: Shar Dung Ri, elevation 5,588 m), the highest peak of the Minshan range. However, Xuebaoding mountain itself is 5.3 km to the NW of the locality, and not visible from the locality, separated by several high ridges and the mountain Sigenxiangfeng (5,360 m). Instead, the peak visible to the immediate NW (Fig. 1a, top left), is Little Xuebaoding (5,443 m) and situated above the valley to the western terminus of the locality area. The confusion between the two peaks, including confusion about first ascents of these mountains persisted well into the 1980s, and until today by the local population, as a reflection of the complex topography and extreme relative relief of up to 4000 m between the valley floors and adjacent peaks, with many of similar altitude.
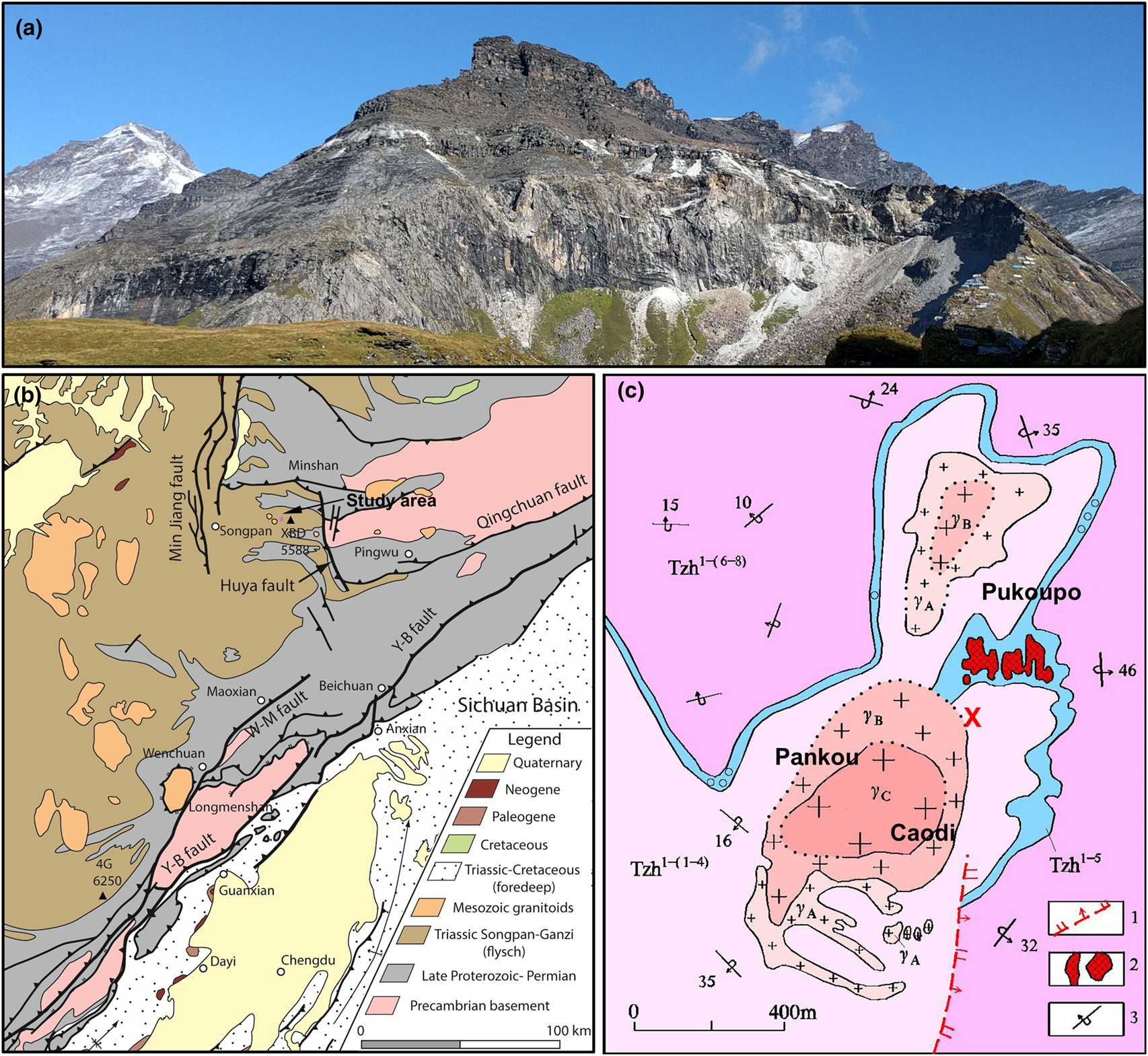
Fig. 1. (a) Xuebaoding locality, with Little Xuebaoding (5,443 m, left) in the background. Mineralised zones along a nearly horizontal marble band (white mine tailings) and in schist, along and below both sides of ridge to the right (mining camp at bottom right). (b) Generalised geological map of eastern margin of Tibetan plateau with Xuebaoding (XBD) mountain west of the Huya fault and to NE of the Longmen Shan range and uplift zone defined by Yingxiu–Beichuan (Y-B) and Wenchuan–Maoxian (W-M) faults; major peaks of Siguniangshan (4G) and Xuebaoding (XBD) indicated (modified with permission after Kirby and Ouimet, Reference Kirby, Ouimet, Gloaguen and Ratschbacher2011). (c) Geological map of the main locality area (pink rectangle in (b)) associated with granite body outcrops Pankou and Caodi (defining the cirques in (a)) and Pukoupo. Legend: 1: normal fault, 2: highly mineralised quartz vein containing beryl, scheelite and cassiterite, 3: overturned dip and strike (modified after Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002). Rock units: Tzh1-(1–4) quartz schist, marble, biotite-quartz schist; Tzh1–5 ore-bearing strata, marble; Tzh1-(6–8) calcareous quartz schist, sericite quartz schist; γA-C granite margin through transition to core zone. The W–Sn–Be mineralisation is concentrated throughout schist and marble within <100 m from the granite intrusions. Approximate sample locations in vicinity and south of reference position (red cross: lat. 32.6121987°N, long. 103.9418417°E).
Tectonically, the area is situated in the Songpan–Ganzi orogenic belt to the west of the Sichuan Province, near the contact with the Qinling orogenic belt (north) and the western Yangtze block (east and south-east) (Fig. 1b). The Songpan–Ganzi orogenic belt is the result of two stages of tectonic movements. Following the late Indochina tectonic epoch with long-term intracontinental convergence and associated series of magmatic and metamorphic events, the subsequent Himalayan movement starting at ~80 Ma resulted in a pronounced twist in the Songpan–Ganzi orogenic belt (Xu et al., Reference Xu, Hou, Wang, Fu and Huang1992; Kirby and Ouimet, Reference Kirby, Ouimet, Gloaguen and Ratschbacher2011). Along the Minshan (or Min Mountains) the active fault system of the N–S trending Huya fault, a steeply west-dipping reverse fault, contributes to the ongoing uplift of the Minshan. The Huya fault separates Precambrian basement and overlying late Proterozoic to Permian rock units to its east, from the Triassic Songpan–Ganzi flysch sequences to its west (Kirby and Ouimet, Reference Kirby, Ouimet, Gloaguen and Ratschbacher2011). The numerous intruded granites in the eastern margin of the Tibetan plateau have been dated at 220 to 188 Ma (Roger et al., Reference Roger, Malavieille, Leloup, Calassou and Xu2004, Reference Roger, Jolivet and Malavieille2010; Xiao et al., Reference Xiao, Zhang, Clemens, Wang, Kan, Wang, Ni and Liu2007).
Geology of the Xuebaoding deposit
The Xuebaoding mineralisation is associated with several Mesozoic leucogranite intrusions, of a few 100 m to 1 km in extent in outcrop (e.g. Pankou, Pukou and Wuzhutang), of similar spatial separation, and dated at 201–194 Ma (Fig. 1c) (Ye et al., Reference Ye, Qi, Luo, Zhou and Pei2001; Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002; Cao et al., Reference Cao, Jianbin, An and Youguo2004; Jiang et al., Reference Jiang, Zhou, Guo, Cao and Xu2007; Li et al., Reference Li, Liu, Wang and Fu2007). Whether these different outcrops are of a single intrusion, or represent separate intrusions, and their relationship is yet unknown. However, their similarities in rock type and associated mineralisation suggests at least a close magmatic relationship.
The granites intruded into the upper Triassic Zhuwo series of deep-marine sedimentation origin, accompanied by slate, schist and marble lenticules, as well as layers of marble of shallow metamorphism of late Indosinian orogeny to early Yanshan age (Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002; Cao et al., Reference Cao, Jianbin, An and Youguo2004; Ottens, Reference Ottens2005; Li et al., Reference Li, Liu, Wang and Fu2007). The sequence of layers of sericite, quartz and carbonaceous schist, mudstone, marble and local skarn, are overturned, with shallow dip to the west, yet steeply dipping to the east of the granite contacts (Fig. 1c).
Mineral assemblage
The Xuebaoding locality is a globally significant occurrence of scheelite and cassiterite, in combination with beryl, all of unusually large size, gem colour, and crystal quality. It is a greisen-type deposit of quartz and muscovite. Albeit locally variable in relative distribution, the joint mineralisation of scheelite, cassiterite and beryl suggests that it is derived from a highly fractionated and evolved magma in the apical section of the granitic body (Cao et al., Reference Cao, Zheng, Li, Ren, Xu, Wang, Shoji, Kaneda and Kabayashi2002, Reference Cao, Jianbin, An and Youguo2004; Li et al., Reference Li, Liu, Wang and Fu2007; Zhang et al., Reference Zhang, Peng, Coulson, Hou and Li2014; Zhu et al., Reference Zhu, Raschke and Liu2020). Highly enriched in volatiles and metallic elements this gives rise to simple mineralisation of large, mostly common, W, Sn, Be, F and P minerals. The mineralisation of muscovite, with fluorite and apatite in addition to the W and Sn mineralisation is typical for an S-type granite derived system. The mineralisation is fracture-controlled and occurs in sheeted veins and stockwork, emanating from the greisenised granite into the country rock. However, associated post-magmatic alteration of the host rock by volatiles and solutions derived from the cooling granitic intrusion, as is otherwise typical for greisen, is not very pronounced (Aurisicchio et al., Reference Aurisicchio, Grubessi and Zecchini1994).
Forming interconnected networks of quartz (with muscovite and fluorite), well-defined mineralised veins with a N–S trend are found primarily in marble, with a more stockwork type in the surrounding schist. This reflects that the marble is more brittle forming fractures for fluids to circulate and deposit, with the schist more ductile. The different ore veins in granite, marble and schist as host rock with different generations and conditions with spatial and temporal variations in mineralisation exhibit large variations in mineral assemblage – with locally spatial ring fractures, replacement mineral bodies, distal vein emplacement, hydrothermal breccia boiling and hydrothermal alterations.
Xuebaoding beryl
The global occurrence of beryl, especially in large crystals, is typically associated with granitic pegmatites. Tungsten and tin mineralisations often occur simultaneously and are associated, but typically without beryl. Mineralisation of W and Sn together with Be is generally uncommon, which makes the combination of W, Sn and Be mineralisation of the Xuebaoding occurrence, and not of a pegmatite-type but rather of a hydrothermal/greisen-type, make Xuebaoding unusual.
Despite decades of research since the discovery of the localities in the 1950s (Guo et al., Reference Guo, Wang, Xu, Fontan, Luo and Cao2000a; Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b; Cao et al., Reference Cao, Zheng, Li, Ren, Xu, Wang, Shoji, Kaneda and Kabayashi2002; Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002; Liu et al., Reference Liu, Deng, Shi and Sun2012; Zhang et al., Reference Zhang, Ruan and Rao2012), only limited field studies have been undertaken and the ore-forming processes and source of the fluid are not yet clear. Further, despite several ages reported for granite and mineralisation, some are inconsistent, and it is unknown if they reflect the date of mineral formation.
Xuebaoding beryl is mostly colourless, transparent and rarely with a hint of blue. It typically exhibits a simple morphology, yet of characteristic tabular habit shortened in the c direction, with the most common basal pinacoid (0001), prisms (10 $\bar{1}$0) and (11
$\bar{1}$0) and (11 $\bar{2}$1) faces as shown in Fig. 2. The habit is rarely columnar, and if so has a low aspect ratio (Fig. 2c).
$\bar{2}$1) faces as shown in Fig. 2. The habit is rarely columnar, and if so has a low aspect ratio (Fig. 2c).

Fig. 2. Examples of representative Xuebaoding beryl habits: (a) cluster of tabular beryl on muscovite matrix (largest crystal 25 mm wide); (b) individual clear tabular crystal (40 mm wide); (c) rare columnar habit (on albite, 25 mm wide), light blue, with typical dislocation texture on (0001); (d) schematic of tabular habit.
Numerous studies covering a wide range of techniques have addressed selected aspects of the crystal chemistry of beryl from Xuebaoding (Guo et al., Reference Guo, Wang, Xu, Fontan, Luo and Cao2000a; Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b; Cao et al., Reference Cao, Zheng, Li, Ren, Xu, Wang, Shoji, Kaneda and Kabayashi2002; Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002; Liu et al., Reference Liu, Deng, Shi and Sun2012; Zhang et al., Reference Zhang, Ruan and Rao2012). Specifically, several studies using bulk chemical analysis, electron microprobe analysis (EPMA), and laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) identified enrichment in Na+ (Na2O = 0.035–1.66 wt.%) and Li+ (Li2O = 0.63–3.04 wt.%) with variable amounts of Cs+ (Cs2O = 0.11–0.92 wt.%), and minor K+ (K2O = 0.01–1.13 wt.%), Mg2+ (MgO = 0.03–0.13 wt.%), Ca2+ (CaO = 0.01–0.88 wt.%) and Fe3+ (FeOT = 0.06–0.54 wt.%) (Liu et al., Reference Liu, Zhang and Yu2001b, Reference Liu, Deng, Cai, Zhou, Wang, Zhou, Gao, De xiu, Fu yu and Zhu2005, Reference Liu, Deng, Shi and Sun2012; Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b; Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002; literature data summarised in Supplementary Table S1). While some analyses are inconsistent, they confirm the trend that defines Xuebaoding beryl as a lithium–sodium or ‘tetrahedron’ beryl (Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988, Reference Aurisicchio, Grubessi and Zecchini1994)
A comprehensive EMPA of beryl, whole rock analysis of granite, and fluid inclusion study suggested a post-magmatic pneumatolytic–hydrothermal solution as the source of the beryl mineralisation (Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002). An X-ray fluorescence and LA-ICP-MS trace-element analysis, in combination with XRD, and surface textural features, identified Li+ substitution at the tetrahedral Be2+ site together with divalent cation substitution for Al3+ with associated charge balancing by other alkali on the vacancy site (Liu et al., Reference Liu, Deng, Shi and Sun2012). The high alkali content (0.255–0.327 apfu, not including Li+) was hypothesised to be responsible for the tabular crystal growth (Liu et al., Reference Liu, Deng, Shi and Sun2012). In contrast, a detailed analysis of surface defects on different crystal phases in combination with a growth rate model as a function of supersaturation (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b) suggested interstitial deformation and dislocation sliding responsible for the different growth rate of basal vs. prismatic crystal faces giving rise to the tabular morphology (Zhang et al., Reference Zhang, Ruan and Rao2012).
Several fluid-inclusion studies have been performed for quartz, beryl and scheelite identifying homogenisation temperatures covering a wide range of 214–288°C (Cao et al., Reference Cao, Zheng, Li, Ren, Xu, Wang, Shoji, Kaneda and Kabayashi2002), 158–310°C (Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002), 250–292°C (Chen et al., Reference Chen, Wan, Lu and Wang2002), 265–315°C (Lin et al., Reference Lin, Cao, Liu, Li, Zhang and Ying2000), 188–373°C (Zhang et al., Reference Zhang, Ruan and Rao2012) and 266–389°C (Liu et al., Reference Liu, Deng, Cai, Zhou, Wang, Zhou, Gao, De xiu, Fu yu and Zhu2005), suggesting a shallow hydrothermal quartz vein type system. These works imply a volatile-rich pneumatolytic–hydrothermal solution derived from post-magmatic fluid as a source of the mineralisation (Cao et al., Reference Cao, Zheng, Li, Ren, Xu, Wang, Shoji, Kaneda and Kabayashi2002; Zhou et al., Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002). Related work suggests a temperature range of 250–292°C, at high pressures (~86 MPa) with high CO2 density (0.662 g/cm3), and low salinity (6.191 wt.% NaCl) during the formation of beryl (Chen et al., Reference Chen, Wan, Lu and Wang2002). This is consistent with more recent work (Zhang et al., Reference Zhang, Ruan and Rao2012) suggesting a temperature of beryl formation of ~300°C.
Material and analytical methods
For this study selected single crystals and rock-forming beryl were chosen from different veins and collected in situ. The rock-forming sample hosted in quartz schist Tzh1-(1–4) has been extracted from an active mine on the east side of Pankou (thin section 1702B2, for details see Supplementary Fig. S1). Further, from clefts in veins hosted in marble Tzh1–5 small single crystals of 0.5–1.5 cm in size were obtained and cut into thin sections, with two samples cut parallel and four perpendicular with respect to the c axis (1901B1 and 1901B2, for details see Fig. S1). Lastly several single crystals were collected from open space in veins in the northern area of Pankou (#Q1–Q7, see Fig. S1). The different crystal faces were first studied by atomic force microscopy (AFM, AIST Inc., noncontact mode, probe tips from μMasch Inc., spatial resolution ~10 nm and sample scanner resolution 2 nm), prior the preparation of slabs of 2–3 mm thickness for EMPA cut along different crystallographic directions perpendicular to (0001), (10 $\bar{1}$0) and (11
$\bar{1}$0) and (11 $\bar{2}$1) (#Q7, #Q3 and #Q6), as shown in Fig. 3. Subsequently for IR transmission spectroscopy and imaging, the samples were thinned to ~0.1 mm double sided polished slabs.
$\bar{2}$1) (#Q7, #Q3 and #Q6), as shown in Fig. 3. Subsequently for IR transmission spectroscopy and imaging, the samples were thinned to ~0.1 mm double sided polished slabs.

Fig. 3. Beryl samples as cut and studied from larger single crystal specimens: (a) #Q7, (0001) faces; (b) #Q6, (11 $\bar{2}$1) faces; and (c) #Q3, (10
$\bar{2}$1) faces; and (c) #Q3, (10 $\bar{1}$0) faces. Indicated are positions of LA-ICP-MS transects (white arrows, Fig. 7), BSE imaging (white rectangles, Fig. 7), micro-IR transects and mapping (red dashed line and rectangles, Figs. 10–12), XRD (chips cut at dashed green triangles) and EMPA (blue dots).
$\bar{1}$0) faces. Indicated are positions of LA-ICP-MS transects (white arrows, Fig. 7), BSE imaging (white rectangles, Fig. 7), micro-IR transects and mapping (red dashed line and rectangles, Figs. 10–12), XRD (chips cut at dashed green triangles) and EMPA (blue dots).
Micro-scale compositional mapping and quantitative micro-analyses were undertaken with a JEOL JXA-8230 electron microprobe at the University of Colorado, USA (Department of Geological Sciences). It is a five-spectrometer instrument equipped with argon X-ray detectors (P-10 mixture) on spectrometer 1 and 2, and xenon X-ray detectors on spectrometers 3 to 5. Analytical conditions were 15 kV and 20 nA with a 5 μm beam diameter (samples XBD 19-01-B1 and 17-02-B2). Background positions were chosen to avoid interferences. In order to improve precision, a few additional analyses were performed at 50 nA and 10 μm beam diameter (samples #Q7, #Q6 and #Q3). The mean atomic number background correlation was used for all analyses (Donovan et al., Reference Donovan, Singer and Armstrong2016). Counting time was optimised to improve the detection limit. The ZAF matrix correction from Armstrong (Reference Armstrong and Newbury1988) using the FFAST mass absorption coefficient table (Chantler et al., Reference Chantler, Olsen, Dragoset, Chang, Kishore, Kotochigova and Zucker2005) was applied throughout. Typical detection limits are ~0.01 wt.% for main and minor elements.
Minor and trace elements were measured by LA-ICP-MS at the USGS Denver Federal Center, USA. Three beryl crystals with representative primary and secondary crystal phases based on the EMPA were selected. A Photon Machines Analyte G2 LA system (193 nm and 4 ns excimer) was coupled to a PerkinElmer DRC-e ICP-MS. Spot-lined analyses were carried out for known locations, crossing the primary and secondary phase zones. A wavelength of 193 nm was used. Ablation was carried out using an 80-μm square spot size at 12 J/cm2. Single spot analyses were ablated using 15 pulses/sec (15 Hz). Signals were calibrated using USGS synthetic basalt glass GSE-1g. Silicon (29Si) was used as the internal standard element (using an average Si wt.% content from EMPA data) for concentration calculations (Pauly, Reference Pauly2019).
For X-ray diffraction and structure refinement, fragments were cut from crystals #Q3, #Q6 and #Q7. Single-crystal X-ray diffraction data were acquired on a Brucker P4 four-circle diffractometer (University of Colorado, USA) equipped with a point detector, an APEX II CCD detector, and a rotating Mo-anode generator operating at 50 kV and 250 mA. Unit-cell parameters were refined using the point-detector. The program SHELXL-97 (Sheldrick, Reference Sheldrick2008) was used to analyse the site occupancies.
Micro-Raman spectroscopy on polished sections and oriented crystals was performed on a setup (XploRA Nano, Horiba Jobin Yvon) based on a confocal Raman microscope, using 532 nm excitation, a 50× objective (NA = 0.5 and focus size ≈1 μm diameter), with power on sample of ~3 mW. Spectra are acquired using a grating of 1800 grooves/mm providing for a spectral resolution of 0.5 cm–1, with a thermoelectrically cooled CCD camera. The phonon response of silicon at 520.7 cm–1 was used to calibrate the spectrometer.
Unpolarised micro-IR spectroscopy and imaging was performed in transmission, reflection and attenuated-total reflection mode (FTIR microscope LUMOS, Bruker Inc.). Background spectra were recorded before the sample measurements in air and subtracted to obtain absorbance spectra of the samples. Measurements were performed in transmission on the double-sided polished crystal slabs of 120 μm thickness or less, with 50 μm spatial resolution, and 2 cm–1 spectral resolution from 800 cm–1 to 6000 cm–1. Point spectra, line scans and images were acquired after EMPA and LA-ICP-MS and in areas of close proximity to correlate the CO2 and H2O IR spectra with the elemental crystal composition. For infrared peak frequencies and mode assignment see Table S2.
Results
Surface texture
Optical microscopy and AFM were used to study the beryl surface texture, notably of the (0001), (10 $\bar{1}$0) and (11
$\bar{1}$0) and (11 $\bar{2}$1) crystal faces. As established previously (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b), the (0001) face shows hexagonal protrusions representing screw dislocations (Fig. 4a, e). Most of the hexagonal screw dislocations in (0001) display finely spaced growth steps. The (10
$\bar{2}$1) crystal faces. As established previously (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b), the (0001) face shows hexagonal protrusions representing screw dislocations (Fig. 4a, e). Most of the hexagonal screw dislocations in (0001) display finely spaced growth steps. The (10 $\bar{1}$0) faces are dominated by left-handed or right-handed screw dislocations of nearly rhombohedral shape with long axis approximately parallel with respect to (0001) (Fig. 4b, f). The screw dislocations grow from the nucleation site in spirals outwards (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b). The lower density of the screw steps and kinks on (10
$\bar{1}$0) faces are dominated by left-handed or right-handed screw dislocations of nearly rhombohedral shape with long axis approximately parallel with respect to (0001) (Fig. 4b, f). The screw dislocations grow from the nucleation site in spirals outwards (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b). The lower density of the screw steps and kinks on (10 $\bar{1}$0) compared to (0001) indicates a more rapid crystal growth in the (10
$\bar{1}$0) compared to (0001) indicates a more rapid crystal growth in the (10 $\bar{1}$0) lattice plane compared to (0001) leading to the tabular crystal habit (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b). In some (11
$\bar{1}$0) lattice plane compared to (0001) leading to the tabular crystal habit (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b). In some (11 $\bar{2}$1) lattice planes of beryl (Fig. 4c, g), the stacking fault defects grow layer-by-layer, until they cover the entire crystal face. Eventually, a step source that no longer disappears is formed at the protrusion of the stacking fault. The stacking fault also acts as the nucleus of the contract twin (Fig. 4k) (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b). Occasionally, dispersed heart-shaped step bunching is observed. The dislocation step spirals outwards from the protrusion and forms a closed spiral (Fig. 4d, h). A detailed future study of defects, dislocations and other structural bulk and surface features may possibly relate these features to crystal morphology and growth environment (Sunagawa, Reference Sunagawa1984; Scandale et al., Reference Scandale, Lucchesi and Graziani1990; Scandale and Lucchesi, Reference Scandale and Lucchesi2000; Tempesta et al., Reference Tempesta, Scandale and Agrosì2011).
$\bar{2}$1) lattice planes of beryl (Fig. 4c, g), the stacking fault defects grow layer-by-layer, until they cover the entire crystal face. Eventually, a step source that no longer disappears is formed at the protrusion of the stacking fault. The stacking fault also acts as the nucleus of the contract twin (Fig. 4k) (Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b). Occasionally, dispersed heart-shaped step bunching is observed. The dislocation step spirals outwards from the protrusion and forms a closed spiral (Fig. 4d, h). A detailed future study of defects, dislocations and other structural bulk and surface features may possibly relate these features to crystal morphology and growth environment (Sunagawa, Reference Sunagawa1984; Scandale et al., Reference Scandale, Lucchesi and Graziani1990; Scandale and Lucchesi, Reference Scandale and Lucchesi2000; Tempesta et al., Reference Tempesta, Scandale and Agrosì2011).

Fig. 4. Surface texture of different crystal faces of Xuebaoding beryl. (a–d) Unpolarised optical microscope images of #Q7, #Q3, #Q6 and #Q5, respectively. (e-f) AFM images of areas indicated in (a–d) (red square). (i–l) Differential interference microscope images of similar defects on corresponding faces [modified after Qi et al., Reference Qi, Ye, Xiang, Pei and Luo2001a]. (a, e, i) are (0001) faces, (b, f, j) are (10 $\bar{1}$0) faces, and (c, d, g, h, k, l) are (11
$\bar{1}$0) faces, and (c, d, g, h, k, l) are (11 $\bar{2}$1) faces.
$\bar{2}$1) faces.
Chemical composition
Figure 5 shows optical and EMPA of the narrow vein (thin section 1702B2) containing scheelite at the schist contact and beryl in muscovite (Fig. 5a plain light, Fig. 5b UV with scheelite grain fluorescence). The beryl exhibits complex zoning as seen in the example of Fig. 5c (corresponding back-scattered electron (BSE) image Fig. 5d). Quantitative compositional element mapping of Al, Na, Fe and Cs (Fig. 5e–h, in oxide wt.%) indicates the negative correlation of Al and Fe, representing the substitution of Al3+ for M 2+/3+ cations, and the corresponding positive correlation of Al3+ with Na+ and Cs+ for charge balance.

Fig. 5. Narrow vein of muscovite in schist with scheelite and beryl (thin section 1702B2): (a) plane light; (b) UV light; (c) beryl under crossed polarised light (red area from a); (d) corresponding BSE image. Quantitative EMPA mapping (scale bar oxide wt.%) of Al2O3 (e), Na2O (f), FeO (g) and Cs2O (h) of yellow region indicated in (c) with zoning and coupled substitution. Data for EMP point analyses (open circles and squares in (f)) in Table 1. For BSE and EMPA data of second area (green rectangles in (a) and (b)) see Fig. 6.
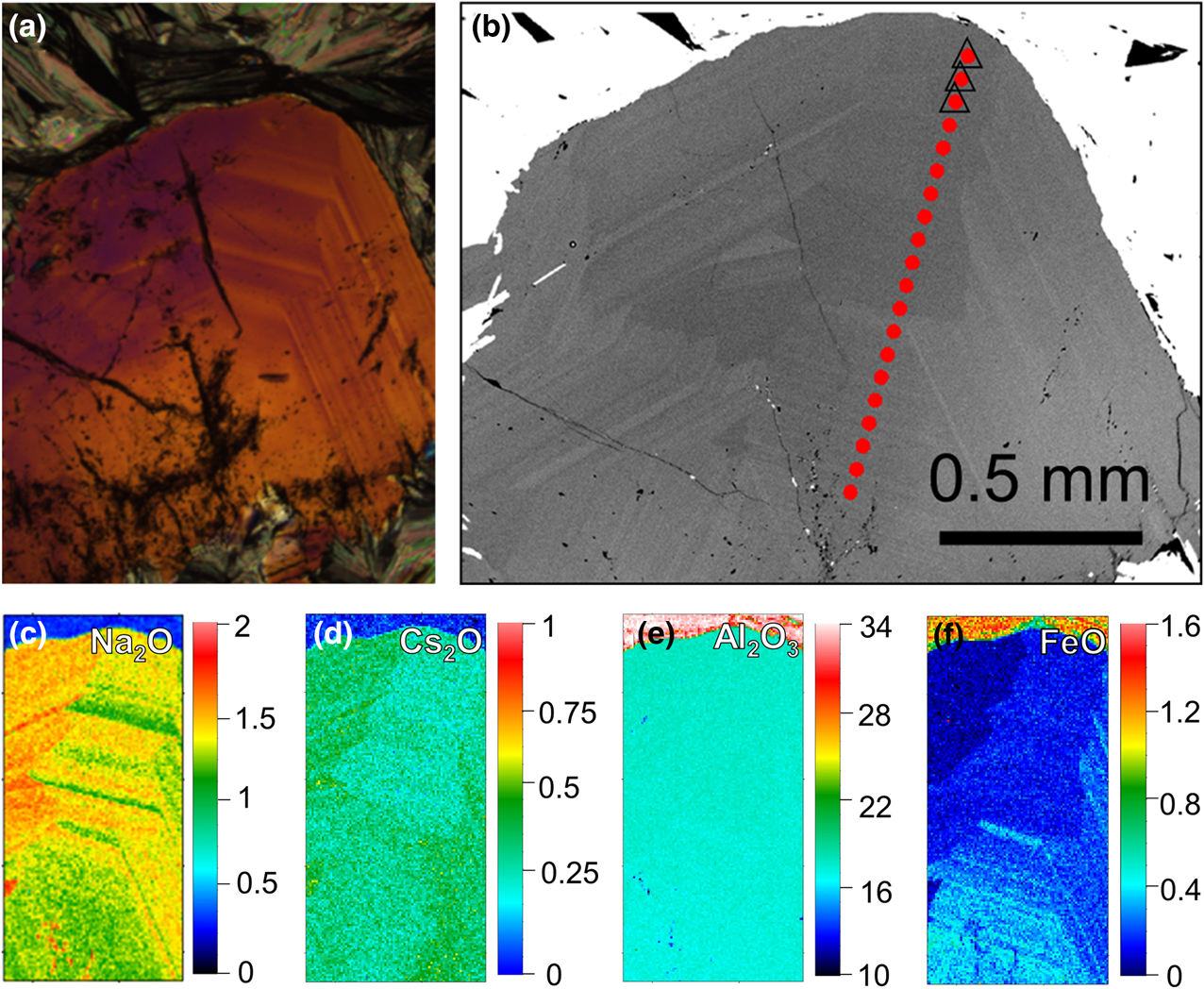
Fig. 6. Concentric zoning of beryl of thin section sample 1702B2 (green rectangle in Fig. 5a and 5b). (a) Optical microscope image under crossed polarisation; and (b) BSE image with EMPA transect (red dots), triangles indicate the selected EMPA points listed in Table 1. (c–f) Quantitative mapping of selected area for Na2O, Cs2O, Al2O3 and FeO, respectively (scale bar oxide wt.%).
Table 1. Quantitative elements content obtained by EMPA for thin sections (TS) and single crystals (SC)*
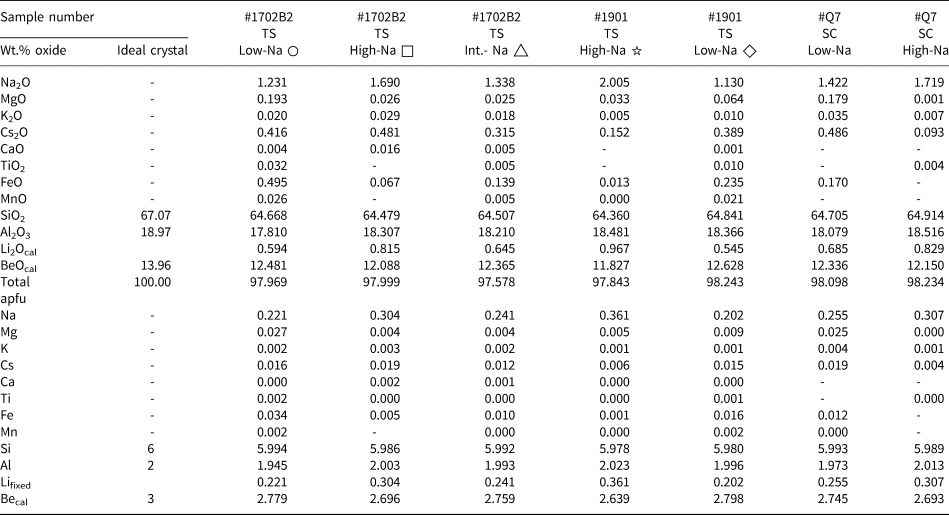
* Every value represents the average of three analyses. The measured points for ○ and □ are shown in Fig. 5, △ is shown in Fig. 6, ☆ and ◇ are shown in Fig. S2. –: close or below the detection limit.
Lifixed: Li set to the Na value based on correlation derived from LA-ICP-MS; ‘cal’: calculated value, Becal = 3 – Li.
Results of EMPA of seven representative zones averaged over three points of 1702B2 (for details see Fig. 5 and 6: low Na+, circles; high Na+, squares; intermediate Na+, triangle), as well as analyses of samples 1901B and #Q7 (see analysis points in Figs S2, S3) are summarised in Table 1 and cover the range of compositional variations observed. SiO2 values range from 64.22 to 65.04 wt.% (ideal 67.07 wt.%) and Al2O3 values range from 17.81 to 18.55 wt.% (ideal 18.97 wt.%). Of the minor elements, Na2O dominates, ranging from 1.23 wt.% up to 2.01 wt.% and Cs2O up to 0.49 wt.%. Iron as FeOT (total Fe reported as FeO) ranges up to 0.50 wt.%, and MgO up to 0.19 wt.%. Traces of K+, Ca2+, Ti4+ and Mn2+ are close to the detection limit of ~0.01 wt.%.
The EMPA calculations (Table 1) are based on 18 O apfu and 3 [Be + Li]. LA-ICP-MS revealed a close correlation between Li+ and Na+, allowing for the use of Na+ as a proxy for Li+ to a good approximation. Be2+ is thus assumed to be 3 – Li+. For comparison, calculations on the basis of either 18 O and 3 Be, or 6 Si are shown in Table S3 and S4, respectively (Groat et al., Reference Groat, Giuliani, Marshall and Turner2008; Fridrichová et al., Reference Fridrichová, Bačík, Bizovská, Libowitzky, Škoda, Uher, Ozdín and Števko2016; Lum et al., Reference Lum, Viljoen, Cairncross and Frei2016).
With Si4+ ranging from 5.978 to 5.994 apfu this indicates almost full 6 Si T1 site occupation without Si–Al substitution. Na+ ranges from 0.20 to 0.36 apfu, which is much higher than Mg2+ (up to 0.027), K+ (0.001–0.004), Cs+ (0.004–0.019), Ca2+ (<0.002), Ti4+ (<0.002), Fe3+ (up to 0.034) and Mn2+ (<0.002). Na+ typically occupies the channel site to balance the charge difference by coupled substitution.
Data from LA-ICP-MS was used to determine Li+ and other minor and trace elements. Representative results of LA-ICP-MS line scans in samples #Q7, #Q6 and #Q3 are shown in Figure 7, highlighting regions of primary concentric zoning dissected by secondary veinlets (#Q7, Fig. 7a), typical primary concentric zoning (#Q6, Fig. 7b), or irregular but zoned regions near a crystal surface (#Q3, Fig. 7c), with corresponding LA-ICP-MS transects (Fig. 7d–f) as indicated. A characteristic and correlated increase in Na+ and Li+ is observed in the secondary phases, with corresponding depletion in K+, Rb+, Cs+, Mg2+, Mn2+ and Fe3+. This effect is reflected in the BSE contrast in primary as well as secondary growth darker areas corresponding to greater light element content of Li+ and Na+, with depletion in the heavy elements.

Fig. 7. Representative BSE images of single crystal samples #Q7 (a), #Q6 (b) and #Q3 (c), with LA-ICP-MS transects (d–f, dashed lines in (a) and (c)): transect (d) location is ~0.3 mm just left of field of view in (a) at extension of meandering secondary feature. Transect (e) is not related to (b) with location indicated in Fig. 3b. Transect (f) shown in (c) as indicated, the full position is shown in inset of Fig. 10b. The LA-ICP-MS elemental pattern reflects primary and secondary beryl phases, with characteristic positive correlation of Na and Li, both negatively correlated with all other trace elements. Area in (c) (red dashed rectangle) indicates micro-IR mapping #Q3-2 as shown in Fig. 12.
Representative quantitative LA-ICP-MS results are listed in Table 2, with calculated apfu values based on assuming 6 Si. Consistent with EMPA results, Na+ ranges from 0.25 to 0.37 apfu, compared to minor Cs+ (0.0035–0.016), Mg2+ (0.000–0.017), Fe3+ (0.000–0.015) and only traces of other elements. In the primary zones, Na+ (0.246–0.268) is considerably less than in the secondary zones (0.354–0.371), yet correlated closely with the amount of Li+ in primary (0.254–0.272) and secondary beryl (0.347–0.368).
Table 2. Representative minor- and trace-element composition of beryl determined by LA-ICP-MS.*
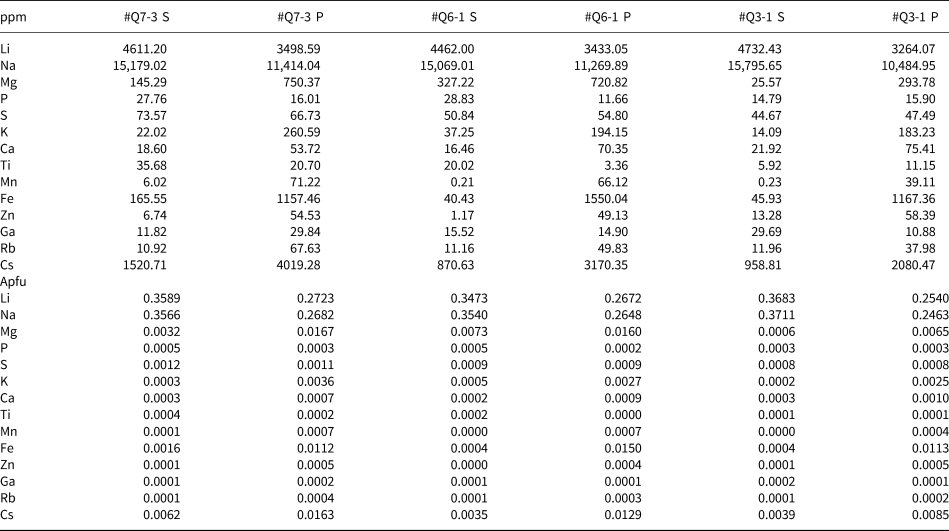
* S indicates the secondary phase of beryl while P indicates the primary phase.
In summary, these results show the presence of only a weak Al3+ A-site for 2+ cations substitution vector, but a dominating Li+ T2-site for Be2+ substitution vector (see discussion below).
Crystallography and structure refinement
The refined crystal structure parameters as well as data collection parameters, are summarised in Table 3. The unit-cell parameters were refined from centring angles of 60 strong reflections with two between 10 and 28° using a point detector system on a Bruker P4 diffractometer. The a-axis values of the lattice range from 9.2161(2) to 9.2171(4) Å, while c-axis values range from 9.2178(2) to 9.2219(4) Å, and the unit-cell volume ranges from 678.03(3) to 678.48(7) Å3. The c/a ratio ranges from 1.0002 to 1.0005 and is consistent with T2 type beryl, with the unit-cell controlled primarily by Be2+ substitution (Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988, Reference Aurisicchio, Grubessi and Zecchini1994). Intensity data were measured using a Bruker APEX II CCD detector out to a two-theta angle of ~80° (Table 3). Atom position, displacement parameters and cation occupancies were refined using SHELXL (Sheldrick, Reference Sheldrick2008) based on ionised atom scattering factors for cations (Cromer and Mann, Reference Cromer and Mann1968) and O–2 (Azavant and Lichanot, Reference Azavant and Lichanot1993). Atom position, displacement and occupancy parameters are given in Supplementary Table S5. The crystallographic information files have been deposited with the Principal Editor of Mineralogical Magazine and are available as Supplementary material (see below).
Table 3. Crystal structure refinement parameters for beryl from Xuebaoding (#Q7, #Q6, and #Q3) compared to reported data.
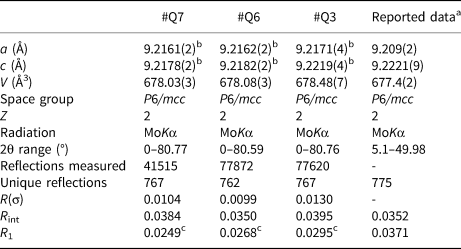
aGuo et al. (Reference Guo, Wang, Xu, Fontan, Luo and Cao2000).
bUnit cell refinement obtained from point detector data.
cUsing ionised atom scattering factors for cations (Cromer and Mann, Reference Cromer and Mann1968) and for O2– (Azavant and Lichanot, Reference Azavant and Lichanot1993).
Selected bond distances derived from refinements using ionised-atom scattering factors are shown in Table 4 (Guo et al., Reference Guo, Wang, Xu, Fontan, Luo and Cao2000a; Robinson et al., Reference Robinson, Gibbs and Ribbe1971) (further details and atomic position coordinates and anisotropic displacement parameters are listed in Table S5). Here, O1 indicates oxygen linked with Si, whereas O2 indicates the oxygen shared by Si, Al and Be. Si–O has the shortest (1.61 Å) while Al–O has the longest bond length (1.91 Å).
Table 4. Selected interatomic distances (Å) for beryl from Xubaoding in comparison to reported data.

aGuo et al. (Reference Guo, Wang, Xu, Fontan, Luo and Cao2000); b Robinson et al. (Reference Robinson, Gibbs and Ribbe1971)
The derived crystal structure is shown in Fig. 8a (projected onto (0001)) and Fig. 8b with its edge-linked distorted tetrahedrally coordinated T2 (Be) and octahedrally coordinated A (Al) sites. Crystal channels are characterised by the wide C1/2a site between the rings and narrow C2/2b site of the interior of the tetrahedral [Si6O18]12– ring with diameters of ~5.2 Å and ~2.6 Å, respectively. Figure 8c shows C2 site occupation with Na+ and associated type IId, IIs and I water configurations, with the distinct coordination of two water molecules to Na+ (type IId), one water molecule and optionally C1 site occupation with CO2 (type IIs), or the alkali cation free-water configuration (type I) (Fridrichová et al., Reference Fridrichová, Bačík, Bizovská, Libowitzky, Škoda, Uher, Ozdín and Števko2016).

Fig. 8. Crystal structure of beryl. (a) Along orientation parallel with respect to the c axis; (b) perpendicular with respect to the c axis, with octahedral A (Al), tetrahedral T2 (Be), Si, and two channel sites at the C1/2a (H2O, large alkali) and C2/2b (Na) positions; and (c) type IId, type IIs and type I H2O molecule configuration, with corresponding Na+ and CO2 positions on channel sites (after Fridrichová et al., Reference Fridrichová, Bačík, Bizovská, Libowitzky, Škoda, Uher, Ozdín and Števko2016).
Generally similar site occupancy values for the different lattice sites are found for all three crystals analysed. The A, T1 and T2 sites are almost fully occupied. The site population calculation is in good agreement with the EMPA and LA-ICP-MS data with a significant Li+ substitution for Be2+ (T2) and minor M 2+ substitution for Al3+ (A), charge balanced predominantly by Na+ as the only alkali occupying the small C2 site, and the larger alkali (K+, Cs+, Rb+) and Fe2+/3+ together with water and minor CO2 on the C1 site.
Raman spectroscopy
Representative polarisation-resolved Raman spectra, reflecting the different crystal orientations and type I and II water orientation, are shown in Fig. 9. Here, E inc and E R represent incident fundamental pump polarisation and Raman scattered polarisation, respectively. All spectra of different sample locations have negligible background indicating high structural integrity and overall low defect/inclusion density. The peaks below 1400 cm–1 are associated with beryl lattice phonon modes (For Raman peak frequencies and mode assignment see Table S6 (Kim et al., Reference Kim, Bell and McKeown1995; Qi et al., Reference Qi, Pei, Zhou, Shi and Luo2001b). The strong peaks at 685 cm–1 and 1066 cm–1 are due to Si–O vibrations. The modes at 322 cm–1 and 396 cm–1 are Al–O bands. The two bands at 524 cm–1 and 1009 cm–1 represent Be–O vibrations. In the water stretch region the two and only partially resolved peaks are due to type II (3594 cm–1) and type I (3604 cm–1) water.

Fig. 9. Polarised Raman spectra of beryl. (a,b) k ⊥ c axis and Einc // c axis and ER // c axis, (c,d) k ⊥ c axis and Einc // c axis and ER ⊥ c axis, (e,f) k ⊥ c axis and Einc ⊥ c axis and ER ⊥ c axis. k is the incident wave-vector, Einc and ER are the polarisation directions of the fundamental and Raman light, respectively. The crystallographic z axis is parallel to optical c axis. A1 g indicates the non-degenerate symmetric modes, E1 g / E2 g indicate the doubly degenerate symmetric modes. Peak assignments are listed in Table S6.
The non-degenerate symmetry modes (A1g) which have the strongest Raman response are shown in Fig. 9a, b. The doubly degenerate symmetry modes (E1g) are presented in Fig. 9c, d. The Raman emission of water, polarised perpendicular with respect to the c axis of beryl, is weak when the incident field is polarised parallel with respect to the c axis, as expected and shown in Fig. 9d. Doubly degenerate symmetric modes E2 g are presented in Fig. 9e, f, in which 398 cm–1 and 684 cm–1 are due to Si–O and Al–O vibrations, respectively, and with a corresponding water peak (Kim et al., Reference Kim, Bell and McKeown1995; Hagemann et al., Reference Hagemann, Lucken, Bill, Gysler-Sanz and Stalder1990). The combination of Fig. 9b and 9f confirm the expected molecular orientation of type I and type IIs water (Hagemann et al., Reference Hagemann, Lucken, Bill, Gysler-Sanz and Stalder1990). Type I is expected to be more polarisable along the c axis, and thus stronger in Fig. 9b, while type II is expected to be more polarisable perpendicular with respect to the c axis and thus stronger in the polarisation combination of Fig. 9f, as observed. Polarised Raman spectra with the incident wave vector perpendicular with respect to the (10 $\bar{1}$0) face and unpolarised Raman spectra are shown in Fig. S4.
$\bar{1}$0) face and unpolarised Raman spectra are shown in Fig. S4.
Micro-IR spectroscopy and mapping of water content
A representative IR point spectrum is shown in Fig. 10 from (a) transect #Q3-7 and (b) acquired parallel with respect to the LA-ICP-MS line on #Q3 in Fig. 7 (see also inset Fig. 10b). Infrared peak frequencies and mode assignments are listed in Table S2 (Charoy et al., Reference Charoy, De Donato, Barres and Pinto-Coelho1996; Qi et al., Reference Qi, Ye, Xiang, Pei and Shi2001d; Łodziński et al., Reference Łodziński, Sitarz, Stec, Kozanecki, Fojud and Jurga2005; Adamo et al., Reference Adamo, Pavese, Prosperi, Diella, Ajò, Gatta and Smith2008; Della Ventura et al., Reference Della Ventura, Radica, Bellatreccia, Freda and Cestelli Guidi2015; Mashkovtsev et al., Reference Mashkovtsev, Thomas, Fursenko, Zhukova, Uskov and Gorshunov2016). The spectrally narrow bending  ${\rm \lpar \nu }_ 2^{\rm I} {\rm \rpar \;}$and asymmetric stretch
${\rm \lpar \nu }_ 2^{\rm I} {\rm \rpar \;}$and asymmetric stretch  ${\rm \lpar \nu }_ 3^{\rm I} \rpar $ of type I (1600 and 3694 cm–1), bending
${\rm \lpar \nu }_ 3^{\rm I} \rpar $ of type I (1600 and 3694 cm–1), bending  ${\rm \lpar \nu }_ 2^{{\rm IIs}} \rpar $ and symmetric stretch
${\rm \lpar \nu }_ 2^{{\rm IIs}} \rpar $ and symmetric stretch  ${\rm \lpar \nu }_ 1^{{\rm IIs}} \rpar $ of type IIs water (1637 and 3588 cm–1), and asymmetric CO2 stretch (2358 cm–1) modes can be discerned. The broad peak at ~1950 cm–1 has been assigned to the overtone absorption of a lattice phonon mode (Charoy et al., Reference Charoy, De Donato, Barres and Pinto-Coelho1996). The detailed transects for the type I and II stretch modes in Fig. 10b show an inversion in relative type I and II water content at the transition from primary to secondary beryl. Further, the type II peak is split as seen in the magnified region of Fig. 10c, with the main peak
${\rm \lpar \nu }_ 1^{{\rm IIs}} \rpar $ of type IIs water (1637 and 3588 cm–1), and asymmetric CO2 stretch (2358 cm–1) modes can be discerned. The broad peak at ~1950 cm–1 has been assigned to the overtone absorption of a lattice phonon mode (Charoy et al., Reference Charoy, De Donato, Barres and Pinto-Coelho1996). The detailed transects for the type I and II stretch modes in Fig. 10b show an inversion in relative type I and II water content at the transition from primary to secondary beryl. Further, the type II peak is split as seen in the magnified region of Fig. 10c, with the main peak  ${\rm \nu }_ 1^{{\rm IIs}} $ at ~3588 cm–1 corresponding to type IIs, and the shoulder at ~3605 cm–1 corresponding probably to a combination of the symmetric stretches of type I
${\rm \nu }_ 1^{{\rm IIs}} $ at ~3588 cm–1 corresponding to type IIs, and the shoulder at ~3605 cm–1 corresponding probably to a combination of the symmetric stretches of type I  ${\rm \lpar \nu }_ 1^{\rm I} \rpar $ and type II water
${\rm \lpar \nu }_ 1^{\rm I} \rpar $ and type II water  ${\rm \lpar \nu }_ 1^{{\rm IId}} \rpar $. The minor peak at about 3651 cm–1 is due to the antisymmetric stretch of type IIs water
${\rm \lpar \nu }_ 1^{{\rm IId}} \rpar $. The minor peak at about 3651 cm–1 is due to the antisymmetric stretch of type IIs water  ${\rm \lpar \nu }_ 3^{{\rm IIs}} \rpar $.
${\rm \lpar \nu }_ 3^{{\rm IIs}} \rpar $.
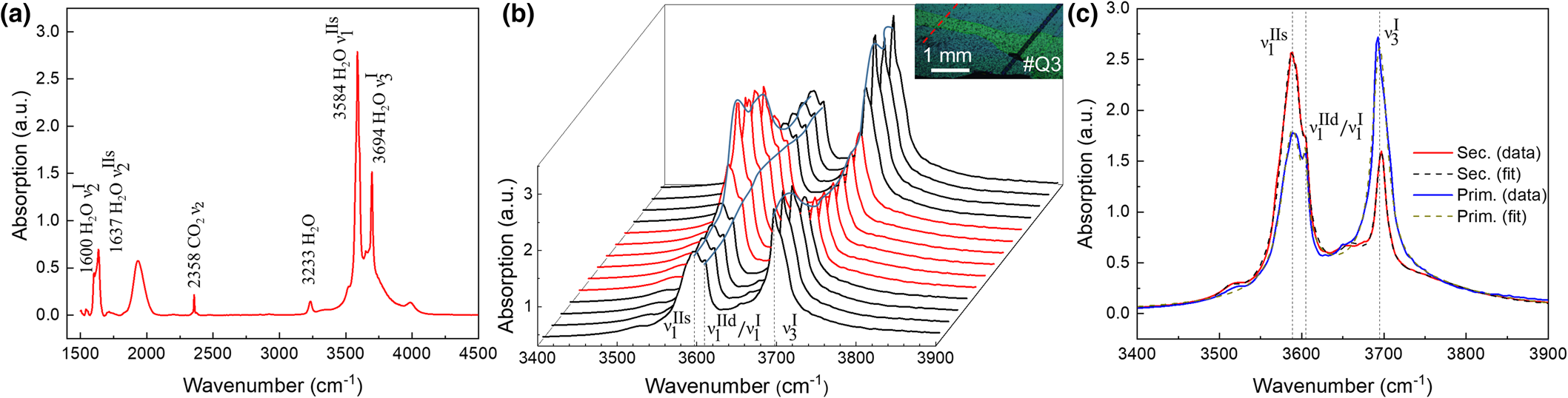
Fig. 10. IR point spectrum and micro-IR transect #Q3-7 (for location see Fig. 3c). (a) Peak assignment with type I water (1600 and 3694 cm–1), type II water (1637 and 3584 cm–1) and CO2 (2358 cm–1). (b) IR spectral transect with spectra centred on the water stretch region crossing secondary beryl zone (red) with primary beryl on either side (black) (inset: optical micrograph, with IR transect in red and LA-ICP-MS line in black). (c) Representative spectra from primary (blue) and secondary (red) zone with intense negative correlation of type I and type II water, with Lorentzian line fits (dashed) for type IIs (3588 cm–1), mixed type IId/I (3605 cm–1) and type I (3694 cm–1) (for details see also Fig. S5).
To determine relative peak intensities, the spectra have been fitted using simple Lorentzian lineshapes, with 5-6 peaks (dashed lines in Fig. 10c, see also supplement Fig. S5), with centre frequencies of  ${\rm \nu }_ 1^{{\rm IIs}} $ and
${\rm \nu }_ 1^{{\rm IIs}} $ and  ${\rm \nu }_ 3^{\rm I} $ of 3588 cm–1 and 3694 cm–1, respectively. The frequencies vary by less than ±1 cm–1 between primary and secondary crystal phases.
${\rm \nu }_ 3^{\rm I} $ of 3588 cm–1 and 3694 cm–1, respectively. The frequencies vary by less than ±1 cm–1 between primary and secondary crystal phases.
The spatial variation of the relative IR peak absorption for type I and IIs water and CO2 across the primary and secondary phase is shown in Fig. 11a. The increase in spectral intensity of  ${\rm \nu }_ 1^{{\rm IIs}} $ (orange) is observed in the secondary phase (1.0 to 1.7 mm), while
${\rm \nu }_ 1^{{\rm IIs}} $ (orange) is observed in the secondary phase (1.0 to 1.7 mm), while  ${\rm \nu }_ 3^{\rm I} $ (brown) decreases. The mixed shoulder at ~3605 cm–1 (black) correlates with the
${\rm \nu }_ 3^{\rm I} $ (brown) decreases. The mixed shoulder at ~3605 cm–1 (black) correlates with the  ${\rm \nu }_ 3^{\rm I} $ intensity, suggesting that it is associated primarily with
${\rm \nu }_ 3^{\rm I} $ intensity, suggesting that it is associated primarily with  ${\rm \nu }_ 1^{\rm I} $. Additionally, the CO2 stretch (purple) slightly decreases in the secondary phase.
${\rm \nu }_ 1^{\rm I} $. Additionally, the CO2 stretch (purple) slightly decreases in the secondary phase.

Fig. 11. IR and ICPMS transect analysis of #Q3-7 (see Fig. 3c and Fig. 7c). (a) IR-absorbance from Lorentzian peak fits showing qualitative correlated and anti-correlated variations of CO2, type I and IIs water across primary and secondary zones. (b) Quantitative comparison of type I and type IIs (type IId negligible) water (apfu) derived from absolute IR absorption cross sections, in relation to Na and Fe content from LA-ICP-MS.
The corresponding absolute amount of water can be estimated based on the Lambert–Beer law A = ɛcd, where A is the absorbance, ɛ the molar absorption coefficient (L/mol⋅cm), c the water concentration (mol/L), and d the sample thickness (cm) (Fukuda et al., Reference Fukuda, Shinoda, Nakashima, Miyoshi and Aikawa2009). In our case, the sample thickness of #Q3 is ~120 μm. We use reported molar absorption coefficients of type I (3696 cm–1) and type IIs (3588 cm–1) water modes of ɛI = 197 L/mol⋅cm (Charoy et al., Reference Charoy, De Donato, Barres and Pinto-Coelho1996), and ɛIIs = 256 L/mol⋅cm, respectively (Goldman et al., Reference Goldman, Rossman and Dollase1977). Though the value for ɛIIs is for cordierite, we expect it to equally apply for beryl to a good approximation given the similarity in water structure and index of refraction in the IR. Absorption peak intensity A for each mode is approximated by the ratio of fitted peak area and line width. Using a density value of beryl calculated from XRD of ρ = 2.75 g/cm3, with its variation with Na+ content negligible for our case, the amount of water can be obtained as H2Otot (wt.%) = c×M×10–1/ρ where M is the molar mass of water. An ideal and maximum 1 apfu water on C1 corresponds to 3.24 wt.% for an ideal beryl with M = 537.47 g/mol (Hawthorne and Černý, 1977; Pankrath and Langer, Reference Pankrath and Langer2002).
The resulting apfu values based on the Lambert–Beer law for  ${\rm \nu }_ 1^{{\rm IIs}} $ and
${\rm \nu }_ 1^{{\rm IIs}} $ and  ${\rm \nu }_ 3^{\rm I} $ from Fig. 11a and their sum are shown in Fig. 11b. Type IIs water (orange) increases from 0.20 apfu in the primary phase to 0.38 apfu in the secondary phase, correlates positively with Na+ (red), and correlates negatively with Fe3+ (green) based on the co-localised LA-ICP-MS transect. Meanwhile, type I water (brown) decreases from 0.21 apfu in the primary phase to 0.12 apfu in the secondary phase. It appears that the ~50% decrease of type I water in the secondary zone is more than compensated by a corresponding increase in type IIs water, with the total water content (
${\rm \nu }_ 3^{\rm I} $ from Fig. 11a and their sum are shown in Fig. 11b. Type IIs water (orange) increases from 0.20 apfu in the primary phase to 0.38 apfu in the secondary phase, correlates positively with Na+ (red), and correlates negatively with Fe3+ (green) based on the co-localised LA-ICP-MS transect. Meanwhile, type I water (brown) decreases from 0.21 apfu in the primary phase to 0.12 apfu in the secondary phase. It appears that the ~50% decrease of type I water in the secondary zone is more than compensated by a corresponding increase in type IIs water, with the total water content ( ${\rm \nu }_ 1^{{\rm IIs}} $+
${\rm \nu }_ 1^{{\rm IIs}} $+  ${\rm \nu }_ 3^{\rm I} $) (blue) increasing from 0.42 apfu in the primary phase to 0.50 apfu in secondary beryl.
${\rm \nu }_ 3^{\rm I} $) (blue) increasing from 0.42 apfu in the primary phase to 0.50 apfu in secondary beryl.
The amount of water of 0.42 apfu determined for the primary phase by IR absorption is in good agreement with the value of 0.40 apfu derived from XRD (Table 5). The XRD result further suggests that the C1 site is almost half occupied with a combination of the trace amounts of alkali K+, Cs+ and Rb+, together with water (see Table 5). The approximately equal total amount of water in primary and secondary beryl is interesting to note, with primarily a change in ratio of type I to type II water, which is controlled by the different Na+ content (occupying the C2 site).
Table 5. Site-scattering refinement with site scattering and site population assignment from single crystals #Q7, #Q6 and #Q3, compared to the results of EMPA and LA-ICP-MS analysis.

epfu – electrons per formula unit
H2O* is estimated by site scattering (epfu) except sum of K+, Cs+ and Rb+.
Corresponding spatial images are shown in Fig. 12 of type I water (a, d), type II water (b, e) and CO2 (c, f) peak integrated values for the two crystal regions #Q3-2 (a–c) and #Q3-9 (d–f) as indicated in Fig. 3c and acquired in proximity and an either side of the LA-ICP-MS trace (images of minor band centred at 3605 cm–1 is shown in Fig. S6 and full width at half maximum of type I in S7). Akin to the IR line trace analysis above, the same negative correlation of type I and type II water, and positive correlation of type I water with CO2 stand out.

Fig. 12. Micro-IR imaging on primary and secondary beryl of #Q3-2 (a–c) and #Q3-9 (d–f) (area indicated in Figs 3c, 7c), based on peak intensities of (a and d) type I water (3694 cm–1), (b and e) type IIs water (3584 cm–1) and (c and f) CO2 (2358 cm–1). Type I water is less (blue area in (a and d)), while type IIs water is more in the secondary phases (red area in (b and e)). CO2 (c and f) correlates with type I water.
Discussion
Coupled substitution
In the following we analyse the role of the two primary substitution vectors for the octahedrally coordinated A and tetrahedrally coordinated T2 site (Belov, Reference Belov1958; Wood and Nassau, Reference Wood and Nassau1968; Bakakin et al., Reference Bakakin, Rylov and Belov1969; Hawthorne and Černý, 1977; Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988; Sherriff et al., Reference Sherriff, Grundy, Hartman, Hawthorne and Černý1991) with
 $$3^{{\rm T}2}{\rm B}{\rm e}^{2 + } + \squ \;\to \lpar {3\ndash n} \rpar {\rm B}{\rm e}^{2 + } + n^{{\rm T}2}{\rm L}{\rm i}^ + { + } n^{\rm C}{\rm R}^ + $$
$$3^{{\rm T}2}{\rm B}{\rm e}^{2 + } + \squ \;\to \lpar {3\ndash n} \rpar {\rm B}{\rm e}^{2 + } + n^{{\rm T}2}{\rm L}{\rm i}^ + { + } n^{\rm C}{\rm R}^ + $$and
 $$2^{\rm A}{\rm A}{\rm l}^{3 + } + \squ \;\to \lpar 2\ndash k\rpar {\rm A}{\rm l}^{3 + } + g{\rm X}^{2 + } + \lpar {k\ndash g} \rpar {\rm Y}^{3 + } + g^{\rm C}{\rm R}^ + $$
$$2^{\rm A}{\rm A}{\rm l}^{3 + } + \squ \;\to \lpar 2\ndash k\rpar {\rm A}{\rm l}^{3 + } + g{\rm X}^{2 + } + \lpar {k\ndash g} \rpar {\rm Y}^{3 + } + g^{\rm C}{\rm R}^ + $$where R+ represent alkali ions, X2+ the divalent cations, Y3+ trivalent cations, with respective coefficients n, k, and g.
Summarising the data from all EMPA and LA-ICP-MS in Fig. 13 shows the corresponding correlations for A and T2 site substitution and indicates only a weak coupling between the two substitution vectors.
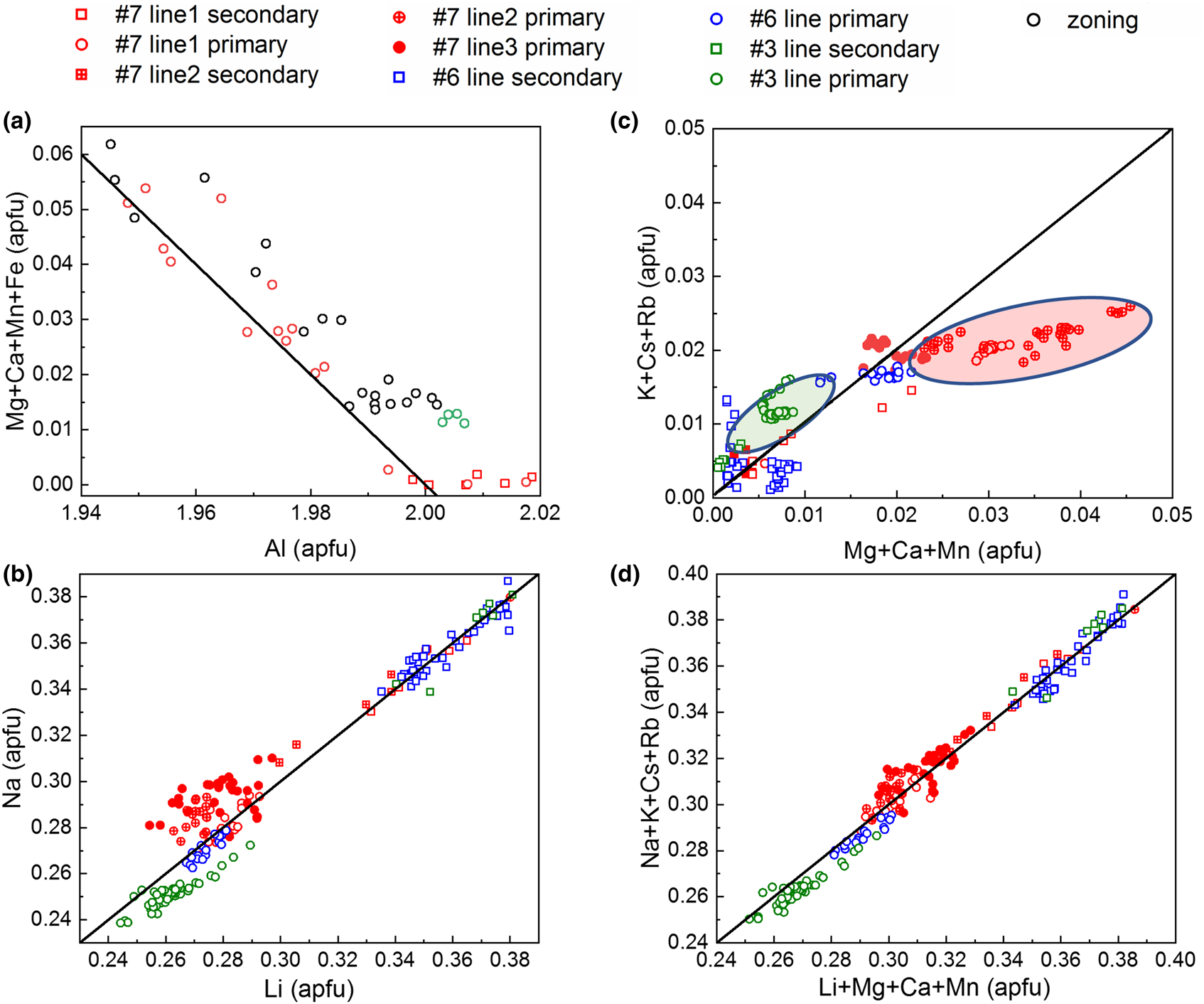
Fig. 13. Correlations and trends in major-element variations. (a) Linear correlation between A-site Al and corresponding X2+ (divalent ions at the A site) substituents. (b) Correlation between T2 Li+ substitution for Be2+ with Na+ charge balancing on the C2 site. (c) R+ – Na+ vs. X2+ with inversely correlated deviations of some data groups compared to (b), indicating weak coupling between the two substitution vectors. (d) Combined A and T2 substitution expressed as R+ (monovalent alkali ions in channel) vs. Li+ + X2+ – Fe3+ with linear correlation. (a) Based on microprobe data, (b–d) LA-ICP-MS data. Black lines represent the ideal substitution or charge balance relationship, Al3+ + X2+ = 2 (a); Li+ = Na+ (b); R+ – Na+ = X2+ (c) and R+ = Li+ + X2+ – Fe3+ (d).
Figure 13a shows the negative correlation of 2+/3+ cations with Al3+ content based on sets of EMPA values from different samples (#Q3, #Q6 and #Q7), in comparison with the theoretical behaviour based on equation 2 with slope –1. Here we assume that all Fe2+/Fe3+ ions occupy the A site. The small deviation of the data from the ideal behaviour is probably due to slightly too high Al EMPA values, as supported by XRD suggesting Al < 2 apfu, or that Fe is dominated by Fe3+ (see also below). Overall, the role of A-site substitution is small, with the sum of divalent trace elements ranging from as low as ~0.001 apfu to a maximum value not exceeding ~0.06 apfu.
Figure 13b shows the positive correlation of Na+ with Li+ closely following the ideal behaviour with slope 1. This shows that the substitution vector equation 1 is dominated by Na+ alone, which ranges from 91.5%–99.7% (apfu) of the sum of all alkali elements, and charge balancing for Li+ as it substitutes for Be2+. This suggests that the remaining alkalis K+, Rb+ and Cs+ are primarily charge balancing the Al3+ substitution for divalent cations at the A site. Note the bimodal distribution with lower Li+ and Na+ content in the primary, and higher Li+ and Na+ values in the secondary phases. Correspondingly this behaviour is to first order inversed for the other alkalis vs. X2+ cations (Fig. 13c). This suggests that in the secondary phase the role of the T2 substitution vector (equation 1) increases, whereas the A substitution vector (equation 2) diminishes. The deviations of certain data groups (green and red) in Fig. 13c with negative correlation in Fig. 13b suggests a weak coupling between the two substitution vectors through the alkali R+ mixing.
Combining both substitutions of equations 1 and 2 in Fig. 13d shows the sum of the vector Li+ for Be2+ at the T2 site, and the sum of all divalent cations to replace for Al3+ at the A site. The good agreement with the theoretical slope of 1 for all data, in contrast to (b) or (c) suggests that all iron at the A site is trivalent, i.e. not necessitating charge balancing with alkalis (Fe with other divalent cations vs. alkali elements see Fig. S8).
The combined results can then be expressed by the empirical formula:
 $$\eqalign{& ^{T2} \lpar {{\rm B}{\rm e}_{2.62\ndash 2.75}{\rm L}{\rm i}_{0.25\ndash 0.38}} \rpar _{\Sigma 3.00} \cr &{}^{A} \lsqb {{\rm A}{\rm l}_{1.94\ndash 2.00}{\lpar {{\rm Mg}\comma \;{\rm F}{\rm e}^{3 + }\comma \;{\rm Ca}\comma \;{\rm Mn}\comma \;{\rm Ti}} \rpar }_{ \lt 0.06}} \rsqb _{\Sigma 2.00} \cr & ^{\;\;\;T1} {\rm S}{\rm i}_6{\rm O}_{18}{}^{C2} \lsqb \lpar {{\rm N}{\rm a}_{0.25\ndash 0.36}{\lpar {{\rm K\comma \;Rb\comma \;Cs}} \rpar }_{ \lt 0.02}} \rsqb \cdot ^{C1}\!\!\lpar {\rm H}_2{\rm O}_{0.4\ndash 0.5}\lpar {{\rm C}{\rm O}_2} \rpar _{ \lt 0.01}} $$
$$\eqalign{& ^{T2} \lpar {{\rm B}{\rm e}_{2.62\ndash 2.75}{\rm L}{\rm i}_{0.25\ndash 0.38}} \rpar _{\Sigma 3.00} \cr &{}^{A} \lsqb {{\rm A}{\rm l}_{1.94\ndash 2.00}{\lpar {{\rm Mg}\comma \;{\rm F}{\rm e}^{3 + }\comma \;{\rm Ca}\comma \;{\rm Mn}\comma \;{\rm Ti}} \rpar }_{ \lt 0.06}} \rsqb _{\Sigma 2.00} \cr & ^{\;\;\;T1} {\rm S}{\rm i}_6{\rm O}_{18}{}^{C2} \lsqb \lpar {{\rm N}{\rm a}_{0.25\ndash 0.36}{\lpar {{\rm K\comma \;Rb\comma \;Cs}} \rpar }_{ \lt 0.02}} \rsqb \cdot ^{C1}\!\!\lpar {\rm H}_2{\rm O}_{0.4\ndash 0.5}\lpar {{\rm C}{\rm O}_2} \rpar _{ \lt 0.01}} $$In accordance with the International Mineralogical Association Commission on New Minerals, Nomenclature and Classification guidance (Bosi et al., Reference Bosi, Hatert, Hålenius, Pasero, Miyawaki and Mills2019), this formula leads to the end-member composition T2Be3 AAl2 T1Si6O18 (beryl). In summary, the sample studied can be classified as (Li, Na-rich) beryl.
Crystal structure
The Si value close to ideal 6 apfu by EMPA indicates a negligible Si–Al substitution. The tetrahedral volumes are 2.133, 2.133 and 2.136 Å3 for #Q7, #Q6 and #Q3 (Table 4), respectively, values typical for pure Si tetrahedral sites.
The distinct coordination of water with Na+ features a shorter bond length of H–O1 for type IIs compared to type IId (Fukuda and Shinoda, Reference Fukuda and Shinoda2008, Reference Fukuda and Shinoda2011). This results in a correspondingly larger bond strength giving rise to a higher frequency of the IR bending mode. Our type IIs bending mode value of 1637 cm–1 agrees with the range of theoretical peak positions of 1637–1639 cm–1 for type IIs water rather than 1624–1627 cm–1 for type IId water (Fridrichová et al., Reference Fridrichová, Bačík, Bizovská, Libowitzky, Škoda, Uher, Ozdín and Števko2016).
Theoretical Na–O bond lengths of type IIs and IId are 2.227 Å and 2.483 Å, and Na–O1 bond lengths of IIs and IId are 2.558 Å and 2.540 Å, respectively (Fridrichová et al., Reference Fridrichová, Bačík, Bizovská, Libowitzky, Škoda, Uher, Ozdín and Števko2016). The IIs values are in good agreement with our measured values of the average Na–O and Na–O1 band lengths of 2.305 Å and 2.556 Å, respectively. This indicates the Na bound water is primarily type IIs.
Water content
The water estimates based on the absolute IR absorbed intensity, consistent with the results from XRD informed by LA-ICP-MS, indicate that the water occupancy ranges from 0.4 apfu to 0.5 apfu in #Q7, which is close to half of the maximum water occupation with an approximate stoichiometry of Be3Al2Si6O18⋅0.5H2O. Similarly, from XRD, the water occupation is close to 0.5 apfu for #Q6 (0.46), yet slightly low for #Q7 (0.37) and #Q3 (0.40). The Na+ content (C2/2b site) controls the dipole orientation and types of water on the channel site in the centre of the Si rings (C1/2a site) with type II water correlated with Na+ content as expected.
Few other investigations have yet addressed the water content in Xuebaoding beryl. Only Guo et al. (Reference Guo, Wang and Xu2000b) and Lin et al. (Reference Lin, Cao, Liu, Li, Zhang and Ying2000) have estimated the water content using a wet chemical method and obtained values of 1.97 wt.% to 3.56 wt.% corresponding to apfu values ranging from 0.61 to 1.13, with the later value exceeding the maximum possible value of 1 apfu.
From other localities, water content in beryl was determined by wet-chemical analyses (Charoy et al., Reference Charoy, De Donato, Barres and Pinto-Coelho1996; Guo et al., Reference Guo, Wang and Xu2000b; Lin et al., Reference Lin, Cao, Liu, Li, Zhang and Ying2000), thermogravimetric analyses (Artioli et al., Reference Artioli, Rinaldi, Stahl and Zanazzi1993; Pankrath and Langer, Reference Pankrath and Langer2002; Gatta et al., Reference Gatta, Nestola, Bromiley and Mattauch2006; Fridrichová et al., Reference Fridrichová, Bačík, Bizovská, Libowitzky, Škoda, Uher, Ozdín and Števko2016), or based on the empirical equation H2O (in wt.%) = [0.84958×Na2O (in wt.%)] + 0.8373 (Giuliani et al., Reference Giuliani, France-Lanord, Zimmermann, Cheilletz, Arboleda, Charoy, Coget, Fontan and Giard1997; Groat et al., Reference Groat, Giuliani, Marshall and Turner2008; Fridrichová et al., Reference Fridrichová, Bačík, Bizovská, Libowitzky, Škoda, Uher, Ozdín and Števko2016), with values ranging from <1 wt.% to >3 wt.%. However, these methods configure sensitivity to the type of water, or may overestimate including fluid inclusions or water on grain boundaries. This highlights the value of quantitative micro-IR spectroscopy with spatial resolution and site specificity.
Comparison with previous Xuebaoding beryl studies
The results of EMPA and LA-ICP-MS are in agreement with some but not all prior studies of beryl from Xuebaoding which are summarised in Table S1 and S7. In general good agreement is found with Guo et al. (Reference Guo, Wang and Xu2000b), Qi et al. (Reference Qi, Pei, Zhou, Shi and Luo2001b) and Zhou et al. (Reference Zhou, Qi, Xiang, Feng, Wu and Hao2002). Yet results from Liu et al. (Reference Liu, Zhang and Yu2001b), republished in Liu et al. (Reference Liu, Deng, Cai, Zhou, Wang, Zhou, Gao, De xiu, Fu yu and Zhu2005) are inconsistent with ours, reporting high SiO2 (66.04–67.25 wt.%) and low Na2O (0.42–0.83 wt.%). A bulk wet-chemical analysis (Liu et al., Reference Liu, He, Zhang, Yu and Pan2001a; Qi et al., Reference Qi, Xiang, Liu, Pei and Luo2001c) republished in Liu et al. (Reference Liu, Deng, Cai, Zhou, Wang, Zhou, Gao, De xiu, Fu yu and Zhu2005) and again in Liu et al. (Reference Liu, Deng, Li and Shi2007) is in general agreement with our results, except for their third set of data with low Na2O (0.27 wt.%) and high FeOT (0.76 wt.%) (bolded data in Table S7). A later analysis by the same authors using LA-ICP-MS and XRF (Liu et al., Reference Liu, Deng, Shi and Sun2012) is again inconsistent with implausibly high Li+ (Li2O 2.60–3.04 wt.%) that cannot be charge balanced by the other alkali as quantified. A single wet-chemical analysis (Lin et al., Reference Lin, Cao, Liu, Li, Zhang and Ying2000) reports exceptionally high Fe (0.67 wt.%) and Ca (0.88 wt.%).
Our XRD unit-cell parameter derived data are in good agreement with an earlier analysis of Guo et al. (Reference Guo, Wang, Xu, Fontan, Luo and Cao2000a) with a = 9.209(2) Å and c = 9.2221(9) Å with c/a = 1.0001. However, our results are in disagreement with Liu et al. (Reference Liu, Deng, Li and Shi2007) that reports implausibly short a and c values based on small number of reflections and poor R value. Instead, our results are in better agreement with earlier (Liu et al., Reference Liu, Deng, Cai, Zhou, Wang, Zhou, Gao, De xiu, Fu yu and Zhu2005) and later work (Liu et al., Reference Liu, Deng, Shi and Sun2012) by the same authors.
Comparison with beryl of similar structure and chemistry from other localities
The Xuebaoding beryl is overall characterised by a simple chemistry with minor octahedral (A) site substitution with Fe3+ and Mg2+ and negligible Ti4+, Zn2+ or Ca2+ with charge balance through Cs+ and minor Rb+ and K+. Fe as Fe3+ is suggested to substitute for Al3+ on the A site (Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988; Neiva and Neiva, Reference Neiva and Neiva2005; Groat et al., Reference Groat, Giuliani, Marshall and Turner2008, Reference Groat, Rossman, Dyar, Turner, Piccoli, Schultz and Ottolini2010). The low Fe3+ content is consistent with only a light colour trend towards blue (aquamarine) (Mihalynuk and Lett, Reference Mihalynuk and Lett2003; Fridrichová et al., Reference Fridrichová, Bačík, Rusinová, Antal, Škoda, Bizovská and Miglierini2015; Lum et al., Reference Lum, Viljoen, Cairncross and Frei2016). Appreciable Li+ substitution for Be2+ on the tetrahedral (T2) site with Li+ up to 0.38 is on the high end compared to other localities worldwide (Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988) and charge balanced by Na+ incorporation on the channel site. This classifies the Xuebaoding beryl as ‘(Na, Li) beryl’ (Černý, Reference Černý1975) as typically associated with Li-rich pegmatites, and of the crystallographic ‘tetrahedral’ variety, i.e. the dominance of the T2 site Li substitution as opposed to A-site substitution (Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988).
Compared to beryl from other localities worldwide, the chemical composition and structure of beryl from Xuebaoding do not stand out in any particular way (Hawthorne and Černý, 1977; Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988, Reference Aurisicchio, Grubessi and Zecchini1994; Sherriff et al., Reference Sherriff, Grundy, Hartman, Hawthorne and Černý1991; Artioli et al., Reference Artioli, Rinaldi, Stahl and Zanazzi1993; Černý et al., Reference Černý, Anderson, Tomascak and Chapman2003; Groat et al., Reference Groat, Giuliani, Marshall and Turner2008; Lum et al., Reference Lum, Viljoen, Cairncross and Frei2016). The Si4+ content of most beryl is close to 6. Al3+ apfu values generally range from as low as 1.2 in few cases, to 2, with the majority of data above 1.5 apfu, compared to Xuebaoding beryl with Al3+ close to 2 apfu. Be2+ ranges from 2.5 to 3.0 apfu (Hawthorne and Černý, 1977; Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988, 1994; Sherriff et al., Reference Sherriff, Grundy, Hartman, Hawthorne and Černý1991; Artioli et al., Reference Artioli, Rinaldi, Stahl and Zanazzi1993; Černý et al., Reference Černý, Anderson, Tomascak and Chapman2003), similar to Xuebaoding beryl varying between 2.62 and 2.75 apfu. Be2+ substitution for Li+ and correlated with Na+ is observed frequently with Li+ ranging from 0 to 0.5 apfu (with Xuebaoding Li+ of 0.25 to 0.4 apfu in the middle of that range).
With site substitution controlling the unit-cell parameters (Pankrath and Langer, Reference Pankrath and Langer2002), the values for the a axis range from 9.202 to 9.228 Å and the c axis from 9.185 to 9.240 Å (Hawthorne and Černý, 1977; Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988; Sherriff et al., Reference Sherriff, Grundy, Hartman, Hawthorne and Černý1991; Artioli et al., Reference Artioli, Rinaldi, Stahl and Zanazzi1993; Černý et al., Reference Černý, Anderson, Tomascak and Chapman2003). Corresponding values for Xuebaoding beryl fall within the middle of that range. Notably similar in composition and unit-cell parameter to Xuebaoding beryl #Q7, is beryl from the Mount Bity region, Madagascar (pale pink sample #18, Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988), with Si4+ = 6 and Al3+ = 2; Li+ = 0.27 substituting Be2+ at the T2 site; and Na+ = 0.18 and Cs+ = 0.02 occupying the channel to balance the charge – resulting in a = 9.215 Å and c = 9.218 Å, closely resembling the Xuebaoding beryl values of a = 9.2161(2) Å and c = 9.2178(2) Å. The beryl from Pala, California, USA (pale pink sample #28, Aurisicchio et al., Reference Aurisicchio, Fioravanti, Grubessi and Zanazzi1988) with a = 9.213 Å and c = 9.212 Å, and with similar composition of Si4+ = 5.996, Al3+ = 2.004, but lower Li+ = 0.17 and Na+ = 0.14 matches Xuebaoding sample #Q7 closely.
Comparison with tabular beryl worldwide
A particular feature of Xuebaoding beryl is the tabular habit. Similar tabular habits are observed in ‘morganite’ (pink beryl) (Cook, Reference Cook2011) from San Diego County, California, USA, yet crystals exhibit dominance of A-site substitution (Mn2+) with charge balanced by Cs+ (on C1) and low water content. Clear beryl (goshinite) from lithium-rich microcline–albite pegmatites from Minas Gerais, Brazil (Cook, Reference Cook2011) sometimes occur as tabular crystals, with high Li+ and Na+ content and are water rich (Fukuda and Shinoda, Reference Fukuda and Shinoda2011). Similarly, tabular beryl from the Bikita rare-element granitic pegmatite, Zimbabwe, exhibit high Li+ content and T2-site substitution (Černý et al., 2003) with alkali charge balancing, and only minor A-site substitution. The beryl-group member pezzottaite with maximum lithium substitution on T2 (ideally Cs(Be2Li)Al2Si6O18, Yakubovich et al., Reference Yakubovich, Pekov, Steele, Massa and Chukanov2009) also exhibits a tabular habit. The crystal chemistry of the Xuebaoding beryl, while not deterministic with regards to the crystal habit, may still give some insight into the evolution of the ore-forming fluids (Černý, Reference Černý and Grew2002; Černý et al., Reference Černý, Anderson, Tomascak and Chapman2003; Uher et al., Reference Uher, Chudík, Bačík, Vaculovič and Galliová2010; Sardi and Heimann, Reference Sardi and Heimann2014). The alkali and ferromagnesian content can reflect the evolution and composition of the host fluid (Černý, Reference Černý1975).
Summary and implications
Overall, the Xuebaoding beryl exhibit very similar tabular habit throughout the deposit, only rarely columnar, or etched. This suggests generally similar and stable conditions during the crystallisation of the veins. The low Fe3+ and Mg2+ content is consistent with the low degree of Fe3+ mineralisation in general (minor pyrite). The secondary phases in crystal sections or metasomatic replacement with decrease in Fe3+ and Mg2+, yet increase in Na+, indicate a further evolution and fractionation of the fluid in its late stage. This would support the hypothesis of early crystallisation of aquamarine with goshenite being late. These observations relate to the suggestion that goshenite is associated with highly evolved fluids, e.g. in pegmatites (Černý et al., Reference Černý, Anderson, Tomascak and Chapman2003; Neiva and Neiva, Reference Neiva and Neiva2005; Wang et al., Reference Wang, Cheng Che, Dong Zhang, Lan Zhang and Cheng Zhang2009) and with the limited Fe3+ and A-site substitution not being of magmatic origin. These results are further consistent with the understanding that the Xuebaoding W–Sn–Be–F–P mineralisation is primarily hydrothermal in origin and derived from a highly evolved fluid emerging from the Pankou and Pukouling albite leucogranites that are highly fractionated, alkali, peraluminous, Li–F-enriched (Zhu et al., Reference Zhu, Raschke and Liu2020) giving rise to the beryl, cassiterite, scheelite, fluorite and apatite mineral assemblage.
The tabular habit is therefore unlikely to be related to chemical composition and alkali content. However, the factors that control crystal habit in this and other localities remain unknown. More plausible than a chemical effect is the role of pressure and temperature, giving rise to anisotropy in lattice compressibility and thermal expansion. That may control, not only the size of lattice sites affecting substitution, but also the free energy of and preferential growth along particular crystallographic directions. Understanding the relationship of crystal habit to chemical, pressure and temperature conditions of the source fluid would be a subject of considerable general importance.
Acknowledgements
PW and MBR thank Chen Meng, Chen Yangang, Yin Xianyong, Wu Shunjin and Zeng Taiyun from the local community enabling field work and providing sample material. We thank Zhong Qian, Lai Meng, and Xu Xing for help with the micro-IR measurement. We acknowledge Mark Jacobsen, Bert Ottens, Alfred Schreilechner and Philip Persson for valuable discussions. We are indebted to the reviewers Junichi Fukuda and Gioacchino Tempesta for valuable input. In addition, we thank miners, mountain guides and area residents for their hospitality, dedication and invaluable support.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2021.13

























 Open access
Open access