Introduction
Multiple sclerosis (MS) is a chronic demyelinating disease of the central nervous system (CNS) characterized by inflammation, demyelination, and neurodegeneration.Reference Compston and Coles 1 , Reference Frohman, Racke and Raine 2 The disease is heterogeneous in its clinical manifestation and progression. The key pathological mechanism involves infiltration of autoreactive immune cells into the CNS where, with varying degrees of severity, they cause demyelination, gliosis, neuronal loss, and eventually cerebral atrophy.Reference Compston and Coles 1 - Reference Broux, Gowing and Prat 4
Since the publication of the Canadian MS Working Group Updated Recommendations regarding treatment optimization in MS,Reference Freedman, Selchen and Arnold 5 several new therapies have become available while others are imminent. The advent of new therapeutic options has created a need for guidance on treatment sequencing, including algorithms to assist clinicians when deciding on therapeutic approaches for a particular patient. The selected approach should provide optimal disease management while not limiting future therapeutic options based on safety concerns. It should also recognize potential future treatments and the possibility of combinations.
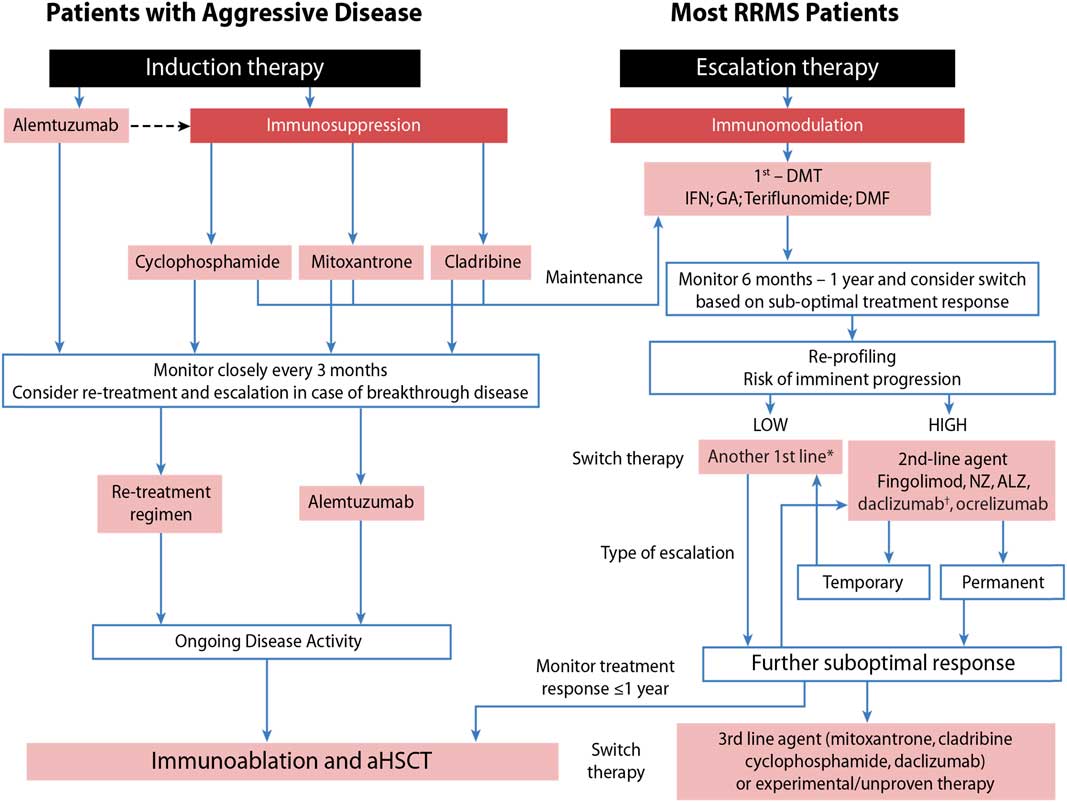
Taking this into consideration, we developed an approach that assesses patients’ risk for imminent progression (low vs. high) and then evaluates their response to treatment (Figure 1) as outlined by Freedman et al.Reference Freedman, Selchen and Arnold 5
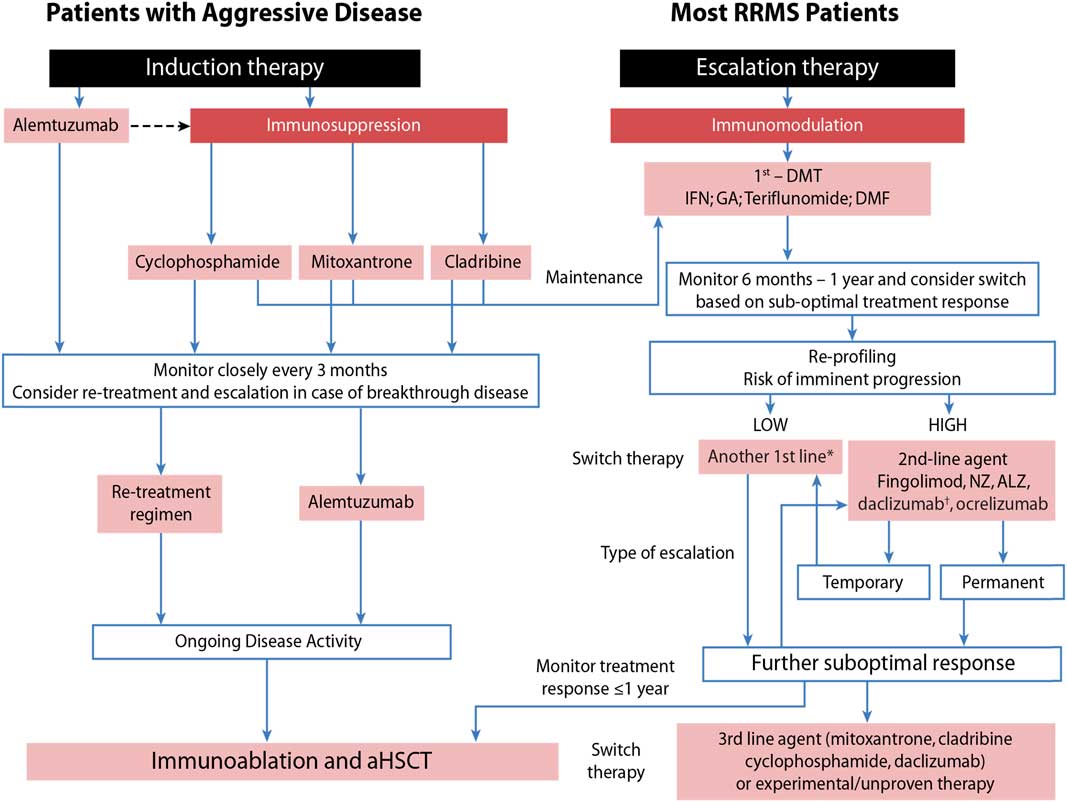
Figure 1 Sequencing algorithm. No monitoring and laboratory tests at baseline is requested when initiating or switching to glatiramer acetate (GA) and only thyroid testing, complete blood count and liver function test are necessary when starting or lateral switching patients to interferons (IFNs). *Tolerability is often a reason for a lateral switch. †Daclizumab was voluntarily removed from the market March 2, 2018 due to safety concerns. 102 aHSCT = autologous hematopoietic stem-cell transplantation; ALZ=alemtuzumab; DMF=dimethyl fumarate; DMT=disease-modifying therapies; MS=multiple sclerosis; NZ=natalizumab; RRMS=relapsing remitting multiple sclerosis. Adapted with permission from Freedman et alReference Freedman, Selchen and Arnold 5 and Rush et al.Reference Rush, MacLean and Freedman 9
Treatment Initiation
The clinical course of MS is highly variable, ranging from patients with relatively mild disease even years after the diagnosis to those with an early aggressive course and rapid accumulation of disability. Before initiating MS therapy, clinicians must consider the nature of the disease and long-term treatment plan, which is likely to be influenced by patient- and disease-related characteristics. Depending on the anticipated course of the disease, two different therapeutic approaches can be considered for treatment initiation: induction and escalation. In addition, as the treatment course may involve more than one agent, the optimal initial treatment should be one that does not limit subsequent therapeutic options.
Aggressive MS, affecting about 10% of MS patients, can be defined as disease leading to disability within 5 years from symptom onset and rapid transition to secondary progressive MS.Reference Gholipour, Healy, Baruch, Weiner and Chitnis 6 , Reference Menon, Shirani and Zhao 7 As early aggressive disease activity drives long-term disability, it is extremely important that it be identified quickly and treated effectively (Table 1).Reference Freedman and Rush 8 , Reference Rush, MacLean and Freedman 9 Treatment of aggressive MS requires an aggressive approach (Figure 1) usually using agents more capable of eliminating disease-causing cells (immunosuppressive therapy) instead of those that only alter their functioning (immunomodulation) or prevent them from entering the brain without altering either their function or their numbers (anti-trafficking agents).
Table 1 Clinical and radiological factors suggestive of aggressive multiple sclerosis (MS)Reference Freedman and Rush 8 , Reference Rush, MacLean and Freedman 9

EDSS=Expanded Disability Status Scale.
Adapted with permission from Rush et al.Reference Rush, MacLean and Freedman 9 Copyright© 2015 MacMillan Publishers Limited.
The selection of treatment(s) for even typical relapsing multiple sclerosis (RMS) has become very challenging because of the number and availability of therapies, including new agents with more complex mechanisms of action and greater risks of adverse effects that may also impact subsequent therapies. Although much remains unknown about the effects of moving between immunosuppressive and other disease-modifying therapies (DMTs), factors such as presence of co-morbidities, desire for pregnancy, previous use of other immunosuppressants (for MS or other conditions), John Cunningham virus (JCV) antibody seropositivity, geographical parameters, health insurance coverage, and patient/neurologist preferences can influence the treatment selection and might also inform the sequencing of therapies. As the order in which treatments are used might predispose patients to increased long-term toxicity, all MS treatment strategies must be monitored to gather more safety data.
Based on the way they affect disease-causing cells, currently available therapies for MS can be divided into: (1) immunomodulators (interferon beta [IFN-β], glatiramer acetate [GA], dimethyl fumarate [DMF], teriflunomide [also a mild cell-depleting agent], daclizumab); (2) anti-trafficking agents (natalizumab, fingolimod); and (3) immune cell-depleting agents (mitoxantrone, cyclophosphamide, cladribine, ocrelizumab, alemtuzumab) (Table 2).Reference Coles, Twyman and Arnold 10 - Reference Chris and Randomized 25
Table 2 Immunosuppressants and immunomodulators in the treatment of relapsing remitting multiple sclerosis (RRMS): potential implication of differences in mechanisms of action
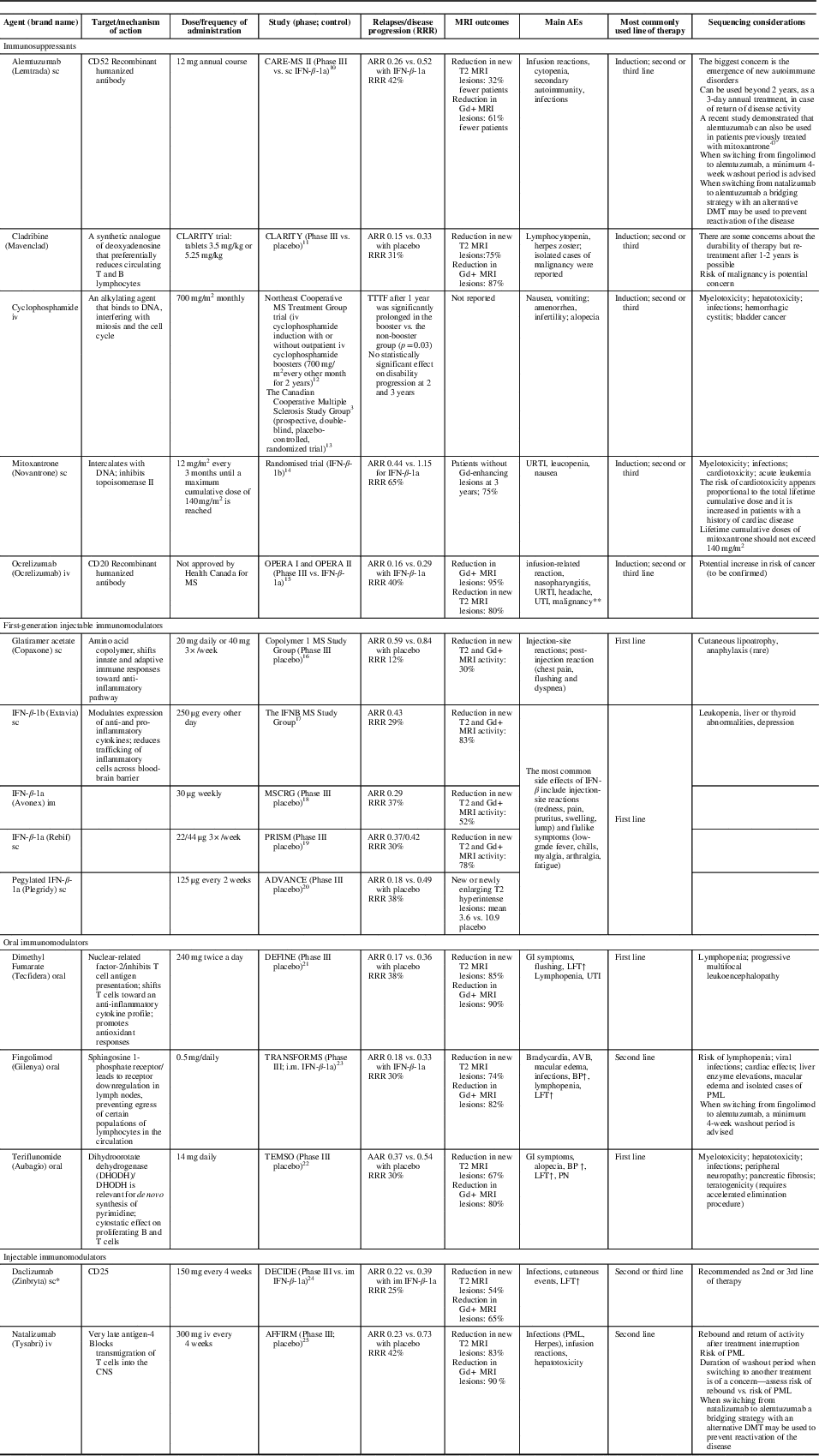
*Daclizumab was voluntarily removed from the market March 2, 2018 due to safety concerns. 102 **The true risk of malignancy, particularly breast cancer, requires further data. ARR=annualized relapse rate; BP=blood pressure; CD=cluster of differentiation; DMF=dimethyl fumarate; DMT=disease-modifying therapy; Gd=gadolinium; GI=gastrointestinal; im=intramuscular; INF=interferon beta; iv=intravenous; LFT=liver function tests; PML=progressive multifocal leukoencephalopathy; PN = peripheral neuropathy; RRR=relative risk reduction; sc=subcutaneous; TTTF=time to treatment failure; URTI=upper respiratory tract infections; UTI=urinary tract infection.
The mechanisms of action of different agents may also contribute to the potential risk of specific toxicities.Reference Gajofatto and Benedetti 26 , Reference Warnke, Olsson and Hartung 27 In the context of sequencing, some mechanisms of action might be considered complementary whereas others might indicate increased risk. For example, the use of natalizumab confers a greater risk of progressive multifocal leukoencephalopathy (PML),Reference Outteryck 28 so possibly using it after another drug that has PML as a potential side effect such as DMF may confer additive PML risk. Contrast that with the use of teriflunomide, both an immunomodulator and mild cell-depleting agent, together with IFN-β, a combination shown to be both safe and synergistic.Reference Freedman, Wolinsky and Wamil 29 However, if the mechanism of action of an agent like GA is to produce regulatory cells that need to reach the CNS in order to work, then concomitant use of an anti-trafficking drug might counteract that ability.Reference Farina, Weber, Meinl, Wekerle and Hohlfeld 30 - Reference Lalive, Neuhaus and Benkhoucha 32 On the other hand, based on their unique mechanisms of action, one could speculate that fingolimod followed by GA might be an “ideal sequence.” Fingolimod might act first to round up all disease-causing cells and sequester them in lymph nodes,Reference Cohen and Chun 33 , Reference Chun and Hartung 34 where GA has its greatest action and shifts T cells from a mostly pro-inflammatory T helper (Th) 1 pattern of cytokine secretion to a mostly anti-inflammatory Th2 pattern.Reference Comi, Amato and Bertolotto 35
Induction Therapies: Cell-Depleting Agents and their Impact on Sequential Therapeutic Approaches
Induction therapy refers to the use of powerful cell-depleting therapy capable of eliminating disease-causing autoimmune cells up front in patients presenting with early active aggressive disease(~10 % of MS patients).Reference Lazibat, Nevajda, Grahovac and Brinar 36 Evidence from experimental models of MS and other immune-mediated diseases suggests that this type of approach might “reset” the immune system to prevent epitope spreading and control inflammatory disease activity more effectively than immunomodulation, thus preventing early structural damage, controlling disease progression, and preserving brain function.Reference Cornaby, Gibbons and Mayhew 37 However, the duration of the induction treatment is often limited by specific toxicity and by cumulative doses.Reference Morrissey, Le Page and Edan 38 Serious side effects related to potent immunosuppressants such as mitoxantrone and cyclophosphamide are dose related, which is often a limiting factor.Reference Morrissey, Le Page and Edan 38 In the case of mitoxantrone, the risk of cardiotoxicity appears proportional to the total lifetime cumulative dose and it is increased in patients with a history of cardiac disease. In addition to cardiotoxicity, blood malignancies have been associated with mitoxantrone. Consequently, a complete blood count is recommended before each dose and each year after mitoxantrone treatment. Due to these serious safety concerns, lifetime cumulative doses of mitoxantrone should not exceed 140 mg/m2. Mitoxantrone exerts its therapeutic effect in MS through the inhibition of proliferation of B and T lymphocytes and macrophages.Reference Morrissey, Le Page and Edan 38 In addition, several other immunosuppressive effects have been described, such as decreased secretion of IFN-β, tumor necrosis factor-α, and interleukin (IL)-2.Reference Morrissey, Le Page and Edan 38 A full course of mitoxantrone treatment can stabilize patients with aggressive MS for 5 years or more.Reference Le Page, Leray and Taurin 39 However, due to its long-term safety concern, lifetime exposure to mitoxantrone is limited.Reference Edan, Comi and Le Page 40 If maximal lifetime doses are reached, patients may require maintenance therapy with a DMT. It has been suggested that a short course of mitoxantrone followed by a first-generation immunomodulator (GA or IFN) after clinical stabilization of the disease may be an appropriate treatment strategy.Reference Le Page, Leray and Taurin 39 - Reference Vollmer, Panitch and Bar-Or 41 However, due to potential toxicity and the emergence of other options mitoxantrone is now rarely used.
Alemtuzumab, a humanized monoclonal antibody that reduces circulating cluster of differentiation (CD) 52-positive cells, is another reasonable induction strategy.Reference Minagar, Alexander and Sahraian 42 It can be used for a set number of courses and re-treatment is needed only in the context of return of disease activity.Reference Havrdova, Horakova and Kovarova 43 With alemtuzumab, CD52 cell depletion is sustained for up to 1 year, followed by lymphocyte repopulation and homeostatic resetting of the immune system. The biggest concern with this agent is the emergence of new autoimmune disorders, which occur in approximately one-third of patients and can develop several years after the last dose.Reference Cossburn, Pace and Jones 44 Although the reason for this is unknown, it is speculated that it takes time for regulatory cells to return to normal.Reference Hill-Cawthorne, Button and Tuohy 45 Most autoimmune events with alemtuzumab are thyroid related and are generally mild or moderate and manageable with timely detection and treatment.Reference Dayan, Cuker and LaGanke 46 Unlike other agents with maximal lifetime exposure limits, alemtuzumab can be used beyond 2 years, as a 3-day annual treatment, in case of return of disease activity. A recent study demonstrated that alemtuzumab can also be used in patients previously treated with mitoxantrone.Reference Le Page, Deburghgraeve, Lester, Cardiet, Leray and Edan 47
Other approaches with high efficacy and more selective lymphocyte depletion include agents such as cyclophosphamide. Cyclophosphamide is an alkylating agent that binds to deoxyribonucleic acid, interfering with mitosis and the cell cycle.Reference Patti and Lo Fermo 48 , Reference Awad and Stüve 49 Although extensive experience with cyclophosphamide in MS patients has been accumulated over the past 40 years, conclusive efficacy data are lacking.Reference Lugaresi, di Ioia, Travaglini, Pietrolongo, Pucci and Onofrj 50 Yet, because of the differential risk for leukemia with mitoxantrone, cyclophosphamide may be preferred to mitoxantrone as induction therapy in rapidly progressing patients.Reference Martinelli, Cocco and Capra 51 Cyclophosphamide should also be considered in the event that approved first- and second-line drugs fail or when fingolimod, natalizumab, or mitoxantrone are contraindicated.Reference Patti and Lo Fermo 52 It is important to remember, however, that previous use of non-selective immunosuppressive drugs increases the risk of PML in natalizumab-treated patientsReference Outteryck 28 and may also confer risk of PML with other agents such as DMF or fingolimod.Reference D’Amico, Zanghì, Leone, Tumani and Patti 53 - Reference Gyang, Hamel, Goodman, Gross and Samkoff 55
Cladribine, a synthetic purine nucleoside and antimetabolite that acts as an antineoplastic agent with immunosuppressive effects, might be used in a similar manner to alemtuzumab.Reference Spurgeon, Yu, Phillips and Epner 56 , Reference Sipe 57 Animal data suggest that the drug is capable of crossing the blood-brain barrier,Reference Liliemark 58 and it was found to be beneficial for patients with RMS. 13 , Reference Giovannoni, Comi and Cook 59 , Reference Cook, Vermersch and Comi 60 Cladribine is an option for patients with aggressive MS,Reference Martinez-Rodriguez, Cadavid, Wolansky, Pliner and Cook 61 although the cladribine tablet formulation was previously rejected in 2011 because of malignancy concerns.Reference Gandey 62 A recent meta-analysis of Phase III trials of licensed DMTs for MS and the CLARITY trial did not support an increased cancer risk from cladribine in the doses used in CLARITY.Reference Pakpoor, Disanto and Altmann 63 The latest data from the extension phase of CLARITY showed that two to four short oral courses (one dose per day for 4-5 days) of cladribine in year 1 and two short courses in year 2 were associated with durable efficacy.Reference Giovannoni, Comi and Cook 64 There are some data concerning the durability of therapy but re-treatment after 1-2 years is possible. Cladribine tablets have now been approved in Europe and Canada for RMSReference Brooks 65 , 66
Escalation Therapies: Immunomodulators and their Impact on Sequential Therapeutic Approaches
Due to the limitations of immunosuppressive/ablative therapies, disease-causing cells may not be completely eliminated in many cases, resulting in breakthrough disease. In some instances, re-treatment with another course of the same therapy may be possible (e.g. alemtuzumab, cladribine). Given the risk of disease return, an appropriate alternative is to consider long-term maintenance following induction therapy, generally with safer immunomodulatory agents, to extend the benefit obtained with aggressive treatment; but such an approach has not been subjected to any form of rigorous study to date. The concept of escalation and maintenance therapy represents a strategy that gives precedence to safety over efficacy and, if necessary, to sequentially advance in the treatment pyramid.Reference Fenu, Lorefice, Frau, Coghe, Marrosu and Cocco 67 - Reference Du Pasquier, Pinschewer and Merkler 69 In this approach, individuals start with first-line agents and are switched to second- or even third-line agents if they exhibit breakthrough disease that pushes them from low to high in terms of the risk for imminent disease progression. For the purpose of this paper, the definitions and categorization of MS medications as first-, second-, and third-line drugs previously outlined in Freedman et alReference Freedman, Selchen and Arnold 5 have been used. It should be noted that the designation of first, second, and third-line agents is not evidence-based as all of the available treatments licensed for MS have been studied primarily as first-line agents. Escalation and maintenance therapy is appropriate for most patients with non-aggressive RMS, provided that they are closely monitored to detect suboptimal response or disease progression.
Sequential DMT monotherapy is currently the most common treatment strategy for RMS with injectables such as IFN-β or GA, along with teriflunomide or DMF being the most frequently used first-line therapies. Injectable IFN-β or GA as well as teriflunomide and cladribine have approved indications not only for patients with RMS but also for those with early MS, including patients with clinically isolated syndrome who are at high risk for the development of more active MS.Reference Freedman and Rush 8 Recent evidence indicates that early initiation of DMT is beneficial over the long term, resulting in significant reduction of long-term disability.Reference Freedman, Selchen and Arnold 5 , Reference Lazibat, Nevajda, Grahovac and Brinar 36 , Reference Havrdova, Horakova and Kovarova 43 , Reference Fenu, Lorefice, Frau, Coghe, Marrosu and Cocco 67 - Reference Kieseier and Jeffery 70
Although the injectable mode of administration of IFN-β or GA may be inconvenient for some patients, it is important to keep in mind the wealth of experience with these drugs in regard to dosing and toxicity management. Newer formulations have attempted to increase convenience with a pegylated version of IFN-β used every 2 weeksReference Bhargava and Newsome 71 and a new formulation of GA taken just thrice weekly.Reference Khan, Rieckmann and Boyko 72 With DMF, for example, the management of severe flushing and/or gastrointestinal toxicities continues to be a problem and efforts continue to reduce these side effects.Reference Phillips, Hutchinson, Fox, Gold and Havrdova 73 The DEFINE study suggested that half dose for 1 month (240 mg daily dose)Reference Gold, Kappos and Arnold 74 is more appropriate in regard to achieving desirable gastrointestinal tolerability than the extended titration (120 mg 3×/day 360 mg daily dose).Reference Kappos, Gold and Miller 75 However, doses lower than 240 mg daily are not as effective in reducing brain MRI activity.Reference Kappos, Gold and Miller 76
First-generation immunomodulators also have well-established short- and long-term safety profiles. Therefore, even if new oral first-line drugs such as teriflunomideReference Garnock-Jones 77 and DMFReference Burness and Deeks 78 may appear more convenient from the patient perspective and have a positive effect on treatment adherence and the patient’s quality of life,Reference Curtin and Hartung 79 it is important to keep in mind that there are still significant uncertainties regarding their long-term effects.
Several cases of PML have been reported with DMF, some of which were not related to previous use of immunosuppressants.Reference D’Amico, Zanghì, Leone, Tumani and Patti 53 , 54 In most, but not all cases, the patients had low lymphocyte counts (grade 2 or 3 lymphopenia).Reference D’Amico, Zanghì, Leone, Tumani and Patti 53 , Reference Nieuwkamp, Murk and van Oosten 80 Lymphopenia is a frequent outcome of DMF therapy and in clinical practice lymphopenia appears to be more common than reported in clinical trials. A retrospective cohort study of 221 patients prescribed DMF at a single academic medical center suggested that the cumulative incidence of grade 3 lymphopenia exceeds 20% in adults older than 55 years; combined grade 2 and 3 lymphopenia developed in more than 40% of patients in this age group.Reference Longbrake, Naismith and Parks 81 A higher risk of lymphopenia was noted in patients switching to DMF from natalizumab therapy and in those with lower baseline lymphocyte counts. Fingolimod and natalizumab are involved in immune cell trafficking that impedes lymphocyte migration across the blood-brain barrier (natalizumab) and/or from exiting lymph nodes into the circulation (fingolimod), preventing CNS inflammation.Reference Brück, Gold and Lund 82 While these therapies may offer substantial efficacy as a consequence of their mechanism of action, their alteration of lymphocyte distribution may influence immune surveillance and increase the risk of infections and PML.
There have been very few “head-to-head” studies using DMTs. High-dose IFN-β1a given subcutaneously three times per week (tiw) has been proven superior to the once-weekly intramuscular IFN-β1a.Reference Schwid and Panitch 83 Fingolimod was shown to be superior to intramuscular once-weekly IFN-β1a,Reference O’Connor, Wolinsky and Confavreux 23 daclizumab is superior to once-weekly IFN-β1aReference Chris and Randomized 25 and both alemtuzumab 84 and ocrelizumabReference Hauser, Bar-Or and Comi 85 were proven superior to subcutaneous IFN-β1a tiw. Although natalizumab is believed to be more potent on clinical and MRI outcomes than first-line injectable immunomodulators,Reference Putzki, Yaldizli and Buhler 86 this has never been shown in properly controlled studies, but is substantiated only with large, “real world” observational data sets.Reference Spelman, Kalincik and Zhang 87
With respect to escalation from either IFN-β or GA to natalizumab, retrospective and observational studies have indicated that switching is associated with a significant reduction in clinical and radiological activity.Reference Prosperini, Gianni and Leonardi 88 - Reference Krysko and OConnor 90 A switch from IFN-β to fingolimod has also been shown to produce significant reductions in clinical and radiological activity.Reference Khatri, Barkhof and Comi 91 However, no large head-to-head studies or evidence-based criteria exist to guide the choice between fingolimod and natalizumab. Thus, apart from the presence/absence of anti-JCV antibodies (researched only in the context of natalizumab) the choice is empirical, although some small, observational, uncontrolled studies suggest that natalizumab may be superior to fingolimod in RMS patients not responding to first-line agents.Reference Baroncini, Ghezzi and Annovazzi 92 - Reference Kalincik, Horakova and Spelman 95 According to the recent report from the MSBase registry, alemtuzumab and natalizumab seem to have similar effects on annualized relapse rates in RMS. While alemtuzumab was superior to fingolimod and IFN-β in mitigating relapse activity, natalizumab was superior to alemtuzumab in enabling recovery from disability. Thus, according to the investigators, treatment decisions between alemtuzumab and natalizumab should be primarily governed by their safety profiles.Reference Kalincik, Brown and Robertson 96
Ocrelizumab is a humanized monoclonal antibody that selectively targets CD20. The superior efficacy of ocrelizumab over IFN-β1a was proven in two Phase III clinical trials OPERA 1 and OPERA 2 that included ~800 patients with RMS.Reference Edan, Comi and Le Page 14 However, four neoplasms (two breast cancers, one renal cancer, and one melanoma) occurred in patients treated with ocrelizumab (0.5%) versus two occurrences in the IFN-β1a group (0.2%). Five additional malignancies were also detected during the open-label extension phase. In the ORATORIO trial involving patients with primary progressive MS, 11 neoplasms were reported, four of which were breast cancer.Reference Montalban, Hauser and Kappos 97 Thus, long-term follow-up with larger numbers of patients will be necessary to assess these risks more fully. Similar to malignancies observed with cladribine, it is unknown whether the cancer risk is idiosyncratic or related to the duration of therapy. Ocrelizumab was recently approved by Health Canada for the treatment of adult patients with RMS, with active disease defined by clinical and imaging features. 98
Daclizumab is a humanized IgG1 monoclonal antibody directed against CD25, the alpha subunit of the high-affinity IL-2 receptor.Reference Shirley 99 As demonstrated in the Phase III DECIDE trial, 25mg once-monthly subcutaneous daclizumab was superior to once-weekly intramuscular IFN-β-1a in reducing the clinical relapse rate and radiological measures of disease in patients with RMS. In addition, daclizumab has demonstrated efficacy in reducing disability progression and in improving health-related quality of life. Toxicity profile includes hepatic, infectious, and cutaneous events. Despite its availability, the place of daclizumab, functioning mainly as an immunomodulator in MS treatment remains to be fully determined. Based on available evidence, EMA advised against using daclizumab in patients with pre-existing hepatic disease or hepatic impairment and in patients with autoimmune diseases other than MS, and they urge caution when using the drug in combination with drugs that can damage the liver.Reference Brooks 100 In Canada, daclizumab was approved as second- or third-line option. 101 However, as of March 2, 2018, Biogen and AbbVie voluntarily removed daclizumab from the market worldwide due to safety concerns voiced by the European Medicines Agency after 8 patients in Europe presented with encephalitis and/or menigoencephalitis. 102
Suggested Strategies to Decrease the Risk of PML with Natalizumab
The increased risk of the development of PML with different DMTs, especially natalizumab, is well established.Reference D’Amico, Zanghì, Leone, Tumani and Patti 53 As of December 7, 2017, there have been 756 confirmed PML cases (753 MS, three Crohn’s Disease). 103 As of November 30, 2017, ~177,800 patients received natalizumab, yielding an incidence of 4.22 in 1000 treated patients. The global overall incidence of PML in natalizumab-treated patients is 4.22 per 1000 patients (95% confidence interval 3.93-4.54 per 1000 patients). 103 The risk is particularly high in patients treated with natalizumab for longer than 12 months, those who had previous exposure to immunosuppressive therapies, and those with a high anti-JCV antibody index. 103 - Reference Plavina, Subramanyam and Bloomgren 105 With the availability of several other DMTs, it is now possible to reduce the risk of PML by switching JCV-seropositive patients treated with natalizumab to another agent. One caveat is that previous exposure to natalizumab may have prolonged effect risk, also referred to as “carryover PML.”Reference Killestein, Vennegoor and van Golde 106 - Reference Martinelli, Colombo and Dalla Costa 108 Carryover PML is the result of the complex pathogenesis of PML, which develops over months to years.Reference Bloomgren, Richman and Hotermans 104 One strategy to reduce the risk of carryover PML is to washout natalizumab before starting another DMT, allowing immune reconstitution of the CNS.Reference Killestein, Vennegoor and van Golde 106 The obvious downside of this strategy is rebound effect and return of disease activity.Reference Martinelli, Colombo and Dalla Costa 108 , Reference Larochelle, Metz and Lécuyer 109 To overcome this risk, several approaches have been tried.Reference Borriello, Prosperini and Mancinelli 110 - Reference Cohan, Edwards and Chen 113 Yet, currently the only effective method to prevent rebound appears to be either fingolimodReference Fragoso, Adoni and Alves-Leon 112 or teriflunomideReference Cohan, Edwards and Chen 113 without any washout period. It is important to be aware that the risk of PML after stopping natalizumab is unlikely to disappear completely.
As JCV reactivation in the CNS is not fully understood, clinical pharmacovigilance is warranted as there might be a risk from either current or previous therapy. Furthermore, it appears that, despite stratification algorithms and the available test for JCV serology, the incidence of PML in natalizumab-treated MS patients has continued to increase, likely because patients are kept on the drug too long.Reference Cutter and Stüve 114 , Reference Mowry and McArthur 115 In their recent editorial, Mowry and McArthurReference Mowry and McArthur 115 suggest the development of Apps or online calculators for PML risk, as has been done for cardiovascular risk. Clinicians need to acknowledge that the incidence of PML remains high and not to rely on cutpoints, but rather appreciate that risk continues to accumulate with increased treatment duration. Given that PML may develop even after natalizumab discontinuation, it is possible that the longer a patient waits after becoming JCV antibody-positive before switching, the higher the risk of PML will become.Reference Mowry and McArthur 115
Course to Follow When Considering Treatment Modification
Factors to Consider When Moving to the Next Line of Therapy
The excitement surrounding new effective DMTs is tempered by concerns about both known and unknown additional risks. Although in many instances the rationale after relapse is to try a DMT with a different mechanism of action, the impact of a previous therapy on potential toxicity profile needs to be considered. In addition, washout period and potential for rebound effect present additional challenges when moving to the next line of therapy. For example, as fingolimod traps lymphocytes in the lymph nodes,Reference Antel 116 efficacy of subsequent agents that target circulating lymphocytes such as alemtuzumabReference Hu, Turner and Shields 117 may be compromised. Fingolimod has a pharmacologic half-life of 6-9 days and lymphocytes would be expected to normalize 2-4 weeks after discontinuation.Reference Johnson, Shames and Keezer 118 Therefore, it is recommended to stop fingolimod for 4 weeks, checking peripheral lymphocyte counts to ensure that they are returning towards normal before giving the first course of alemtuzumab, in order to maximize its effect on circulating lymphocytes.Reference Giovannoni, Marta and Davis 107 From a safety standpoint, moving from natalizumab or fingolimod (and probably DMF)—agents that have proven in monotherapy to cause PML—to alemtuzumab requires additional consideration. Should latent PML (i.e. active copies of JCV) be present and a patient switched to alemtuzumab, the patient will be immunocompromised and the PML can be life threatening. Such patients should probably undergo a lumbar puncture, with measurement of CSF for JCV polymerase chain reaction, before starting alemtuzumab or any other potent immunosuppressant, as a precautionary measure.
Managing pregnancy-related issues in patients with RMS
The theoretical potential for teratogenicity is relevant for all DMT when considering the treatment of female patients of childbearing age, but the risk varies among agents. In general, patients should be advised to discontinue DMTs before conception although there are suggestions that IFN-β Reference Amato, Portaccio and Ghezzi 119 and GAReference Giannini, Portaccio and Ghezzi 120 can be continued throughout pregnancy in patients with severe or highly active disease. Fingolimod should be discontinued 2-3 months before the cessation of contraception,Reference Lu, Wang and Guimond 121 and evidence regarding the discontinuation of natalizumab is not clear considering the high risk of disease recurrence even during pregnancy.Reference De Giglio, Gasperini, Tortorella, Trojano and Pozzilli 122 Teriflunomide is associated with a theoretical risk of teratogenicity, despite little evidence for this in the treatment of MS,Reference O’Connor, Li and Freedman 123 and the Food and Drug Administration regulation changed in regard to labeling all pharmaceuticals regarding the risk during pregnancy. 124 Most of the warnings for teriflunomide derive from those tagged to the parent drug, leflunomide, used for the treatment of rheumatoid arthritis. 125 Without going through an accelerated 11-day elimination procedure, 125 teriflunomide is eliminated slowly from plasma and it can take up to 2 years to achieve systemic clearance at a level where teratogenicity is theoretically unlikely. Across nine Phase II/III clinical studies with teriflunomide a total of 71 pregnancies were reported.Reference Henson, Stuve, Kieseier, Benamor and Benzerdjeb 126 Upon learning of their pregnancies, patients were instructed to discontinue their study treatment and undergo an accelerated elimination procedure (cholestyramine or activated charcoal). In total, 44 of the 71 pregnancies were reported in patients exposed to teriflunomide; the remaining pregnancies occurred in patients treated with placebo, IFN-β, or blinded therapy. In the 17 live births, including 12 exposed to teriflunomide, no structural defects or functional deficits have been reported. The duration of fetal exposure was up to 11 weeks and newborns had birth weights between 2780 and 4150 g. Thus far there is no signal for teratogenicity in newborns with prenatal teriflunomide exposure following accelerated elimination although more prospective data are needed with respect to pregnancy outcomes. The recent report on the outcomes of pregnancies occurring in the teriflunomide clinical program (n=69 pregnancies) and in the post-marketing setting (n=171 pregnancies) confirmed these observations.Reference Vukusic, Coyle and Jurgensen 127
Tests to Obtain Before Attempting Treatment Modification
Detection of anti-JCV antibodies in serum or plasma using a two-step enzyme-linked immunosorbent assay has been proposed to stratify the risk of PML. Testing for JCV antibody status is recommended for all natalizumab candidates before treatment initiation as well as before starting a new therapy after having taken natalizumab, DMF, or fingolimod. Natalizumab is generally not recommended in patients who are JCV antibody-positive and have received prior immunosuppressants, such as mitoxantrone or cyclophosphamide; however, the risk of other agents such as cladribine, alemtuzumab, or ocrelizumab are unknown.Reference Mowry and McArthur 115
Patients considered for fingolimod should be assessed for cardiovascular disease, diabetes, and liver dysfunction. As fingolimod can also increase low-density lipoprotein and total cholesterol, evaluation of serum lipids should be part of routine monitoring for patients taking fingolimod.Reference Traboulsee, Simon and Stone 128 As increased susceptibility to serious viral infections was noticed during the TRANSFORMS trial,Reference O’Connor, Wolinsky and Confavreux 23 patients considering fingolimod should probably be vaccinated against the varicella zoster virus and immunity confirmed before initiating therapy.Reference Traboulsee, Simon and Stone 128 Furthermore, due to the immunosuppressive mode of action of fingolimod, an increased awareness of infectious complications is required.Reference Haars, Schmidt and Orthgieß 129 , Reference Stecchi, Scandellari, Gabrielli and Lazzarotto 130 Cases of cryptococcal infections, including cryptococcal meningitis, and systemic meningitis, have been reported in patients taking fingolimod.Reference Rudnicka, Czerwiec and Grywalska 131 , Reference Ward, Jones and Goldman 132
Although a recent study found no increased risk of early relapse (within the first 6 months) following switch of patients previously stable on injectable therapy to oral treatment, 133 the longer-term (>6 months) risk of relapse is unknown. The long-term adverse event profile of most oral agents has yet to be established compared with the injectable immunomodulators IFN-β and GA.
Washout Period
Transitioning from one particular DMT to another can be complex, and a washout period may be required in certain circumstances. There is a paucity of data regarding the optimal washout period when switching from one agent to another. Furthermore, it is often unclear for which treatment transitions a washout period is required, how long it should last, or what long-term safety surveillance procedures should be implemented. Nevertheless, concomitant treatment within the washout period of the previous DMT is not advisable, owing to the potential risk of carry-over PML from previous treatment and additive effects on the immune system. Consequently, it is important to consider the half-life as well as the mechanism of action of the previous DMT when transitioning from one therapy to another.
In general, a washout period is not required when switching from first line injectable to any other treatment. As mentioned, an accelerated elimination protocol may be advisable when switching from teriflunomide to another agent due to the long half-life of the drug. 125
When switching from fingolimod to alemtuzumab, a minimum 4-week washout period is advised. 101 , 134 , Reference Hassoun, Eisele, Thomas and Ziemsse 135 Recent reports of patients treated with fingolimod who experienced significant rebound of disease activity when switched to alemtuzumab indicate that one must wait for lymphocytes to desequester in order to get the most out of the first 5-day course of alemtuzumab.Reference Spelman, Mekhael and Burke 134 , Reference Hassoun, Eisele, Thomas and Ziemsse 135 Because of the potential for PML in patients on fingolimod alone, it is imperative that PML be ruled out before initiating alemtuzumab for reasons outlined below. An “exit MRI” along with CSF for JCV PCR is strongly recommended.
The proposed mechanism of action of DMF (activation of the nuclear factor erythroid 2-related factor 2) would generally not foresee any issues with rapid transition to another therapy,Reference Spelman, Mekhael and Burke 134 although DMF has also been shown to have a lymphopenic effect in certain patient populations.Reference Willis, Pearson and Illes 136 Thus, similar to fingolimod, a washout period might be advisable when transitioning from DMF to alemtuzumab.Reference Fox, Kita and Cohan 137 , Reference Miller 138 For both drugs, however, the time period over which lymphocytes return to normal is variable and can take many months, during which time the patient is at risk of relapse.Reference Fox, Kita and Cohan 137 This is a significant challenge in sequencing from DMF. As DMF has also had cases of PML associated with it, an “exit MRI” and CSF for JCV PCR is recommended before initiating alemtuzumab (see below).
Teriflunomide is also associated with a reduction in lymphocytes and neutrophil counts. However, patients receiving teriflunomide have the opportunity to undergo an accelerated elimination procedure, potentially allowing the initiation of alemtuzumab relatively quickly after stopping teriflunomide.
As natalizumab does not reduce circulating lymphocyte counts, rather it blocks their entry to the CNS resulting in only mild lymphocytosis,Reference Fox, Kita and Cohan 137 , Reference Miller 138 there may be limited benefit in delaying initiating treatment with another DMT following natalizumab discontinuation.Reference Miller 138 It has also been suggested that starting a new treatment immediately after stopping natalizumab (without washout period) may be more beneficial because the risk of developing PML is lower than the risk of a severe relapse.Reference Polman, O’Connor and Havrdova 139 , Reference Gallo and Van Wijmeersch 140
However, despite preferred outcomes in the absence of a prolonged washout period after natalizumab, it is important when switching to confirm JCV status and exclude any PML carryover in JCV-positive patients by MRI and by cerebrospinal fluid examination for JCV DNA, before treatment initiation. This is of particular relevance when patients are switched from natalizumab to alemtuzumab as the effects of alemtuzumab cannot be reversed in the short term, and, if carry-over PML develop following alemtuzumab treatment, it is unlikely that full immune cell repopulation can occur and that patients will be unable to clear the virus. Thus, when switching from natalizumab to alemtuzumab, either this should occur swiftly, as soon as an “exit MRI” and CSF study ruling out JCV by PCR are performed, or a bridging strategy with an alternative DMT may be required in the interim to prevent reactivation of the disease.
Given that there was a PML case described with ocrelizumab as a carryover from natalizumabReference Cliford 141 switching from natalizumab to ocrelizumab should be done the same way as alemtuzumab with the same precautionary CSF analysis and MRI.
When switching from natalizumab to fingolimod, a washout period of>3 months is associated with an increased risk of disease reactivation.Reference Clerico, Schiavetti and De Mercanti 142 - Reference Fox, Cree and De Seze 144 The latest evidence suggests that no washout period is recommended.Reference Cohan, Edwards and Chen 113 However, since both drugs carry a risk of PML, it would be important to perform an “exit MRI” and CSF for JCV PCR before initiating fingolimod as there have been many cases of “carry-over” PML from natalizumab-treated patients initiated on fingolimod.Reference Sinnecker, Othman and Kühl 145 , Reference Putzki 146
Treatment Discontinuation
Sometimes RMS patients receiving DMT may decide to interrupt their therapy due to various personal reasons (e.g. travel, pregnancy). These patients should continue to be periodically monitored both clinically and by MRI. Pregnancy planning requires DMT discontinuation (unless the benefit to the mother outweighs the risk to the fetus) with the appropriate timing according to the drug-specific pharmacokinetics.Reference Houtchens and Kolb 147 Discontinuation of anti-trafficking agents presents significant concern for a rebound syndrome.Reference Hatcher, Waubant, Nourbakhsh, Crabtree-Hartman and Graves 148 Cases of fulminant relapse after stopping natalizumab, leading to death despite intensive care and immunosuppressive therapy, have been reported.Reference Martinelli, Colombo and Dalla Costa 108 Rebound effect and the development of tumefactive demyelinating lesions were also reported in patients who stopped fingolimod in order to conceive.Reference Faissner, Hoepner, Lukas, Chan, Gold and Ellrichmann 149 , Reference Salam, Mihalova and Siripurapu 150
Therefore, patients should be cautioned that some medications pose additional issues upon discontinuation. This is important especially in pregnancy planning as a bridging strategy to prevent rebound might be initiated before discontinuing anti-trafficking agents. If disease worsening is detected in patients who stopped their DMT, the decision to restart the treatment should be revisited.
Combination Therapy
Combination therapy, allowing for treatments with different mechanisms of action to accomplish better disease control, could be an effective strategy for MS. However, limited evidence, concerns regarding potential additive effects, increased risk of toxicity, and cost concerns are all obstacles to wider uptake of combination therapy in MS. According to Phase II data, some combinations are both highly effective and safe, at least in the short term.Reference Freedman, Wolinsky and Wamil 151 , Reference Goodman, Rossman and Bar-Or 152 Combined teriflunomide and IFN-β produce additive responses, especially on MRI.Reference Goodman, Rossman and Bar-Or 152 The combination of natalizumab and GA appeared safe and well tolerated during 6 months of therapy.Reference Goodman, Rossman and Bar-Or 152 Currently, cost is one of the main reasons prohibiting combination therapy, despite the fact that some combinations might well afford synergy due to their different mechanisms of action.
Evaluating Treatment Response
A full review of methodologies to evaluate treatment response to disease-modifying medication is beyond the scope of this paper. However, recommendations for the clinical assessment of relapses, which comprise relapse rate, severity, and recovery, have been outlined previously by Freedman et al (Table 3).Reference Freedman, Selchen and Arnold 5 Critical factors affecting the decision of changing the current DMT are listed in Table 4.Reference Gajofatto and Benedetti 26 A new MRI with gadolinium is recommended 6-12 months after treatment initiation or switching to establish a new “reference” MRI for future comparison.Reference Freedman, Selchen and Arnold 5 , Reference Henson, Stuve, Kieseier, Benamor and Benzerdjeb 126 Typically another MRI is recommended within a year of the new “reference” scan for full evaluation of treatment response.
Table 3 Level of concern according to the criteria for relapse, Expanded Disability Status Scale (EDSS) progression, and MRI findingsReference Freedman, Selchen and Arnold 5

ADL=activities of daily living
When considering a switch in therapy, whether it is a lateral switch between two agents in the same line of therapy (e.g., interferon beta [IFN-β] to glatiramer acetate [GA]) or treatment escalation to a second-line agent, clinicians should first determine if suboptimal treatment response warrants a switch in treatment. This can be done by determining the level of concern according to the criteria for relapse, EDSS progression, and MRI.
The level of concern should be reassessed and determined at regular follow-up intervals, typically every 6 or 12 months. Routine follow-up MRI with gadolinium (Gd) is recommended 6-12 months after initiating therapy for relapsing remitting multiple sclerosis (or in clinically isolated syndrome if therapy is not initiated). New T2 lesions that are also enhancing on the same scan are only counted once as unique active lesions. The presence of Gd-enhancing lesions is more reliable than new T2 lesion counts. New T2 lesion counts require high-quality comparable MRI scans and interpretation by highly-qualified individuals.
* If EDSS progression alone is used to assess response to treatment, any change requires subsequent confirmation at 3-6 months.
** Timed 25-foot walk (T25FW) tested at baseline with aid, if required
Table 4 Factors affecting the decision to modify disease-modifying therapies (DMT) for multiple sclerosisReference Gajofatto and Benedetti 26
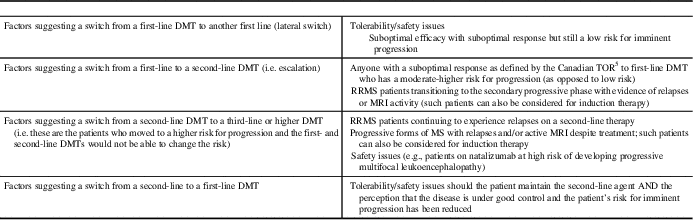
RRMS=relapsing remitting multiple sclerosis; TOR = Treatment Optimization Recommendations.
Adapted from: Gajofatto et al.Reference Gajofatto, Bianchi and Deotto 94
Conclusion
The availability of new treatment options for MS provides an opportunity for improving long-term clinical outcomes. However, it has also made patient management more complex. The suggestions regarding the sequencing of MS therapies outlined in this manuscript were developed to assist clinical neurologists in their management of RMS patients.
When switching the RMS patient from one therapy to another, it is essential to consider cumulative toxicities. At the same time risk of disease reactivation and rebound effect need to be taken into consideration should too much time pass between stopping one agent and starting the next. The patient should undergo full general and neurological examinations and laboratory measurements, as well as getting either an “exit MRI” (in the case of PML causing agents) or a new “reference” MRI (done 3-6 months after starting the new agent to allow time for it to be effective). All patients should undergo regular monitoring and follow-up so that, if needed, appropriate measures can be taken in a timely manner to offset unexpected toxicities or to treat suboptimal disease response, which is much more likely in patients already showing a lack of good treatment response to a first-line therapy.Reference Antel 116
Acknowledgments
Teva, the funding sponsor, offered unrestricted support to the development of the manuscript and did not have any part in creating this document. The funding also provided the authors with the services of an experienced and qualified medical writer to ensure a professional manuscript. The medical writer, solely under the direction and outline of the authors, assisted in researching the topic and preparing a first draft. At no time did the medical writer have any involvement in determining the content of the manuscript. The authors gratefully acknowledge the contribution of Radmila Day in the drafting of the manuscript.
Statement of Authorship
MSF, PSG, AP, and DS participated in revising the manuscript critically for important intellectual content and approved the final version of the manuscript to be published.
DISCLOSURES
MSF reports grants from Teva, during the conduct of the study; grants and personal fees from Genzyme, personal fees from Sanofi-Genzyme, personal fees from Actelion, personal fees from Bayer HealthCare, personal fees from Biogen Inc., personal fees from Chugai, personal fees from EMD Serono Canada Inc., personal fees from Hoffman La-Roche, personal fees from Merck Serono, personal fees from Novartis, personal fees from Sanofi-Aventis, personal fees from Clene Nanomedicine, outside the submitted work. MSF has been a member of a company advisory board, board of directors, or another similar group for Acetlion, Bayer HealthCare, Biogen Inc., Hoffman La-Roche, Merck Serono, MedDay, Novartis, and Sanofi-Aventis; he also particpated in a Genzyme-sponsored speaker bureau.
PSG reports grants from Teva Canada Innovation, during the conduct of the study; grants and personal fees from Biogen Inc., grants and personal fees from Teva, grants from Alexion, grants and personal fees from Bayer HealthCare, grants from Elan, grants and personal fees from EMD Serono Canada Inc., grants from GlaxoSmithKline, grants from MedDay, grants and personal fees from Novartis, grants from Ono, grants and personal fees from Roche, grants from Sanofi-Aventis, personal fees from NeuroRx, personal fees from Allergan, personal fees from Sanofi-Genzyme, personal fees from Merz, outside the submitted work. PSG has acted as a principal investigator or sub-investigator for clinical trials for Alexion, Bayer HealthCare, Biogen Inc., Elan, EMD Serono Canada Inc., GaxoSmithKline, MedDay, Novartis, Ono, Roche-Genetech, Sanofi-Aventis, and Teva Neuroscience.
AP reports grants from Teva Canada Innovation, during the conduct of the study; personal fees from Teva, personal fees from EMD Serono Canada Inc., personal fees from Merck Serono, personal fees from Biogen Inc., personal fees from Sanofi-Genzyme, outside the submitted work. AP is also a senior Canada Research Chair in MS.
DS reports grants from Teva Canada Innovation, during the conduct of the study; personal fees from Merck Serono, personal fees and grants from Novartis, personal fees and grants from Roche, personal fees from Genzyme, personal fees from Teva, grants from Sanofi-Genzyme, personal fees from Biogen Inc., personal fees from Bayer HealthCare, outside the submitted work.