


1. Introduction
Double-stranded (ds) DNA can adopt multiple conformations, exhibiting ‘polymorphism’, directly related to the physical properties of the molecule and to its biological function. The most well-known forms of dsDNA are the B-, A- and Z-forms. The B-form is the ‘normal’ DNA found in most biological aqueous contexts. Under reduced water conditions the A-form is favoured, and under certain ionic and base sequence conditions the inverted Z-form prevails. Biological roles of both A- and Z-DNA are possible (Arnott et al. Reference Arnott, Chandrasekaran, Millane and Park1986; Brown et al. Reference Brown, Lowenhaupt, Wilbert, Hanlon and Rich2000; Lu et al. Reference Lu, Shakked and Olson2000; Rich et al. Reference Rich, Nordheim and Wang1984). Compared with many other polymers, DNA has a spectacular compactness and high stiffness (‘persistence length’) and with a high negative charge density owing to the presence of close phosphate groups in the backbone. The high persistence length is in part due to the electrostatic repulsion of these moieties, partly to the local steric interactions of the coin pile-stacked nucleobases. The great stability of B-form DNA, needed to securely store the genomic information, is due to mainly the stacking of the bases, which in turn is due to hydrophobic effects (requiring surrounding bulk water) and to anisotropic dispersive forces (Friedman & Honig, Reference Friedman and Honig1995). The base-pair hydrogen bonds, which are important for many biological processes, play only a partial role in the stability of DNA (Guckian et al. Reference Guckian, Krugh and Kool2000) but are strengthened by the hydrophobic environment in the core of the close-packed bases (see below). DNA can be extended, twisted or unwound during diverse biological processes, such as DNA repair, chromatin compaction and gene regulation. Proteins that interact with DNA are able to exploit the unique structural properties of DNA, although it is not clear to what extent the polymorphic phase map is fully exploited. Taking the particular case of homologous recombination, the recombinase proteins RecA and Rad51 organize themselves in a helical manner around DNA, which is unwound and stretched to about 50% in length compared with the ds B-form (dsDNA). RecA and Rad51 play crucial roles in chaperoning homologous recombination and DNA repair by catalyzing strand exchange in, respectively, prokaryotes and eukaryotes. They use similar molecular reaction mechanisms for the strand exchange reaction and bind first to a single-stranded (ss) part of DNA with high cooperativity, in the presence of ATP, to form a filamentous complex in which DNA adopts an extended conformation (Flory et al. Reference Flory, Tsang and Muniyappa1984). The ssDNA–RecA filament then interacts with an incoming dsDNA to form an ssDNA–RecA–dsDNA complex (Kiianitsa & Stasiak, Reference Kiianitsa and Stasiak1997) and, if the two DNAs have the same or similar sequence, strand exchange occurs.
Twenty-five years ago, we studied shear-aligned fibres of Escherischia coli RecA complexes with both ss as well as dsDNA in aqueous solution by small-angle neutron scattering (SANS) and UV linear dichroism (LD) spectroscopy under identical flow conditions (Nordén et al. Reference Nordén, Elvingson, Kubista, Sjöberg, Ryberg, Ryberg, Mortensen and Takahashi1992). The orientation created a well-resolved SANS pattern, where the helical diffraction cross provided exact information about the helix pitch and, most importantly, yielded the flow orientation distribution, making it possible to translate the LD signals into average base-plane orientations (Hagmar et al. Reference Hagmar, Norden, Baty, Chartier, Takahashi, Nordén, Baty, Chartier and Takahashi1992; Nordén et al. Reference Nordén, Elvingson, Kubista, Sjöberg, Ryberg, Ryberg, Mortensen and Takahashi1992). Electron microscopy had revealed that the DNA was extended by approximately a factor of 1·5 (Stasiak & Di Capua, Reference Stasiak and Di Capua1982; Stasiak et al. Reference Stasiak, Di Capua and Koller1981), but surprisingly the base orientation concluded from LD did not show any inclination of the base planes (neither for ss- nor for dsDNA) as would be expected for a continuously stretched and unwound DNA form (Hagmar et al. Reference Hagmar, Norden, Baty, Chartier, Takahashi, Nordén, Baty, Chartier and Takahashi1992; Nordén et al. Reference Nordén, Elvingson, Kubista, Sjöberg, Ryberg, Ryberg, Mortensen and Takahashi1992, Reference Nordén, Wittung-Stafshede, Ellouze, Kim, Mortensen and Takahashi1998). This observation remained puzzling until 2002, when the application of systematically mutated aromatic residues in RecA allowed a three-dimensional model to be constructed for the aqueous solution structures of RecA-dsDNA and RecA-ssDNA (Morimatsu et al. Reference Morimatsu, Takahashi and Nordén2002). From LD results combined with crystal data for RecA (Story et al. Reference Story, Weber and Steitz1992), a model emerged in which the DNA was accommodated in an ordered way inside a helical arrangement of RecA monomers allowing the bases to be perpendicular (Morimatsu et al. Reference Morimatsu, Takahashi and Nordén2002). A later crystal structure of RecA–dsDNA and RecA–ssDNA confirmed our conclusion: a near perpendicular nucleobase orientation and clustering of triplets of bases stacked approximately as in B-form DNA (Chen et al. Reference Chen, Yang and Pavletich2008) (Fig. 1). Using LD spectroscopy together with site-specific tyrosine mutations, we found a similar nucleobase organization, perpendicular to the fibre axis of the complex, for the dsDNA complex with Rad51 in solution (Reymer et al. Reference Reymer, Frykholm, Morimatsu, Takahashi and Nordén2009). For the Rad51 complex with ssDNA, however, a perpendicular base orientation was observed only in the presence of activating factors, such as Ca2+ ion or Swi5/Sfr1 protein (Fornander et al. Reference Fornander, Renodon-Corniére, Kuwabara, Ito, Tsutsui, Shimizu, Iwasaki, Nordén and Takahashi2014). Recent cryo-EM high-resolution structural analyses of activated human Rad51 in complex with DNA have demonstrated conserved features with the RecA system (Xu et al. Reference Xu, Zhao, Xu, Zhao, Sung and Wang2017).

Fig. 1. The DNA structure inside a RecA-dsDNA filament according to crystal structure (left) (Chen et al. Reference Chen, Yang and Pavletich2008) compared with free-solution double-stranded B-form DNA (right).
In 2012, we identified using single-molecule force spectroscopy on short synthetic DNAs, in the absence of recombination proteins, the existence of an overstretched state of DNA, which we shall here return to and study in detail. All evidence indicates it is a thermodynamically well-defined conformation. We will call it Σ-DNA, and it consists of a ca 50% extended stable conformation of ds (base-paired) GC-rich DNA, at a transition force of ca 64 pN applied to the 3′−3′ strands (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012). The Σ-form may be considered a special case of the wider group of stretched DNA forms that have been called ‘S-DNA’ some of which, though, appear less well defined. In contrast to the Σ-form observed for base-paired GC-rich DNA stretched along the 3′–3′ ends, S-DNA is usually observed as a 70% elongated form when stretching a long plasmid or phage DNA (Cluzel et al. Reference Cluzel, Lebrun, Heller, Lavery, Viovy, Chatenay and Caron1996; Williams et al. Reference Williams, Rouzina and Bloomfield2002) and, at least in AT-rich DNA, S-DNA appears to involve denaturation (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012, Reference Bosaeus, El-Sagheer, Brown, Åkerman and Nordén2014). The fact that the Σ-form has so long escaped discovery is thought to be due to that it is first recently stretch experiments on short synthetic DNA have been possible with high accuracy (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012, Reference Bosaeus, El-Sagheer, Brown, Åkerman and Nordén2014).
The degree of extension that we observe for Σ-DNA, compared with B-DNA, is within experimental error the same as that found in complexes with the recombinase enzymes RecA and Rad51. One may thus ask if this is just a coincidence or if this structural distortion is somehow related to biological function. Here we further analyze previous data and perform some new strategic single-molecule and computational experiments in order to assess the possibility that Σ-DNA may have fundamental roles in biology.
Our hypothesis is that Σ-DNA is an inhomogeneous structure consisting of stacked triplets of nucleobases, with base planes arranged perpendicularly as in B-DNA, and that these triplets are separated by empty gaps. Such a triplet structure may have a biological role in enhancing the recognition of complementary base sequence and promote the strand exchange process in gene recombination. We further speculate that the base triplets separated by gaps may be a physical origin of the occurrence of three letters in the genetic code.
2. Materials and methods
See online SI (Sections 1–3) for details of methods, materials and calculations.
3. Results
Mixed-sequence long plasmid or phage dsDNA when pulled mechanically undergoes an elongation by about 70% at 60–70 pN (Cluzel et al. Reference Cluzel, Lebrun, Heller, Lavery, Viovy, Chatenay and Caron1996; Williams et al. Reference Williams, Rouzina and Bloomfield2002), partly involving fuzzy denaturation. This is also what we observed with short synthetic AT-rich DNAs (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012, Reference Bosaeus, El-Sagheer, Brown, Åkerman and Nordén2014). In contrast, we found more GC-rich DNAs to populate a well-defined conformation with a contour length ca 50% longer than B-form DNA (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012, Reference Bosaeus, El-Sagheer, Brown, Åkerman and Nordén2014), i.e. nearly the same degree of extension that DNA has in its complex with RecA (Fig. 1).
Here, we further analyze our earlier results (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012) and perform additional experiments on GC-rich DNA sequences (see online SI Section 1 ‘DNA oligomers sequences’), in order to better characterize the transition to the Σ-DNA conformation and its structure. Critical values of force and extension at the B→Σ transition point, (B→S for 5′-5′ and 5′-3′) together with free energy changes, are collected in Table 1. Figure 2 shows a typical example of response when force is applied to the 3′-ends of a double helix, resulting in an overstretched base-paired DNA conformation, as previously reported (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012). By probing the response when the stretching force instead is applied to the 5′-ends, or to both ends of one strand (3′-to-5′), we aimed to gain further insight about the conformational flexibility of DNA. However, we discovered that the 5′–5′ and 5′–3′ stretch transitions in our short synthetic DNA appear much less distinct and, furthermore, occur at a significantly higher transition force than the 3′–3′ case (Table 1). Although RecA and Rad51 may use intrastrand interactions to stretch DNA (Xu et al. Reference Xu, Zhao, Xu, Zhao, Sung and Wang2017), we therefore here confine ourselves to the 3′–3′ case, which is associated with the distinct Σ conformation in the absence of recombinase enzymes, and analyze this closer.
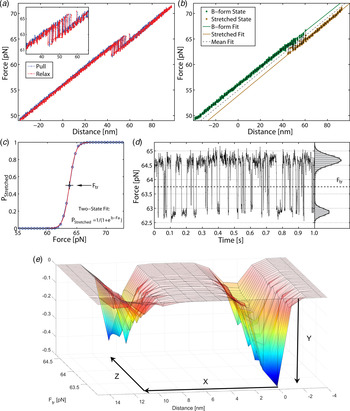
Fig. 2. Stretching a single 60 bp GC molecule (a) force-trap position during pull (blue) and relaxation (red). DNA duplex exhibits bistability at 61–65 pN, with no detectable hysteresis between pull and relax. (b) Force position during single pulling, illustrating two states. (c) Probability of finding DNA in overstretched state as a function of force. Data (circles) pooled from 16 pull-and-relax cycles on one molecule. (d) Time trace of force in overstretch region. Histogram (right) gives distribution during time interval of 1 s (a–d reproduced with permission from (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012)). At bottom (e) an energy landscape is generated based on the experimental data: y = free energy stabilization in B-form ca 1·5 kcal mol−1 base pairs, x = stretch displacement 11·0 nm, z = force ca 64 pN. Elongation: from 21·4 nm (B-DNA) to 32·4 nm (Σ-DNA), i.e. elongation factor 1·51 ± 0·05. The corresponding average elongation with double DNA (122 base pairs) was 1·54.
Table 1. The table lists the force response at room temperature in 1 M NaCl buffer of different DNA constructs based on a double stranded region containing 60% GC (see online SI Section 1 for sequences)
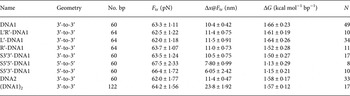
Geometry – positions of the single-stranded DNA handles transmitting the force. F tr, equilibrium transition force; Δx@F tr, molecular extension during transition measured at the transition force; ΔG, free energy difference between states; N, number of molecules in the set.
As shown in Fig. 2, stretching a single 60 bp GC-rich (60% GC) molecule can be well described statistically in terms of a two-state thermodynamic model. As shown by the force-trap position during pull (blue) and relaxation (red), the DNA duplex exhibits bistability at 61–65 pN, with no detectable hysteresis between pull and relax. Data (circles) pooled from 16 pull-and-relax cycles on one molecule (DNA1; for sequence, see online SI Section 1) and time trace of force in overstretch region show a distribution during time intervals of 1 s. A different sequence but with the same total GC content (DNA2) shows within experimental error identical results indicating that the transition to Σ-form is not markedly sequence dependent.
Measurements displaying the force versus distance spectrum contain thermodynamic and kinetic information. One observable is the transition frequency when the force is tuned to the point of symmetric switching between the two conformations, corresponding to equilibrium constant K = 1. Our observation that the frequency is relatively low (typically 100 Hz for 60 bp DNA) indicates that it is not a single-base destacking scenario (expected to be ns) that we are observing but an avalanche involving a large number of bases turning coherently from one structure to another (coherence length estimated to be as large as 50 bp, see below). This is a large cooperativity, part of which may be understood in classical mechanical terms.
Figure 3 shows the force spectrum for a 122 bp DNA concatamer consisting of two identical 60 bp GC-rich sequences connected covalently in series ((DNA1)2; for sequence, see online SI Section 1). The result can be understood in mechanical terms: once one of the two segments yields to force, then the slack will imply decreased force on the second half of the molecule. The transition force for the 122 bp DNA is similar to the force observed in Fig. 2 for one 60 bp DNA (64·2 pN compared with 63·3 pN).
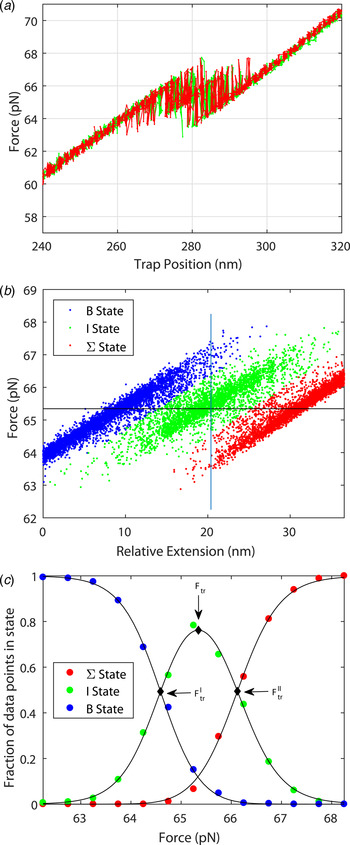
Fig. 3. Stretching a double 60 bp GC-rich DNA molecule ((DNA1)2, 122 bp, for sequence see online SI Section 1). (a) Multiple pull/relax cycles on a dimer in 1 M NaCl. The results show that the DNA-dimer overstretches at about the same force as the DNA monomer and that the extension is twice that of the monomer. F = 64·2 ± 1·56 pN, Δx = 23·8 ± 1·92 nm (L'DNA1: 62·0 + 1·18 pN, 11·5 ± 0·91 nm). (b) For analysis purposes, each measured set of data points during the pull/relax cycles of the molecule is assigned to one of three states based on the proximity to linear fits describing each state. (c) Fractional occupancy of the states as a function of applied force; solid lines show the fitted non-degenerate three-state model (see online SI Section 2 ‘Fitting a three state model’).
The system may be described in terms of three non-degenerate states as shown in Fig. 3b and 3c . From the data in Fig. 3c , one may deduce the cooperative length in the same way as was performed for the result in Fig. 2 (see online SI Section 2): the cooperative length as measured is 8 nm for the first transition and 12 nm for the second transition corresponding to an average extension of 23·8 nm or a cooperative length of 41 base pairs (to be compared with 50 bp for the 60 bp DNA molecule). The exactness by which the system can be described by three thermodynamic states is remarkable (Fig. 3c ). The total stretching of the DNA dimer corresponds to an extension ratio of 1·54 ± 0·04.
Figure 4 shows the two models of stretched DNA that we consider to be feasible, given the deformation and energetics observed: the continuous one with strongly slanted bases (to the right, called Σ′) and the inhomogeneous model with stacked base triplets (Σ, in the middle). In the SI part of (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012) a simple energy estimate was made to compare orthogonal and slanted base arrangements, which we here expand upon. Denoting L the length of DNA helix, N the number of bases, l G the gap size and l B the base rise, r B the base length, and n the number of clustered bases in the inhomogeneous models and A and E the stacking surface and energy, respectivly, one has for the case of slanted bases (right, θ base tilt angle): L = l B N cosθ and E(tilted) = AN[1 – (2 l B tanθ)/r B]. With 50% lengthening L = 1·5 l B N gives θ = 48°. Stacking energy for θ = 48° tilt and base length r B = 10 Å and width l B = 3·4 Å: E T = 0·25 AN. Correspondingly, for the orthogonal, inhomogeneous model (middle) assuming l B = 3·4 Å, one has for 50% lengthening l G = 5 Å with n = 3. Stacking energy E (orthogonal) = 0·50 A for n = 2 and 0·66 A for n = 3.

Fig. 4. Two energetically plausible stretched Σ-DNA structures compared with B-DNA (left): inhomogeneous disproportionation triplet form with perpendicular bases (middle, θ = 75–90°), homogeneous form with inclined bases (right, θ = 48°).
With this crude estimate, we conclude that the inhomogeneous perpendicularly stacked DNA model with base triplets is the most stable one provided that we may assume (know) that the bases are perpendicular. A question is how much more stable is the triplet compared with quadruplet and doublet arrangements? We discard the n = 4 case because of too high backbone strain (estimated to be >5 kcal mole−1 quadruple base pair), while doublets are still conceivable: using the stacking free energy determined by Kool et al. from a single-stacked base, the energy penalties for 50% stretched perpendicular base arrangements are: 9, 6 and 3 kcal per base pair, respectively, for homogeneous perpendicular, doublet and triplet arrangements compared with stacked B-DNA. Thus, the triplet is the energetically most economical conformation. The triplet state may also be concluded to be the energetically optimal cutoff for the many-body van der Waals interactions arising between DNA bases (Distasio et al. Reference Distasio, von Lilienfeld and Tkatchenko2012). We have then not considered stabilization due to entropic contributions which Harris et al. have found is important (Harris et al. Reference Harris, Sands and Laughton2005; Řezáč et al. Reference Řezáč, Hobza and Harris2010; Sutthibutpong et al. Reference Sutthibutpong, Matek, Benham, Slade, Noy, Laughton, Doye, Louis, Harris, JP, Louis and Harris2016). Note that both experiments and steered molecular dynamics (MD) simulations suggest that S-DNA is a strongly extended structure with slanted bases (average θ < 50°) prone to form large denaturation bubbles – in agreement with our observations of the behaviour of AT-rich DNA (where covalent glyoxal probing confirms denaturation) (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012, Reference Bosaeus, El-Sagheer, Brown, Åkerman and Nordén2014). This variation emphasizes the difference between earlier S-forms and our new Σ-DNA conformation. However, accurate quantification of various energy terms in DNA base pairing is still a challenge. Even experiments addressing DNA base stacking have large variations: for instance, values for AA stacking can be between −1·2 kcal mol−1 (Mitchell & Sigel, Reference Mitchell and Sigel1978) and −5·73 kcal mol−1 (Morcillo et al. Reference Morcillo, Gallego and Peral1987), depending on the applied conditions. To address energy components, base stacking was studied earlier both by MD simulations as well as quantum mechanical (QM) calculations (Friedman & Honig, Reference Friedman and Honig1995; Giudice et al. Reference Giudice, Várnai and Lavery2003; Kabeláč et al. Reference Kabeláč, Zendlová, Řeha and Hobza2005; Řezáč & Hobza, Reference Řezáč and Hobza2007; Swart et al. Reference Swart, van der Wijst, Fonseca Guerra and Bickelhaupt2007). As summarized in online Table S1, hydrogen bonds formed between the base pairs have a large contribution to base stability, however, accompanied with a large hydration-free energy per base, which may contribute to destabilization of Watson–Crick (WC) hydrogen bonds. Entropy terms based on QM normal mode analysis are also significant, further decreasing the large stabilization by hydrogen bonds. The non-polar contribution from base stacking was estimated to be similar by several techniques, in the range of ≃5–9 kcal mol−1 (Friedman & Honig, Reference Friedman and Honig1995; Kabeláč et al. Reference Kabeláč, Zendlová, Řeha and Hobza2005). Due to difficulties to obtain remaining entropy and hydration terms as well as non-additive parts of stacking contributions, accurate energetic details remain obscure.
From a more general perspective, the energetic problem is challenged due to the non-symmetric nature of AT and GC base pairs. In online Table S2, we report data for calculations of stacking stabilization using QM computations for a planar benzoic-acid hydrogen-bonded dimer, which we view as a primitive symmetric model for a DNA base pair. We have also considered effects of surrounding environment on hydrogen bond strength, an aspect that previous gas phase QM calculations on DNA base pairs did not address. Our results confirm that the hydrogen bonds are significantly strengthened by a non-polar matrix. We investigated typical pairwise interactions identified between DNA base pairs (Kabeláč et al. Reference Kabeláč, Zendlová, Řeha and Hobza2005), i.e. WC-type H-bonded (WC), intramolecular stacking (S), and interstrand stacking (IS) (online Fig. S1). The WC H-bonds are partially destabilized and 3·0 kcal mol−1 weaker in water than in an apolar, e.g. cyclohexane, environment (online Table S2). In contrast, for intrastrand stacking, S, the stacking of base pairs is more favoured in water by 1·2 kcal mol−1, while interstrand stacking, IS, is nearly identical in aqueous and apolar matrices (ΔΔE = −0·12 kcal mol−1). These results are in line with the increased strength of buried H-bonds (Deechongkit et al. Reference Deechongkit, Nguyen, Powers, Dawson, Gruebele and Kelly2004) and also known from the experimental appearance of strong hydrogen-bonded dimers in polyethylene compared with water solution (Norden, Reference Norden1977).
Further strengthening of base-pair stacking is due to contribution from dispersive forces as shown by π-stacking of two benzoic-acid dimers (online Fig. S1). Similar conclusions are found in the literature (Da̧bkowska et al. Reference Da̧bkowska, Gonzalez, Jurečka and Hobza2005; Friedman & Honig, Reference Friedman and Honig1995). The magnitude and nature of the individual interactions are similar to previous high-level QM calculations performed on DNA base pairs (Kabeláč et al. Reference Kabeláč, Zendlová, Řeha and Hobza2005; Swart et al. Reference Swart, van der Wijst, Fonseca Guerra and Bickelhaupt2007) and also on benzene dimers (Swart et al. Reference Swart, van der Wijst, Fonseca Guerra and Bickelhaupt2007). In the latter study, benzene dimers were also considered and coupled cluster with single double and perturbative triple excitation [CCSD(T)] and Keal & Tozer functional (KT1) calculations resulted in stacking energies close to those determined here for the benzoic acid dimers (for more details, see online SI Section 3 ‘Calculations of DNA stacking energies’). A great advantage of the benzoic acid model is its symmetric nature, where we may also gain insight into relative energy gains of adding an additional stacked dimer into the construct without considering the base-pair variance arising from the asymmetry of the four bases plus their sequential ordering. The obtained energy values for adding on dimers demonstrate, similar to a many-body van der Waals stacking interaction (Distasio et al. Reference Distasio, von Lilienfeld and Tkatchenko2012), that the relative change in energy gain per benzoic acid dimers becomes significantly reduced after the third base pair is added (online Fig. S2). The energy gain upon adding a third benzoic acid pair to a dimer of pairs is nearly double that for adding a fourth pair to a trimer of pairs or a fifth pair to tetramer of benzoic acid pairs (for details, see online SI Section 3).
Regarding the disproportionation mechanism, it appears that the relatively small force (60–70 pN) will allow various stacked regions of an extended DNA, to compete with each other. Then mainly stochastic effects will determine which stacked conformation will break up or lose one stacked pair from the end of a B-DNA fragment. Considering a hypothetical-stacked benzoic acid filament, when the pulling force makes a triplet and a quadruplet compete, then changing to a doublet from a triplet would mean a loss of 2·95 kcal mol−1 stabilization energy, compared with changing from a quadruplet to triplet, which costs a mere 1·45 kcal mol−1. This suggests that in case of DNA, the smaller force applied during pulling will probably render all longer stacked regions to break up, except for those that are in triplet form, where the split into a doublet will be too energy demanding. Again, this supports the formation of Σ-DNA, with triplets, to occur.
The transition force we obtained for Σ conformation (64 pN) corresponds to a free-energy change of ca 1·5 kcal per mol base pair, which compares fairly well with the experimental end-stacking energy of 2–6 kcal mol−1 for an AT base pair (Guckian et al. Reference Guckian, Krugh and Kool2000; Kool, Reference Kool2001). Note that our energy diagram (Fig. 2) does not include any activation barrier for the transition from B to Σ forms. An accurate determination of the activation energy will require precise kinetic data at varied temperatures.
4. Discussion
Laser-tweezers force-spectroscopic data (Bosaeus et al. Reference Bosaeus, El-Sagheer, Brown, Smith, Akerman, Bustamante and Norden2012, Reference Bosaeus, El-Sagheer, Brown, Åkerman and Nordén2014) on short DNA of variable sequence are here used as basis for discussing the structural and energetic properties of Σ-DNA and to put forward a disproportionation hypothesis.
The study of DNA-stretching mechanisms could give us several important clues about DNA physical properties in general and for the homology search of genetic recombination in particular. It is yet to be determined which conformation of pure stretched DNA is more stable: an inhomogeneous conformation (Σ) with perpendicular bases, or a homogeneously stacked structure with slanted bases (Σ′). Our energetic arguments are in favour of the inhomogeneous (disproportionated triplet gap) Σ-form, but this rests on the ad hoc assumption of perpendicular bases for which there is as yet no structural evidence, except for in DNA complexes with RecA (Fig. 1). Nonetheless, the observations of an energetically feasible transition force (consistent with base-pair triplet stacks) as well as the same extension length as observed for DNA in complex with RecA (Fig. 1) strongly argue that the Σ-form of DNA is a metastable structure that can form when alone free in solution.
Because the conformation of stretched DNA may be identical to that promoted by RecA, we would like to speculate about the possible role of DNA stretching and triplet arrangement in RecA-promoted genetic recombination. As suggested by Taghavi and Berryman and supported by their detailed modelling (under consideration in QRB Discovery), the presence of some intercalating cationic ligand could provide an energetic bias favouring perpendicularity and the triplet kind of disproportionation in DNA. In our LD-based model for the Rad51-dsDNA structure, we suggested one tyrosine to be intercalated as its orientation would be consistent with such a structure (Reymer et al. Reference Reymer, Frykholm, Morimatsu, Takahashi and Nordén2009), and this is clearly an attractive mechanism to explain the triple base stack. However, full intercalation might not be a necessary condition for disproportionation to occur, as MD simulations of Rad51-DNA complexes have shown that although a positive-charge moiety (tyrosine) is positioned close to the gap between the two base triplets in the starting structure, the evolving structure stays essentially stable (over 10 ns) and the tyrosine does not enter the potential intercalation slot (A. Reymer, unpublished data). For both RecA and Rad51, the presence of ATP (or non-hydrolyzable ATPγS) is needed for the formation of the extended recombinase–DNA complexes, both for ssDNA as well as for dsDNA, although with ssDNA a different, unstructured complex can form in absence of ATP (Williams & Spengler, Reference Williams and Spengler1986). Although ATP is a putative candidate for intercalation, both LD and fluorescence data (for etheno-adenine) indicated that the adenine plane of ATP was not intercalated (Takahashi & Nordén, Reference Takahashi and Nordén1994) and this was also verified in the later X-ray structure (Chen et al. Reference Chen, Yang and Pavletich2008).
We propose that base triplets might serve as recognition elements in the search for homology step of RecA and that physical properties of nucleic acids favouring triplet grouping relate to the triplets of codon/anticodon RNA. It has been proposed, based on dual-molecule manipulation studies, that the distance between the DNA-binding sites governs the fidelity of homology recognition (De Vlaminck et al. Reference De Vlaminck, van Loenhout, Zweifel, den Blanken, Hooning, Hage, Kerssemakers and Dekker2012). An extended, yet locally dsDNA conformation seems to match the way both ssDNA and dsDNA are distorted and line up in parallel inside the RecA filament. The base-pair matching in the case of a stacked triplet package can be anticipated to be more critical and exact than if the bases were moving freely. This is an alignment effect like for the sections of a key which are rigidly oriented perpendicular to the shaft of the key to fit critically in the lock. The unstacking of B form into Σ form will provide some destabilization, but this may be balanced by this local orientational recognition stability. Furthermore, the newly formed duplex DNA inside the RecA tube, after strand exchange, will then also be in Σ form. This means in turn that it is less stable and presumably more sensitive to sequence heterogeneity (mismatch), which may permit quick discrimination and, in absence of acceptable complementarity, promote back-reaction and dissociation. In accord, for homologous DNA, the complex of dsDNA and RecA/ssDNA has a half-life of seconds (Xiao et al. Reference Xiao, Lee and Singleton2006) and for non-homologous DNA even shorter. Molecular modelling suggests that B-DNA is deformed towards a curved form with kinks appearing during homology search by RecA filament (Saladin et al. Reference Saladin, Amourda, Poulain, Férey, Baaden, Zacharias, Delalande, Prévost, Nicolas and Pr2010). Distortion of dsDNA has been observed also in joint complexes of RAD51/ssDNA by scanning force microscopy (Ristic et al. Reference Ristic, Kanaar and Wyman2011).
Our new experiments show that application of force to opposite strands (3′–3′ or 5′–5′) of dsDNA differs from when the force is applied to the same strand (3′–5′) (see Table 1). Quite surprisingly, the 3′–3′ case also differs significantly from 5′–5′, implying that the structure feels the diastereomeric effect of the ribophosphate chirality and the helical winding of the double helix. Such results (also observed by others (Danilowicz et al. Reference Danilowicz, Limouse, Hatch, Conover, Coljee, Kleckner and Prentiss2009)) provide information about interactions of the distorted nucleic acid conformations, and may hint about a broken symmetry, so that the first and third base of a triplet stack would differ from each other. Note that the observation of insignificant differences in transition properties between 5 mM and 1 M NaCl is in agreement with the concept of highly stable stacked triplets surrounded by a high concentration of counterions regardless of added NaCl. The diastereomeric effect should be further analyzed but is beyond the scope of this study.
The high quality of the force-distance data and their nearly perfect quantitative fit to the two-state thermodynamic model for the 60-mer DNA are remarkable observations that demonstrate both accuracy of the experimental setting as well as that the conformational transition is well defined. For example, if the transition path had involved significant amounts of long-lived bulges or other trapped conformational species, they would have been revealed as deviations in the oscillatory read-out (Fig. 2d ). The good fit for the double sequence is also remarkable (Fig. 3), populating the three combinatorial thermodynamic states almost as quantitatively predicted. The high cooperative length (50 bp) for the 60 bp dsDNA is another indication of a well-defined global conformation without any significant, sequence-dependent local conformations. The slightly shorter cooperative length for the double-sequence DNA (41 bp) might reflect a kinetic imperfectness in the way thermal fluctuations populate the three combinatorial states, or is an entropic effect.
No connection between triplet base conformation and origin of the genetic code has been proposed before, as far as we know, while RNA templating of amino acids has been speculated as a possible code origin in an RNA-world context (Koonin & Novozhilov, Reference Koonin and Novozhilov2009; Yarus et al. Reference Yarus, Chen, Yarus and Harris2010). In support of our hypothesis of stacked base triplets as intrinsic recognition elements based on physical properties, we note that base-stacking arrangements also appear in mRNA/ribosome/tRNA complexes (Selmer et al. Reference Selmer, Dunham, Murphy, Weixlbaumer, Petry, Kelley, Weir and Ramakrishnan2006). Along the line of our speculation about a physical (conformational) origin of the genetic code, together with our observation of a broken symmetry for single-molecule 3′–3′ and 5′–5′ stretch experiments, the polarity of triplet base-pair stacks could be discussed. Already Crick postulated the ‘Wobble’ hypothesis to account for the fact that most organisms have <64 (often fewer than 45) species of tRNA: the 5′ base on the anticodon (which binds to the 3′ base of mRNA) is not as spatially confined as the other two bases (Crick, Reference Crick1966) As an example, yeast tRNA(Phe) has the anticodon 5′-GmAA-3′ and can recognize the codons 5′-UUC-3′ and 5′-UUU-3′. The two-out-of-three hypothesis of Lagerkvist (Lagerkvist, Reference Lagerkvist1978) explains the synonyms and degeneracy of the code in terms of structural (possibly chemical) ambiguity of the third base.
The origin of the genetic code is part of the question of origin of life (Lagerkvist, Reference Lagerkvist1978) and, of course, then any progress could be disruptively important. However, here we confine ourselves to the role of base stacking, which is the most important stabilizing energy term in DNA double-helix motifs (Kool, Reference Kool2001), and if recognition of triplet base aggregates by hydrogen bonding and steric effects may be responsible for high-fidelity self-recognition mechanisms, in RecA and generally. Returning to the question how RecA exploits the triplet DNA base stack, there are no indications of any preferred sequence of three bases, but the triplets are stochastically composed along the genetic sequence in complex with RecA. RecA will guarantee sequence neutrality by not interacting significantly with the nucleobases but binding only to the phosphate backbone. Still the stacking will contribute to stability, so that any mechanism testing homology will benefit from the freedom of dealing with each triple-base stack as a discrete unit, and at the same time have decreased base-pairing stability.
In conclusion, despite the importance of recombinases in many contexts (e.g. human diseases and treatments) and many years’ intense research, their mechanistic functions in performing search for homology and executing strand exchange are not yet understood at an atomic level. Thus, while nucleobase recognition, in replication and translation contexts, has been studied extensively and is mechanistically relatively well understood, much remains to unveil about recombination reactions in general and the role of extension of DNA in particular. Here the knowledge from single-molecule force spectroscopy could add importantly to our mechanistic understanding. Thermodynamic studies confirm that with both DNA and RNA, the stabilizing free energy is dominated by hydrophobic and dispersive nucleobase–stack interactions, while base-pair hydrogen bonding, electrostatic and stereochemical effects, crucial for recognition fidelity, are less prominent or even repulsive (Kool, Reference Kool2001). Our results show that the Σ conformation in GC-rich DNA is characterized by a clear two-state transition with a substantial cooperative length, emphasizing a model where a disproportionated triplet base structure may have a leading role in recombinase function and possibly also in other nucleic acid recognition contexts.
5. Speculation box
We speculate that the base triplets constituting the genetic code may have their origin in the physical preference of triplet base stacking in stretched nucleic acids, represented by the Σ conformation of DNA. Such a metastable but distinct conformation may improve discrimination between match and mismatch base pairing in homology search reactions, present already in the earliest organisms.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0033583517000099.
Acknowledgements
The authors acknowledge the Swedish Research Council (AR, BN and PW-S) and the Wallenberg foundation (PW-S) for financial support. The support for TB-S by the Hungarian Momentum programme (LP2016-2), by the National Competitiveness and Excellence Program (NVKP_16-1-2016-0007) and by the BIONANO_GINOP-2·3·2-15-2016-00017 project is greatly appreciated.
Conflict of interest
None.








 Open access
Open access