


1. Introduction
Since the first crystal structure of myoglobin (Kendrew et al. Reference Kendrew, Bodo, Dintzis, Parrish, Wyckoff and Phillips1958), the three-dimensional (3D) reconstruction image of T4 bacteriophage tails by electron microscopy (EM) (De Rosier & Klug, Reference De Rosier and Klug1968), and the solution nuclear magnetic resonance (NMR) structure of proteinase inhibitor IIA (Williamson et al. Reference Williamson, Havel and Wuthrich1985), structural biology has made tremendous strides toward revealing intimate atomic level details that guide the function of biological molecules. We live at a time when we know the structures of more than 120 000 (and counting – Source: http://www.rcsb.org/pdb/statistics/holdings.do) biological macromolecules, when we can visualize the inner workings of the ribosome (Ben-Shem et al. Reference Ben-Shem, Garreau DE Loubresse, Melnikov, Jenner, Yusupova and Yusupov2011), or the nucleosome interactions that preserve the integrity and identity of our genome (Luger et al. Reference Luger, Mader, Richmond, Sargent and Richmond1997). At the same time, advances in instrumentation engineering have pushed the frontiers of structural biology methodologies and have allowed experiments and accomplishments that would have been unthinkable 30 years ago. Thus, it is now possible to record high-resolution movies of fast protein motions using X rays (Tenboer et al. Reference Tenboer, Basu, Zatsepin, Pande, Milathianaki, Frank, Hunter, Boutet, Williams, Koglin, Oberthuer, Heymann, Kupitz, Conrad, Coe, Roy-Chowdhury, Weierstall, James, Wang, Grant, Barty, Yefanov, Scales, Gati, Seuring, Srajer, Henning, Schwander, Fromme, Ourmazd, Moffat, Van Thor, Spence, Fromme, Chapman and Schmidt2014), obtain cryo-EM electron density maps at sub 3 Å resolution (Campbell et al. Reference Campbell, Veesler, Cheng, Potter and Carragher2015; Merk et al. Reference Merk, Bartesaghi, Banerjee, Falconieri, Rao, Davis, Pragani, Boxer, Earl, Milne and Subramaniam2016), or record multidimensional NMR spectra of protein crystals (Igumenova et al. Reference Igumenova, Mcdermott, Zilm, Martin, Paulson and Wand2004). Yet, the task in front of the structural biologist is getting harder and harder. The wealth of structural, biochemical and biological data has revealed that many mammalian cellular proteins are very large (>50 kD) (Brocchieri & Karlin, Reference Brocchieri and Karlin2005), that they are often part of complex assemblies composed of many interchangeable molecular players, and that their function is often defined and regulated by an intricate layer of post-translational modifications (PTMs). In addition, many disease-related biological macromolecules do not have a defined secondary or tertiary structure at all, and function, instead, through intrinsic disorder and numerous weak, transient interactions (Hyman et al. Reference Hyman, Weber and Julicher2014; Tompa, Reference Tompa2012). To make sense of this complicated, multilayered and often chaotic biological world, the structural biologist will become more and more dependent on the ability of protein engineers to faithfully and efficiently reproduce this complexity in the test tube.
Analogous to the advances in instrumentation design and engineering that have allowed structural biology to travel far, the tools of protein engineering have also become much more sophisticated, efficient and ultimately broader in scope over time. It is now possible to routinely synthesize polypeptide chains that are 50 amino acids long, to stitch them together into much longer chains without leaving any chemical scars (Dawson et al. Reference Dawson, Muir, Clark-Lewis and Kent1994), and to decorate them with PTMs, biophysical probes and chemical moieties that perturb or enhance their function. It is also possible to ‘persuade’ the cellular protein synthesis machinery to produce polypeptide chains incorporating completely unnatural amino acids, thus expanding the genetic code of engineered living organisms (Wang et al. Reference Wang, Brock, Herberich and Schultz2001). The current protein engineering toolbox contains many biocompatible chemical reactions, proteins with unique polypeptide ‘stitching’ abilities, and concepts and ideas that might ultimately prove essential in solving the interesting and relevant structural biology problems of today (Fig. 1). As the structural biologist might not be aware of all the current developments in protein chemistry, we intend this review as a resource that describes the state-of-the-art protein engineering tools, keeping an eye on the past and future to provide context for their limitations and the exciting new possibilities that undoubtedly lie ahead. We start with a very brief overview of recent advances in X-ray crystallography, cryo-EM and NMR, and outline challenges where the tools of protein engineering might be the most impactful. We then describe the contents of the molecular engineering toolbox that allow the construction of large modified proteins and complex macromolecular assemblies. We continue with a discussion of the concepts and ideas that directly concern structural biology methodology development. Our monograph ends with an outlook toward emerging trends in structural and chemical biology and exciting new developments that will guide the two fields in the future.
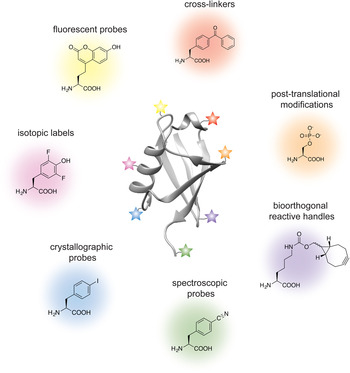
Fig. 1. Molecular engineering toolbox for the structural biologist.
2. Advances and challenges in structural biology
2.1 X-ray crystallography
The workhorse of structural biology, X-ray crystallography, is more than 100 years old and has contributed nearly 90% of the macromolecular structures deposited in the PDB (http://www.rcsb.org/pdb/statistics/holdings.do). Protein engineering has long been part of the everyday life of the crystallographer as mutations, truncations and fusion proteins are often required to ‘trick’ proteins into adopting a crystal form. There are many crystal structures of proteins containing PTMs or their analogs, and chemical approaches are often used to trap interesting functional states, stabilize dynamic interactions or aid the formation of crystals (e.g. racemic crystallography (Yeates & Kent, Reference Yeates and Kent2012)). Yet, the voracious need to test hundreds if not thousands of single crystal growth conditions has certainly challenged the protein chemist to optimize her tools and deliver relevant samples with much greater yields. Recent instrumentation developments such as X-ray free-electron lasers may potentially alleviate this need as these sources allow the acquisition of room-temperature data from easier to obtain micro-, nano- and 2D crystals (Neutze et al. Reference Neutze, Branden and Schertler2015). Currently, there is also a growing demand for the construction of homogeneous, chemically well defined and stable complex biological assemblies such as those relevant for chromatin biology, for example.
2.2 Cryo-EM
Exciting developments in the last few years have propelled cryo-EM into the spotlight and turned this method into a mainstream and vital structural biology technique that can achieve crystallographic resolution (Cheng, Reference Cheng2015; Nogales, Reference Nogales2016). The commercialization of direct electron-detection cameras has allowed the acquisition of images with higher contrast and fast readouts that can overcome beam-induced motion and radiation damage (Brilot et al. Reference Brilot, Chen, Cheng, Pan, Harrison, Potter, Carragher, Henderson and Grigorieff2012; McMullan et al. Reference Mcmullan, Chen, Henderson and Faruqi2009). On the other hand, improvements in data analysis approaches have made it possible to characterize heterogeneous samples and even rare structural states (Fernandez et al. Reference Fernandez, Bai, Hussain, Kelley, Lorsch, Ramakrishnan and Scheres2013; Scheres, Reference Scheres2012). Coupled with other cryo-EM advantages (no need for crystallization and only small amounts of sample required), these advances have made it possible to obtain subnanometer (and in some cases <3 Å) resolution maps of integral membrane proteins (Matthies et al. Reference Matthies, Dalmas, Borgnia, Dominik, Merk, Rao, Reddy, Islam, Bartesaghi, Perozo and Subramaniam2016), biological polymers (von der Ecken et al. Reference Von der Ecken, Muller, Lehman, Manstein, Penczek and Raunser2015), chromatin (Song et al. Reference Song, Chen, Sun, Wang, Dong, Liang, Xu, Zhu and Li2014), as well as biological assemblies such as the transcription and translation initiation complexes (Fernandez et al. Reference Fernandez, Bai, Hussain, Kelley, Lorsch, Ramakrishnan and Scheres2013; He et al. Reference He, Fang, Taatjes and Nogales2013; Plaschka et al. Reference Plaschka, Hantsche, Dienemann, Burzinski, Plitzko and Cramer2016). In this context, protein chemistry and engineering can have a tremendous benefit for the cryo-EM structural biologist in the design and construction of relevant biological samples such as post-translationally modified proteins. Perhaps more importantly, however, chemical biology approaches such as cross-linking can allow the preparation of samples that are more robust and do not fall apart during sample vitrification. The addition of cross-linkers can also be extremely useful in integrated cryo-EM/mass-spec structural approaches for samples where the high-resolution identification of protein–protein interfaces might not be possible (Leitner et al. Reference Leitner, Faini, Stengel and Aebersold2016).
2.3 NMR spectroscopy
NMR spectroscopy detects the magnetic properties of nuclei in molecules, which in turn provide a window into their surrounding chemical environment. Uniquely suited to probe molecular structure and dynamics in solution at physiologically relevant conditions (temperature, pH and salts) and ultimately non-destructive in its readout, NMR spectroscopy has long been battered by its intrinsically low sensitivity. The introduction of ‘NMR-visible’ isotopic labels into biological macromolecules has become a standard practice in the field, and efficient molecular engineering approaches that allow the installation of nuclear isotopes at specific positions within the polypeptide or polynucleotide chain are highly desirable. Recent advances such as (methyl)-TROSY and dark-state exchange saturation transfer experiments have pushed the molecular size limits of solution NMR into the MDa regime (Fawzi et al. Reference Fawzi, Ying, Ghirlando, Torchia and Clore2011; Pervushin et al. Reference Pervushin, Riek, Wider and Wuthrich1997; Tugarinov et al. Reference Tugarinov, Hwang, Ollerenshaw and Kay2003), while the rapid instrumentation and pulse sequence developments in magic angle spinning NMR have made it possible to pursue the structures of large biological polymers such as amyloid fibrils (Fitzpatrick et al. Reference Fitzpatrick, Debelouchina, Bayro, Clare, Caporini, Bajaj, Jaroniec, Wang, Ladizhansky, Muller, Macphee, Waudby, Mott, De Simone, Knowles, Saibil, Vendruscolo, Orlova, Griffin and Dobson2013; Lu et al. Reference Lu, Qiang, Yau, Schwieters, Meredith and Tycko2013; Wasmer et al. Reference Wasmer, Lange, Van Melckebeke, Siemer, Riek and Meier2008), bacterial secretion needles (Loquet et al. Reference Loquet, Sgourakis, Gupta, Giller, Riedel, Goosmann, Griesinger, Kolbe, Baker, Becker and Lange2012), membrane proteins embedded in their native lipid environments (Cady et al. Reference Cady, Schmidt-Rohr, Wang, Soto, Degrado and Hong2010; Wang et al. Reference Wang, Munro, Shi, Kawamura, Okitsu, Wada, Kim, Jung, Brown and Ladizhansky2013), or even the molecular composition of bones (Chow et al. Reference Chow, Rajan, Muller, Reid, Skepper, Wong, Brooks, Green, Bihan, Farndale, Slatter, Shanahan and Duer2014). Thus, molecular engineering approaches can have a profound impact on the assembly of homogeneous, isotopically labeled and yet native substrates for structural investigation by in vitro NMR. Also, uniquely suited to probe structure and dynamics in the cellular milieu (Burz et al. Reference Burz, Dutta, Cowburn and Shekhtman2006; Frederick et al. Reference Frederick, Michaelis, Corzilius, Ong, Jacavone, Griffin and Lindquist2015; Inomata et al. Reference Inomata, Ohno, Tochio, Isogai, Tenno, Nakase, Takeuchi, Futaki, Ito, Hiroaki and Shirakawa2009; Sakakibara et al. Reference Sakakibara, Sasaki, Ikeya, Hamatsu, Hanashima, Mishima, Yoshimasu, Hayashi, Mikawa, Walchli, Smith, Shirakawa, Guntert and Ito2009), NMR spectroscopy can benefit tremendously from chemical and molecular biology techniques that allow the specific isotopic labeling of macromolecules in the cell.
3. Molecular engineering toolbox for complex biological samples
Before we delve into the chemistry, it is important to note that the methods described below complement the well-established molecular biology framework that allows the manipulation of protein sequences at the genetic level. Such manipulations can now be achieved in several different organisms ranging from bacteria (Escherichia coli, Lactococcus lactis), yeast (Saccharomyces cerevisiae, Pichia pastoris), insect cells, and stable mammalian expression cell lines such as HEK293 and CHO. Thus, we will start with review of the methodologies that can selectively modify natural amino acids introduced at specific protein positions with site-directed mutagenesis. We will then describe tools that can be used to ligate modified peptides and proteins into longer polypeptide chains, including native chemical ligation (NCL), inteins and transpeptidases. We will then discuss the molecular engineering toolbox afforded by incorporation of unnatural amino acids by amber suppression. These chemical and genetic tools give chemists the ability to position bio-orthogonal reactive handles into polypeptide chains with extraordinary precision and control, and we will end this section with discussion of bioconjugation approaches that take advantage of this rapidly evolving expertise.
3.1 Cysteine chemistry
The field of protein chemistry would have been very different in a world without cysteines. Indeed, many of the protein chemistry tools that exist today have been developed to exploit the reactivity of the cysteine sulfhydryl group that uniquely stands out among a sea of other protein side-chains. Cysteines are relatively rare in Nature (<2% abundance), and their high nucleophilicity makes them good candidates for the development of selective chemical reactions that work well in protein compatible conditions (aqueous solution, physiological pH and temperature). Generally, these reactions can be divided into three types: (1) cysteine alkylations, (2) oxidations and (3) desulfurization reactions, each providing a unique way to exploit the reactivity of this amino acid and a pathway to build proteins with distinct and desirable properties (reviewed in (Chalker et al. Reference Chalker, Bernardes, Lin and Davis2009; Spicer & Davis, Reference Spicer and Davis2014)) (Fig. 2).
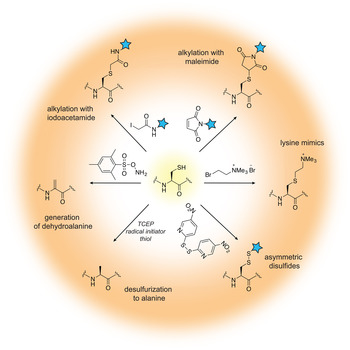
Fig. 2. Chemical modification of cysteine residues.
Alkylation reactions have long been used to modify cysteine containing proteins and rely on familiar reagents such as iodoacetamide and maleimide. By careful control of the buffer pH, these electrophiles can selectively react with the cysteine sulfhydryl group and tether additional functionalities to the protein of interest. Iodoacetamides and other α-halocarbonyls, for example, have been used to attach carbohydrate moieties to proteins and create glycoprotein mimics (Macmillan et al. Reference Macmillan, Bill, Sage, Fern and Flitsch2001; Tey et al. Reference Tey, Loveridge, Swanwick, Flitsch and Allemann2010). Maleimides, on the other hand, are commercially available and user-friendly means to add spectroscopic probes to proteins, including fluorescent, electron paramagnetic resonance (EPR) and paramagnetic relaxation enhancement (PRE) labels. Aminoethylation of cysteines also provides a cheap and convenient way to generate lysine methylation analogs, available in the mono-, di- and tri-methylation states (Simon et al. Reference Simon, Chu, Racki, De La Cruz, Burlingame, Panning, Narlikar and Shokat2007). In recent years, the chemist's attention has turned toward the development of more advanced cysteine bioconjugation protocols that circumvent, for example, the reversibility of maleimide additions in the presence of external bases and thiols (Lyon et al. Reference Lyon, Setter, Bovee, Doronina, Hunter, Anderson, Balasubramanian, Duniho, Leiske, Li and Senter2014), or provide the opportunity to conjugate additional functionalities (such as aryl groups) under an expanded range of reaction conditions (Vinogradova et al. Reference Vinogradova, Zhang, Spokoyny, Pentelute and Buchwald2015).
Cysteine oxidation in the protein context is usually associated with the formation of disulfide bonds, a unique structural transformation with often dramatic consequences to protein function. In the test tube, disulfide bonds are very easy to build – all that is required is basic pH and exposure to air. Furthermore, the reaction is fast and does not require large excess of reagents. Therefore, it comes as no surprise that protein chemists often exploit disulfide bonds as means to attach useful functionalities to proteins of interest. The challenging aspect of such protocols is to ensure that only the desired disulfide bonds are formed and the final product is not a mixture of homo- and heterodimer species. A common strategy used to alleviate this problem relies on controlled cysteine activation and disulfide exchange based on the lower pKa of aromatic thiols. In this case, a selected cysteine side-chain can be activated (and protected) at low pH with the aromatic thiol, and upon addition of the other thiol-containing component and increase in pH, the aromatic disulfide bond is exchanged with the desired connectivity (Pollack & Schultz, Reference Pollack and Schultz1989; Rabanal et al. Reference Rabanal, Degrado and Dutton1996). This strategy has found diverse applications ranging from the design of cytochrome peptides (Rabanal et al. Reference Rabanal, Degrado and Dutton1996) to the tethering of ubiquitin moieties to various proteins (Chatterjee et al. Reference Chatterjee, Mcginty, Fierz and Muir2010; Chen et al. Reference Chen, Ai, Wang, Haracska and Zhuang2010a; Meier et al. Reference Meier, Abeywardana, Dhall, Marotta, Varkey, Langen, Chatterjee and Pratt2012). Other activating molecules include methanethiosulfonates, glycosyl and allylic thiols with applications in protein glycosylation, prenylation and sulfenylation (Gamblin et al. Reference Gamblin, Van Kasteren, Bernardes, Chalker, Oldham, Fairbanks and Davis2008; Grayson et al. Reference Grayson, Ward, Hall, Rendle, Gamblin, Batsanov and Davis2005; van Kasteren et al. Reference Van Kasteren, Kramer, Jensen, Campbell, Kirkpatrick, Oldham, Anthony and Davis2007).
While the formation of disulfide bonds is fast and facile, they are just as easily destroyed in the presence of common biochemical reducing reagents or in the cellular environment where glutathione is present at high concentrations. If compatible linkages are desired, it is possible to convert the disulfide to a thioether bond, which is stable under reducing conditions. This is essentially a desulfurization reaction that proceeds with the formation of a dehydroalanine intermediate. Dehydroalanines, on the other hand, can be useful stepping stones to a vast number of protein PTMs, reactions that will be discussed in more detail in Section 3.7. Another key desulfurization reaction involves the conversion of cysteine to alanine. This transformation, discussed in more detail in Section 3.3, is particularly important for NCL, as it allows the construction of polypeptide chains without cysteine ‘scars’.
Cysteines are relatively rare in polypeptide chains and are usually essential for protein function. Thus, the outstanding challenge for the protein chemist is to find new approaches and reaction conditions that target only the desired residues in a polypeptide chain. Many of these efforts are focused on the search of suitable peptide sequences that can provide the necessary amino acid context for tuning side-chain reactivity. For example, Tsien and co-workers have developed tetra-cysteine motifs that can be selectively targeted with biarsenic reagents, including in living cells (Griffin et al. Reference Griffin, Adams and Tsien1998). Pentelute and co-workers, on the other hand, have reported the so-called ‘π-clamp’ sequence (Phe–Pro–Cys–Phe) that reacts preferentially with perfluoroaromatic moieties in aqueous solvents (Zhang et al. Reference Zhang, Welborn, Zhu, Yang, Santos, Van Voorhis and Pentelute2016a). Excellent selectivity can be obtained by replacing cysteine with selenocysteine (reviewed in (Metanis et al. Reference Metanis, Beld, Hilvert and Rappoport2009; Yoshizawa & Bock, Reference Yoshizawa and Bock2009), a rare natural amino acid that shares many of the desirable properties of the cysteine side-chain, yet is more acidic and substantially more reactive at lower pH (selenol pKa is ~5·2 versus 8·3 for thiols).
3.2 Chemical modification of other amino acids
In addition to cysteine, several other natural amino acids present functional groups that can be targeted for protein modification (reviewed in (Basle et al. Reference Basle, Joubert and Pucheault2010; Spicer & Davis, Reference Spicer and Davis2014)). These include lysine, tyrosine, arginine, glutamate, aspartate, serine, threonine, methionine, histidine and tryptophan side-chains, as well as N-terminal amines or C-terminal carboxyls (Basle et al. Reference Basle, Joubert and Pucheault2010; Hu et al. Reference Hu, Berti and Adamo2016; Lin et al. Reference Lin, Yang, Jia, Weeks, Hornsby, LEE, Nichiporuk, Iavarone, Wells, Toste and Chang2017). The modification of these residues is less precise as they are significantly more abundant than cysteine, yet, some selectivity can be achieved in a context-dependent manner. The primary amines of lysine side-chains are often popular targets due to favorable reaction kinetics that can be achieved with activated esters such as N-hydroxysuccinimide (Kalkhof & Sinz, Reference Kalkhof and Sinz2008) (Fig. 3a ), isothiocyanates (Nakamura et al. Reference Nakamura, Kawai, Kitamoto, Osawa and Kato2009), or aldehydes in a reductive alkylation reaction with sodium cyanoborohydride (Jentoft & Dearborn, Reference Jentoft and Dearborn1979; McFarland & Francis, Reference Mcfarland and Francis2005). Since these reactions usually modify all accessible lysine side-chains, they can be used in applications that require multiple modifications (e.g. therapeutic protein conjugates) or in protein cross-linking for mass spectrometry analysis (Holding, Reference Holding2015). An example of a more discriminating lysine-based modification strategy involves the 6π-aza-electrocyclization reaction with unsaturated aldehyde esters that targets solvent-accessible lysine residues with excellent selectivity and reaction kinetics, and has been used for the attachment of fluorescent or positron emission tomography probes (Tanaka et al. Reference Tanaka, Kitadani and Fukase2011). Yet, further selectivity can be achieved by discriminating the lower pKa of N-terminal amino groups (~8) from the pKa of the ε-amine of a lysine side-chain (~10·5). The identity of the N-terminal amino acid may further change the reactivity of the α-amine, although more general modification strategies have been developed. For example, functionalized ketenes can preferentially react with the α-amine in the context of 13 different N-terminal amino acids (Chan et al. Reference Chan, Ho, Chong, Leung, Huang, Wong and Che2012), while 2-pyridinecarboxyaldehydes provide efficient and specific N-terminal labeling for all amino acids except proline (MacDonald et al. Reference Macdonald, Munch, Moore and Francis2015) (Fig. 3b ). The unique reactivity of the N-terminus is also central to the mechanism of native chemical ligation, discussed in Section 3.3.

Fig. 3. Chemical modification of natural amino acids. (a) Modification of lysine ε-amines with activated esters such as N-hydroxysuccinimide. (b) Modification of terminal α-amines with 2-pyridinecarboxyaldehydes. (c) Three-component Mannich reaction for tyrosine modification at the ortho-position. (d) Coupling of carboxyls and amines with carbodiimides such as EDC.
A set of chemical reactions has also been developed for the specific modification of the aromatic electron-rich tyrosine side-chain. The selectivity of tyrosine-focused reactions usually exploits the low exposure of this residue on native protein surfaces and its susceptibility to oxidation (ElSohly & Francis, Reference Elsohly and Francis2015; Seim et al. Reference Seim, Obermeyer and Francis2011). In particular, modification of the ortho-position can be achieved with diazonium salts (Schlick et al. Reference Schlick, Ding, Kovacs and Francis2005) or the three-component Mannich-type reaction with aldehydes and anilines (Joshi et al. Reference Joshi, Whitaker and Francis2004; McFarland et al. Reference Mcfarland, Joshi and Francis2008) (Fig. 3c ). While some side-reactions have been known to occur, it has been possible to optimize the selectivity of the Mannich-type reaction to achieve efficient modification of a single tyrosine residue with an EPR spin label in the presence of disulfide and tryptophan functionalities (Mileo et al. Reference Mileo, Etienne, Martinho, Lebrun, Roubaud, Tordo, Gontero, Guigliarelli, Marque and Belle2013). Proteins containing few surface exposed tyrosine residues can also be modified at low concentrations (5 µM) in aqueous solvents with π-allylpalladium reagents (Tilley & Francis, Reference Tilley and Francis2006).
Carboxyl groups in glutamate and aspartate side-chains can be targeted with water-soluble carbodiimides such as N-ethyl-3-N′-N′-dimethylaminopropylcarbodiimide (EDC) (Fig. 3 d). In what is essentially a standard peptide coupling reaction, this reagent pairs carboxyls and amines in a covalent amide bond under aqueous conditions in a pH-dependent manner (Gilles et al. Reference Gilles, Hudson and Borders1990). Commonly employed as a cross-linking reagent for the identification of protein–protein interactions in biochemical assays and mass spectrometry, EDC can also be used to modify protein assemblies such as viral capsids with a variety of functional and biophysical probes (Schlick et al. Reference Schlick, Ding, Kovacs and Francis2005). While EDC-based reactions do not discriminate between side-chain and C-terminal carboxyl groups, unique reactivity at the C-terminus can be generated by replacing the carboxyl functionality with a thioester, as discussed in the sections below.
3.3 Native chemical ligation
The unique reactivity of the cysteine side-chain and the positional control afforded by the protein N- and C-termini are at the heart of a simple, yet powerful, chemical reaction that allows the construction of native polypeptide chains from peptide-building blocks. This methodology, called NCL (Dawson et al. Reference Dawson, Muir, Clark-Lewis and Kent1994), grants unprecedented control over the chemical functionalities that can be introduced in a polypeptide chain and is a fundamental tool in the repertoire of protein chemists. NCL simply requires two components with the following properties: (1) a peptide with a C-terminal thioester, and (2) a peptide with an N-terminal cysteine residue (or functional equivalent). When these components are mixed, the cysteine side-chain attacks the thioester of the other peptide, resulting in the formation of an intermolecular thioester (Fig. 4). This intermediate rapidly and irreversibly rearranges to form a native peptide bond, thus ligating the two fragments. Since both peptides can be produced by solid-phase peptide synthesis, virtually any modified natural or unnatural amino acid, spectroscopic probe, cross-linker or isotopic label can be incorporated at a well-defined position into the ligated sequence (Dawson & Kent, Reference Dawson and Kent2000). Moreover, in contrast to biosynthetic approaches such as amber suppression (see Section 3.6), there is no practical restriction on the number, or type, of unnatural amino acids that can be introduced – this point is driven home by the total synthesis of enantiomeric proteins composed of all D-amino acids (Mandal et al. Reference Mandal, Uppalapati, Ault-Riche, Kenney, Lowitz, Sidhu and Kent2012). The remarkable chemoselectivity of the NCL reaction means that ligations can be performed under aqueous conditions in the presence of internal cysteine residues. NCL is also compatible with protein denaturants or detergents allowing the construction of aggregation prone polypeptide chains or even polytopic membrane proteins (Hejjaoui et al. Reference Hejjaoui, Butterfield, Fauvet, Vercruysse, Cui, Dikiy, Prudent, Olschewski, ZHANG, Eliezer and Lashuel2012; Kwon et al. Reference Kwon, Tietze, White, Liao and Hong2015; Valiyaveetil et al. Reference Valiyaveetil, Leonetti, Muir and Mackinnon2006). The fragment bearing the N-terminal cysteine can also be produced recombinantly, usually preceded by a cleavable tag or fusion protein to avoid N-terminal cysteine processing complications in bacteria. Alternatively, the thioester component can be generated recombinantly with the help of proteins called inteins (Muir et al. Reference Muir, Sondhi and Cole1998) (see Section 3.4). It is also possible to perform sequential NCL reactions, allowing three or more building blocks to be assembled in a regioselective fashion (Mandal et al. Reference Mandal, Uppalapati, Ault-Riche, Kenney, Lowitz, Sidhu and Kent2012; Torbeev & Hilvert, Reference Torbeev and Hilvert2013). These advances thus allow the construction of considerably larger polypeptide chains that would be accessible from two synthetic peptides alone.

Fig. 4. Native chemical ligation at cysteine followed by desulfurization to alanine for the construction of larger polypeptide chains without any ‘scars’.
While NCL results in a native peptide bond, the required cysteine may still produce an unwanted ‘scar’ at the ligation junction – i.e. in cases where the target does not contain a native cysteine at an appropriate ligation point. If the final ligation product does not contain other cysteine residues, then the undesirable cysteine can be converted to alanine in a subsequent desulfurization step. Options for desulfurization reactions include reduction with metals or radical desulfurization mediated by the familiar tris(2-carboxyethyl)phosphine (TCEP) reagent (Dawson, Reference Dawson2011; Wan & Danishefsky, Reference Wan and Danishefsky2007; Yan & Dawson, Reference Yan and Dawson2001). Selective desulfurization reactions are also possible in the presence of other cysteine residues, provided that compatible protecting groups are used on the non-targeted cysteine side-chains (Ficht et al. Reference Ficht, Payne, Brik and Wong2007; Pentelute & Kent, Reference Pentelute and Kent2007). Alternatively, selective ligation and desulfurization of selenocysteine can provide additional sequence positional control (Hondal et al. Reference Hondal, Nilsson and Raines2001; Reddy et al. Reference Reddy, Dery and Metanis2016). Today, desulfurization has become a routine part of NCL protocols and the abundant alanine residue is a commonplace choice for a ligation site. To expand the junction amino acid set, a more advanced strategy involves the incorporation of β- and γ-thio amino acids. These moieties replace the cysteine-like residue at the N-terminal position, and provide a reactive thiol for trans-thioesterification. Desulfurization protocols can then produce the desired native side-chain. While ligations at such sites proceed more slowly due to increased steric hindrance, NCL can now be performed at phenylalanine (Crich & Banerjee, Reference Crich and Banerjee2007), valine (Chen et al. Reference Chen, Wan, Yuan, Zhu and Danishefsky2008; Haase et al. Reference Haase, Rohde and Seitz2008), leucine (Harpaz et al. Reference Harpaz, Siman, Kumar and Brik2010; Tan et al. Reference Tan, Shang and Danishefsky2010), threonine (Chen et al. Reference Chen, Wang, Zhu, Wan and Danishefsky2010b), lysine (El Oualid et al. Reference El Oualid, Merkx, Ekkebus, Hameed, Smit, De Jong, Hilkmann, Sixma and Ovaa2010; Kumar et al. Reference Kumar, Haj-Yahya, Olschewski, Lashuel and Brik2009; Yang et al. Reference Yang, Pasunooti, Li, Liu and Liu2009), proline (Shang et al. Reference Shang, Tan, Dong and Danishefsky2011), glutamine (Siman et al. Reference Siman, Karthikeyan and Brik2012), arginine (Malins et al. Reference Malins, Cergol and Payne2013), tryptophan (Malins et al. Reference Malins, Cergol and Payne2014), aspartate (Thompson et al. Reference Thompson, Chan, Radom, Jolliffe and Payne2013), glutamate (Cergol et al. Reference Cergol, Thompson, Malins, Turner and Payne2014) and asparagine (Sayers et al. Reference Sayers, Thompson, Perry, Malins and Payne2015). Work also continues on more streamlined ligation/desulfurization approaches that remove purification steps and increase the yield of ligated products (Moyal et al. Reference Moyal, Hemantha, Siman, Refua and Brik2013; Thompson et al. Reference Thompson, Liu, Alonso-Garcia, Pereira, Jolliffe and Payne2014).
The preceding discussion highlights just a few of the many refinements and extensions the NCL strategy has undergone since its introduction over 20 years ago (Harmand et al. Reference Harmand, Murar and Bode2014; Malins & Payne, Reference Malins and Payne2015). As a consequence of this massive effort, the technique has become a central tool in protein science, having been applied to literally hundreds of protein targets. Of particular relevance here, it has provided the raw materials for numerous structural biology studies that have employed a broad range of spectroscopic or crystallographic methods (Fig. 5) (Grosse et al. Reference Grosse, Essen and Koert2011; Kent et al. Reference Kent, Sohma, Liu, Bang, Pentelute and Mandal2012; Muralidharan & Muir, Reference Muralidharan and Muir2006). In general, the ability to modify any atom in the protein of interest with the precision afforded by synthetic organic chemistry is enormously powerful for dissecting protein function, especially when combined with high-resolution structural approaches such as NMR spectroscopy and X-ray crystallography. Thus, we imagine that NCL will continue to evolve as an approach and be integrated into structural biology campaigns.

Fig. 5. Examples of constructs prepared by NCL and EPL for X-ray crystallography studies. (a) D-alanine was introduced at position 77 in the sequence of the potassium channel KcsA to elucidate its ion selectivity mechanism (Valiyaveetil et al. Reference Valiyaveetil, Leonetti, Muir and Mackinnon2006) (PDB ID: 2IH3). (b) Acetylated lysine (Ac) was incorporated at postions 401 and 408 in S-Adenosylhomocysteine hydrolase (SAHH) to evaluate the structural basis of enzyme inhibition (Wang et al. Reference Wang, Kavran, Chen, Karukurichi, Leahy and Cole2014b) (PDB ID: 4PFJ). (c) Chemical synthesis of HIV protease afforded the site-specific incorporation of unnatural amino acids such as 2-aminoisobutyric acid to modulate conformational dynamics and catalysis (Torbeev et al. Reference Torbeev, Raghuraman, Hamelberg, Tonelli, Westler, Perozo and Kent2011) (PDB ID: 3IAW). (d) Semi-synthesis of Mxe GyrA and the installation of β-thienyl-alanine instead of the native histidine at position 187 provided a route to trap the branched intermediate of the intein (Liu et al. Reference Liu, Frutos, Bick, Vila-Perello, Debelouchina, Darst and Muir2014b) (PDB ID: 4OZ6).
3.4 Inteins
Inteins (intervening proteins) are a peculiar group of proteins that can excise themselves from a larger precursor polypeptide chain, a process that leads to the formation of a native peptide bond between the flanking extein (external protein) fragments. This auto-processing event, called protein splicing, is analogous to the self-splicing of RNA introns and is spontaneous, i.e. it does not require external factors or ATP. Since they were first discovered in the early 1990s (Hirata et al. Reference Hirata, Ohsumk, Nakano, Kawasaki, Suzuki and Anraku1990; Kane et al. Reference Kane, Yamashiro, Wolczyk, Neff, Goebl and Stevens1990), thousands of putative intein domains have been identified in the genomes of many unicellular organisms and viruses, with some containing multiple inteins in their genomes or even within the same gene (Perler, Reference Perler2002; Shah & Muir, Reference Shah and Muir2014). A small fraction of the known inteins has an even more curious property – the intein is split into two fragments, each fused to a separate extein fragment (the N- and C-exteins) (Wu et al. Reference Wu, Hu and Liu1998). These intein fragments, called split inteins, are transcribed and translated separately, and upon a spontaneous and non-covalent association in the cellular milieu, they carry out protein splicing in trans to unite the extein fragments into a single polypeptide chain. While it is known that many inteins are embedded within essential protein genes (such as DNA or RNA polymerase, ribonucleotide reductase or metabolic enzymes), their evolutionary origins and biological significance remain mysterious, and only a small percentage of the identified intein domains have been carefully characterized (Pietrokovski, Reference Pietrokovski2001; Shah & Muir, Reference Shah and Muir2014). Despite these big gaps in our knowledge, the unique reactivity of inteins has turned them into a versatile and transformative tool in protein chemistry and chemical biology. For a detailed overview of intein applications, we refer the interested reader elsewhere (Shah & Muir, Reference Shah and Muir2014; Topilina & Mills, Reference Topilina and Mills2014; Volkmann & Mootz, Reference Volkmann and Mootz2013; Wood & Camarero, Reference Wood and Camarero2014). Here, we will focus on aspects of intein function that would be of use to the structural biologist looking to install site-specific PTMs, segmentally label proteins with NMR isotopes, or aid the purification of recombinant polypeptides. Inteins have come a long way since their first applications in structural biology (Xu et al. Reference Xu, Ayers, Cowburn and Muir1999; Yamazaki et al. Reference Yamazaki, Otomo, Oda, Kyogoku, Uegaki, Ito, Ishino and Nakamura1998), so we will end this section with a discussion of the current members of the intein toolbox and research directions taken to circumvent their limitations.
3.4.1 The intein splicing mechanism
Despite the low-sequence homology of known intein domains, they share a common protein splicing mechanism that relies on several conserved residues in the intein/extein polypeptide (Fig. 6a ) (Volkmann & Mootz, Reference Volkmann and Mootz2013). One of these key residues is a cysteine (or in some cases a serine) at position 1 of the intein sequence. This nucleophilic side-chain attacks the amide carbon of the N-extein at position −1 (Fig. 6b ) resulting in an N to S(O) acyl shift and the formation of a linear thio(oxy)ester intermediate. This intermediate is subject to a nucleophilic attack by a side-chain (cysteine, serine or threonine) at position +1 on the C-extein leading to trans-(thio)esterification and the generation of a branched intermediate. The branched intermediate is resolved through the cyclization of the C-terminal asparagine of the intein and results in intein excision from the polypeptide chain. Next, the spliced exteins quickly undergo an S(O) to N acyl shift to form a native peptide bond (i.e. identical to the last step in NCL). The protein splicing mechanism is facilitated by several conserved threonine and histidine residues occupying strategic positions in the intein structural fold (Frutos et al. Reference Frutos, Goger, Giovani, Cowburn and Muir2010). The efficiency and kinetics of the splicing mechanism may also depend on the identity of the residues immediately flanking the intein placing important constraints on the choice of ligation junction (Cheriyan et al. Reference Cheriyan, Pedamallu, Tori and Perler2013; Iwai et al. Reference Iwai, zuger, Jin and Tam2006; Shah et al. Reference Shah, Dann, Vila-Perello, Liu and Muir2012).

Fig. 6. Intein structure and mechanism. (a) Intein/extein residues important for splicing. (b) Protein splicing mechanism of contiguous inteins. In some cases, the hydroxyl groups of Ser/Thr act as nucleophiles in the first two steps.
3.4.2 Applications in protein engineering
Expressed protein ligation (EPL) is an extension of NCL that employs a contiguous intein to recombinantly generate a protein bearing a C-terminal thioester (Muir et al. Reference Muir, Sondhi and Cole1998). In this case, the N-extein is fused to a modified intein construct lacking the ability to perform trans-thioesterification. Instead, this step is performed by an exogenously added thiol, resulting in cleavage of the N-extein α-thioester intermediate (Fig. 7a ). The resultant thioester can be used in NCL reactions as described in Section 3.3, while the recombinant origin of this fragment allows the construction of much larger semi-synthetic proteins as compared to total chemical synthesis by NCL. The efficiency of thioester generation rests on the propensity of the intein to avoid unwanted side reactions that result in premature N-extein cleavage and hydrolyzed products, problems that can be alleviated by the use of streamlined EPL protocols (Vila-Perello et al. Reference Vila-Perello, Liu, Shah, Willis, Idoyaga and Muir2013). Alternatively, hydrolysis of the N-extein under slightly basic conditions can be exploited in the so-called tagless protein purification protocols (Batjargal et al. Reference Batjargal, Walters and Petersson2015; Guan et al. Reference Guan, Ramirez and Chen2013; Southworth et al. Reference Southworth, Amaya, Evans, Xu and Perler1999) (Fig. 7b ). In this case, the protein of interest is fused to an intein carrying the appropriate mutations and a suitable purification tag. The tag can be used for affinity column enrichment of the construct, followed by increase in the buffer pH. This results in the release of the tagless protein while the intein remains on the column.

Fig. 7. Protein engineering with inteins. (a) Expressed protein ligation. (b) Tagless protein purification. (c) Protein trans-splicing and recombinant production of segmentally isotopically labeled proteins.
Harnessing the protein trans-splicing (PTS) process mediated by split inteins offers an alternative approach to the ligation of polypeptide building blocks (Fig. 7c ). Natural split inteins are especially attractive in this regard due to the extremely high affinity between the fragments (Shah et al. Reference Shah, Vila-Perello and Muir2011, Reference Shah, Eryilmaz, Cowburn and Muir2013) – this renders the ligation reaction less dependent on reagent concentration as compared to strictly chemical processes like NCL/EPL. Using orthogonal split intein pairs, it is also possible to perform one-pot three-piece ligations (Carvajal-Vallejos et al. Reference Carvajal-Vallejos, Pallisse, Mootz and Schmidt2012; Shah et al. Reference Shah, Vila-Perello and Muir2011; Shi & Muir, Reference Shi and Muir2005) resulting in the regiospecific assembly of the associated extein building blocks. While most natural split inteins have N- and C-fragments that are relatively large, it is possible to generate artificially split inteins that are as short as six or eleven residues (Appleby et al. Reference Appleby, Zhou, Volkmann and Liu2009; Ludwig et al. Reference Ludwig, Pfeiff, Linne and Mootz2006). There is also an efficient natural split intein pair (AceL–TerL) where the N-intein fragment is only 25 amino acids long (Thiel et al. Reference Thiel, Volkmann, Pietrokovski and Mootz2014). Thus, it is now possible to use PTS with both synthetic or recombinant intein fragments and to install a wide range of N- or C-terminal chemical modification, including biophysical probes. One of the most important applications of PTS in structural biology is protein segmental isotopic labeling discussed in Section 4.2. Other applications include the cyclization of proteins and peptides (Lennard & Tavassoli, Reference Lennard and Tavassoli2014; Scott et al. Reference Scott, Abel-santos, Wall, Wahnon and Benkovic1999), conditional protein splicing (Mootz et al. Reference Mootz, Blum, Tyszkiewicz and Muir2003; Schwartz et al. Reference Schwartz, Saez, Young and Muir2007), and protein semi-synthesis in cells (David et al. Reference David, Vila-Perello, Verma and Muir2015).
3.4.3 Toward fast and promiscuous inteins
The first intein tools were introduced in the mid-to-late 1990s. One of the ‘early’ inteins, the 198-residue gyrase A intein from Mycobacterium xenopi (Mxe GyrA) (Southworth et al. Reference Southworth, Amaya, Evans, Xu and Perler1999), still used today, exhibits many desirable properties for applications in protein engineering – it is relatively small, can be efficiently expressed in E. coli, works in moderate concentrations of denaturants and its activity can be controlled with temperature. The first ‘natural’ split intein was discovered in 1998 in the cyanobacterium Synechocystis sp. strain PCC6803 (Ssp) where it was found to ligate two fragments of the catalytic subunit of DNA polymerase III (DnaE) (Wu et al. Reference Wu, Hu and Liu1998). This discovery paved the road to more efficient protein trans-splicing and opened the way to using split inteins in other applications such as the cyclization of proteins and peptides and the creation of large cyclized libraries for potential therapeutic applications (Scott et al. Reference Scott, Abel-santos, Wall, Wahnon and Benkovic1999).
While these first generation intein tools were certainly enabling (Vila-Perello & Muir, Reference Vila-Perello and Muir2010), they were not without their limitations – in retrospect, one could even say they were rather fussy and slow. For example, depending on the fusion partners, splicing (or thiolysis) could take hours to days and was often inefficient (Muralidharan & Muir, Reference Muralidharan and Muir2006). These ‘idiosyncrasies’ constrained the application of inteins in structural biology and fueled the search for faster, more promiscuous and efficient inteins. Several important discoveries in the mid-2000s challenged the view that all natural inteins are inefficient and slow. A genomic study of cyanobacterial genes expanded the DnaE intein family (Caspi et al. Reference Caspi, Amitai, Belenkiy and Pietrokovski2003) and the characterization of one newly discovered member, the DnaE intein from Nostoc punctiforme (Npu) revealed a few surprises. This split intein could perform protein trans-splicing reactions in vitro on a minute timescale and was much more tolerant to sequence deviations on the attached exteins than Ssp ((Iwai et al. Reference Iwai, zuger, Jin and Tam2006; Zettler et al. Reference Zettler, Schutz and Mootz2009). Now we know that many members of the DnaE family are fast (Shah et al. Reference Shah, Dann, Vila-Perello, Liu and Muir2012), thus greatly expanding the choice of natural intein tools for the efficient generation of protein α-thioesters for EPL or the ligation of protein fragments in PTS (Table 1). Furthermore, efficient natural split inteins that are not part of the DnaE family have also been discovered and these include the gp41-1 and gp41-8 inteins (with insertion sites in the gp41 DNA gyrase gene), the IMPDH-1 intein (splitting a gene coding for inosine-5′-monophosphate dehydrogenase), the NrdJ intein (splitting the gene coding for the ribonucleotide reductase subunit NrdJ) (Carvajal-Vallejos et al. Reference Carvajal-Vallejos, Pallisse, Mootz and Schmidt2012), and the AceL–TerL pair (discovered in metagenomics data from the antarctic permanently stratified saline Ace Lake) (Thiel et al. Reference Thiel, Volkmann, Pietrokovski and Mootz2014). These proteins bring more diversity to the intein molecular engineering toolbox, including splicing rates that are an order of magnitude faster than those for NpuDnaE; N- and C-intein fragments that are relatively short and can be made by peptide synthesis rather than recombinantly; the option of utilizing serine instead of cysteine at the +1 position; and the possibility of exploiting orthogonality in one-pot multi-piece ligations.
Table 1. Intein toolbox for protein semi-synthesis

* Optimal splicing kinetics in the presence of native sequences at the immediate intein-extein junctions. Variation from this sequence context can lead to less efficient splicing. See also ref. (Shah & Muir, Reference Shah and Muir2014).
Careful biochemical characterization of newly discovered inteins has provided insights into the principles governing fast splicing and extein tolerance. For example, a batch mutagenesis approach that compared the slow split intein DnaE family member Ssp and the fast Npu split intein revealed that speed is determined by a handful of ‘accelerator’ residues located in the second shell of the folded protein, adjacent to the intein active site (Stevens et al. Reference Stevens, Brown, Shah, Sekar, Cowburn and Muir2016). These residues were used as a filter in an informatics analysis of the DnaE sequence database, leading to the identification of several dozens of other split inteins predicted to support ultrafast splicing. A consensus split intein sequence, termed Cfa, was then derived from this putative fast set and was found to possess quite remarkable properties; in addition to splicing faster than Npu at ambient conditions, Cfa is extremely robust, maintaining efficient activity at 80 °C or in the presence of up to 4 M guanidinium chloride or 8 M urea. As a result of these attributes, Cfa was found to be a superior tool for several PTS applications (Stevens et al. Reference Stevens, Brown, Shah, Sekar, Cowburn and Muir2016).
3.5 Sortases
Sortases are a class of cysteine transpeptidases responsible for the attachment of virulence proteins to the cell wall of Gram-positive bacteria (Mazmanian et al. Reference Mazmanian, Liu, Ton-That and Schneewind1999). They are also involved in the polymerization of pilin subunits to form the pilus structures responsible for bacterial attachment to the host and biofilm formation (Mandlik et al. Reference Mandlik, Swierczynski, Das and Ton-That2008). As important players in bacterial virulence, they have evolved to recognize a specific sorting sequence (LPXTG in the case of Staphylococcus aureus) and to attach the virulence factor to the cell wall using a pentaglycine cross-bridge (Ton-That et al. Reference Ton-That, Mazmanian, Faull and Schneewind2000). Naturally, sortases are of a considerable interest as drug targets, but they have also become an important and versatile protein engineering tool (Mao et al. Reference Mao, Hart, Schink and Pollok2004). Most sortase-based applications utilize the soluble fragment of wild-type or modified sortase A from S. aureus. These enzymes recognize the LPXTG motif and use their catalytic cysteine residue to cleave between the threonine and glycine backbone within the recognition sequence (Fig. 8). The cleavage reaction involves a thioacyl intermediate similar to the intermediates generated by cysteine proteases (Aulabaugh et al. Reference Aulabaugh, Ding, Kapoor, Tabei, Alksne, Dushin, Zatz, Ellestad and Huang2007). Unlike the water molecules employed by proteases, however, sortases use a nucleophilic attack from the N-terminus of an oligoglycine motif to create a peptide bond between the acyl donor and acceptor. This results in the ligation of polypeptide chains that are subsequently connected with a LPXT(G)5 linker. The sortase mechanism also requires binding of Ca2+ to a dynamic loop of the enzyme, an event that slows down the loop motion and allows enough time for the substrate to find the catalytic site (Naik et al. Reference Naik, Suree, Ilangovan, Liew, Thieu, Campbell, Clemens, Jung and Clubb2006). Another peculiarity of sortase-based ligations is their reversibility: the generated ligation site has the recognition sequence LPXTG and can serve as an acyl donor, while the released fragment contains an aminoglycine acyl receptor. Thus, to obtain efficient ligations, the donor or acceptor polypeptide chain typically has to be added in large excess (Guimaraes et al. Reference Guimaraes, Witte, Theile, Bozkurt, Kundrat, Blom and Ploegh2013).

Fig. 8. C-terminal protein labeling with sortase. The acyl donor requires the LPXTG recognition motif, while the acyl acceptor often contains a pentaglycine sequence.
Analogous to intein technology development, sortases have considerably improved as protein engineering tools since their introduction in 2004 (Antos et al. Reference Antos, Truttmann and Ploegh2016). The sortase-based protein engineering toolbox now contains evolved variants that exhibit much faster kinetics or that eliminate Ca2+-dependence (albeit at the cost of slightly reduced enzyme activity) (Chen et al. Reference Chen, Dorr and Liu2011; Hirakawa et al. Reference Hirakawa, Ishikawa and Nagamune2015; Wuethrich et al. Reference Wuethrich, Peeters, Blom, Theile, Li, Spooner, Ploegh and Guimaraes2014). There are also alternatives based on S. aureus sortase A or homologs from other organisms that can recognize variations of the LPXTG motif and/or allow non-glycine amino acids as the acyl acceptor (Antos et al. Reference Antos, Truttmann and Ploegh2016; Dorr et al. Reference Dorr, Ham, An, Chaikof and Liu2014; Glasgow et al. Reference Glasgow, Salit and Cochran2016). To increase the yields of the ligation reaction, several clever strategies have been employed. In situations where the released aminoglycine peptide fragment is relatively small, it can be removed by dialysis or centrifugation while the reaction is proceeding (Freiburger et al. Reference Freiburger, Sonntag, Hennig, Li, Zou and Sattler2015). Affinity immobilization strategies or flow-based platforms have also been used for the selective removal of reaction components (Policarpo et al. Reference Policarpo, Kang, Liao, Rabideau, Simon and Pentelute2014; Warden-Rothman et al. Reference Warden-Rothman, Caturegli, Popik and Tsourkas2013). Alternatively, the equilibrium of the reaction can be controlled by ligation product or by-product deactivation. In the first case, a WTWTW motif was added to the donor and acceptor, and upon ligation this sequence promoted a stable hairpin at the ligation junction, rendering the site inaccessible for cleavage (Yamamura et al. Reference Yamamura, Hirakawa, Yamaguchi and Nagamune2011). In the latter case, the acyl donor glycine was chemically modified such that upon release, chemical rearrangements occurred on the by-product transforming it into a poor nucleophile (Liu et al. Reference Liu, Luo, Flora and Mezo2014a; Williamson et al. Reference Williamson, Webb and Turnbull2014).
One important advantage of sortase-based ligations is that the acyl donor and acceptor polypeptide chains can be very short (only the LPXTG tag is required on the donor, and the oligoglycine motif is necessary on the acceptor) and thus are easily accessible by solid-phase peptide synthesis. Therefore, N- and C-terminal labeling reactions of large proteins are relatively straightforward (assuming by-products are efficiently removed) (Guimaraes et al. Reference Guimaraes, Witte, Theile, Bozkurt, Kundrat, Blom and Ploegh2013; Theile et al. Reference Theile, Witte, Blom, Kundrat, Ploegh and Guimaraes2013). Larger polypeptides, on the other hand, can be expressed recombinantly with the appropriate donor and acceptor tags and the ligation reaction unites them in a single polypeptide chain with an LPXT(G) n ‘scar’. In such cases it is recommended that the ligation junction is chosen on an unstructured region where it will not affect the function and/or fold of the protein and will be accessible to the sortase catalytic site (Guimaraes et al. Reference Guimaraes, Witte, Theile, Bozkurt, Kundrat, Blom and Ploegh2013). Such reactions can be applied to create polymers and cyclized polypeptides (van ‘t Hof et al. Reference Van ‘T Hof, Hansenova Manaskova, Veerman and Bolscher2015), or to stitch together domains into bifunctional or segmentally labeled proteins (Matsumoto et al. Reference Matsumoto, Furuta, Tanaka and Kondo2016; Williams et al. Reference Williams, Milbradt, Embrey and Bobby2016; Witte et al. Reference Witte, Cragnolini, Dougan, Yoder, Popp and Ploegh2012). Sortase-based labeling has also been used in the functionalization of solid supports, nanoparticles, antibodies or cell surfaces, as well as in the labeling of proteins in vivo. We refer the interested reader to several comprehensive reviews on the subject (Popp & Ploegh, Reference Popp and Ploegh2011; Ritzefeld, Reference Ritzefeld2014; Schmohl & Schwarzer, Reference Schmohl and Schwarzer2014).
The success of sortase-based ligations has stimulated efforts to discover other protein ligases with expanded capabilities. A promising candidate is butelase-1, which was isolated from the plant Clitoria ternatea (Nguyen et al. Reference Nguyen, Wang, Qiu, Hemu, Lian and Tam2014). Butelase-1 is the fastest ligase known with catalytic efficiencies as high as 542 000 M−1 s−1. Furthermore, it only requires the recognition sequence NHV on the acyl donor and produces ligation junctions with a minimal ‘scar’ (NX). Currently, the major limitation of this technology is that the enzyme is not available in recombinant form, and therefore has to be extracted and purified from the native plant (Nguyen et al. Reference Nguyen, Kam, Loo, Jansson, Pan and Tam2015). There are, however, evolutionary related ligases that may be more amenable to protein engineering approaches (Yang et al. Reference Yang, Wong, Nguyen, Tam, Lescar and Wu2017).
3.6 Genetic code expansion
After billions of years of evolution, Nature has engineered extraordinary functional diversity into proteins with only 20 amino acid building blocks. Yet, many of these building blocks are often modified post-translationally, clearly indicating the need of living organisms to enhance and modulate their protein repertoire with additional chemical functionalities. There are also organisms from all domains of life that can produce and incorporate other building blocks into their proteins. This includes selenocysteine, often called the 21st amino acid that provides a unique reactive site for precise tuning of biological function in cells. Interestingly, this amino acid is incorporated into proteins by a natural reassignment of the UGA stop codon coupled with the recognition of a specific structural element on the mRNA transcript known as the selenocysteine-insertion sequence (reviewed in (Metanis et al. Reference Metanis, Beld, Hilvert and Rappoport2009; Yoshizawa & Bock, Reference Yoshizawa and Bock2009)). Similarly, there are methane producing Archaea species that have evolved a specialized tRNA/aminoacyl-tRNA synthetase (tRNA/aaRS) pair to exploit the UAG stop codon and insert pyrrolysine site-specifically into certain methyltransferase proteins (Srinivasan et al. Reference Srinivasan, James and Krzycki2002). Exploiting the natural translation machinery, protein engineers have worked hard to ‘persuade’ living organisms to incorporate additional building blocks into polypeptide chains (reviewed in (Liu & Schultz, Reference Liu and Schultz2010)). One approach involves the use of cell lines that are auxotrophic for one of the 20 amino acids, for example methionine, and that will only grow when the missing amino acid is included in the culture medium. Replacing this amino acid with a close structural analog that is utilized by the wild-type aminoacyl-tRNA synthetase, results in incorporation of the unnatural amino acid (UAA) into overexpressed proteins. Since this results in global incorporation of the UAA, this approach is often used for the replacement of rare amino acids with their structural analogs. For example, methionine can be substituted with selenomethionine to provide a heavy atom for phasing crystallographic data (Barton et al. Reference Barton, Tzvetkova-Robev, Erdjument-Bromage, Tempst and Nikolov2006; Yang et al. Reference Yang, Hendrickson, Crouch and Satow1990).
A second approach involves the semi-synthesis of tRNAs that are pre-loaded with the UAA of interest (Hecht et al. Reference Hecht, Alford, Kuroda and Kitano1978; Noren et al. Reference Noren, Anthony-cahill, Griffith and Schultz1989). These tRNAs have been used in in vitro translation systems that bypass the need for a matching aaRS, and since the identity of the UAA is decoupled from the information content of the tRNA, any coding or blank codon can be used for reassignment (Cornish et al. Reference Cornish, Benson, Altenbach, Hideg, Hubbell and Schultz1994; Judice et al. Reference Judice, Gamble, Murphy, De Vos and Schultz1993; Koh et al. Reference Koh, Cornish and Schultz1997). While the semi-synthesis of acylated tRNAs can be technically challenging, more efficient production strategies have been developed. This includes flexizymes, flexible tRNA acylation ribozymes that accept a versatile range of aminoacyl substrates and tRNAs with different sequences (Goto et al. Reference Goto, Katoh and SUGA2011). Pre-loaded tRNAs can also be injected or transfected directly into living cells (England et al. Reference England, Zhang, Dougherty and Lester1999; Kohrer et al. Reference Kohrer, Yoo, Bennett, Schaack and Rajbhandary2003), although the success of such strategies has been limited by their short lifetimes in the cellular environment and challenges associated with in-cell delivery.
Today, UAA incorporation in living cells is almost exclusively performed following the strategy introduced by Peter Schultz and co-workers in 2001 (Wang et al. Reference Wang, Brock, Herberich and Schultz2001). This methodology, commonly referred to as amber suppression, relied on the development of an orthogonal tRNA/aaRS pair that could be expressed in E. coli and was used to incorporate O-methyl-L-tyrosine into dihydrofolate reductase with 99% fidelity. In the 15 years since this landmark study, the unnatural building block palette for genetic incorporation has grown more than 100 amino acids strong (Lang & Chin, Reference Lang and Chin2014; Liu & Schultz, Reference Liu and Schultz2010; Neumann-Staubitz & Neumann, Reference Neumann-Staubitz and Neumann2016). This includes amino acids carrying natural modifications (e.g. phosphoserine or acetyllysine), biophysical and structural probes, cross-linkers, reactive handles for bio-orthogonal reactions, and site-specific protein engineering functionalities that can modify their attendant proteins upon a specific cellular or chemical cue. Here, we describe the basic principles of the technology, review UAAs of particular interest to the structural biologist, and discuss current limitations and efforts to improve the efficiency of UAA incorporation.
3.6.1 Amber codon suppression in living cells
The successful incorporation of an UAA into a protein synthesized by a living cell requires several important considerations and components (Fig. 9a ). First and foremost, the UAA of interest must be chemically and metabolically stable, cell permeable or otherwise biosynthetically accessible in the cellular environment. It also must be tolerated by the ribosome and the cellular elongation factors without being recognized as a substrate by any of the endogenous synthetases. The UAA then requires its own unique codon, with the amber stop codon (UAG) being a popular choice due to its low occurrence in both prokaryotic and eukaryotic systems. The successful site-specific incorporation of the UAA, however, rests on the presence of a dedicated tRNA/aaRS pair that is highly specific for the UAA of interest, yet orthogonal in the context of all endogenous tRNA/aaRS pairs. Developing such pairs for a chemically diverse set of UAA is currently one of the time consuming and difficult steps of this technology. Since tRNA recognition by aaRS is often species specific, it is sometimes possible to import a heterologous pair into the cell of interest and use it as a starting point to build orthogonality and specificity into the system. For example, many UAA incorporation systems in E. coli are based on the heterologous tRNATyr/TyrRS pair from Methanococcus jannaschii, while the tRNATyr/TyrRS and tRNALeu/LeuRS pairs from E. coli have been used in eukaryotic cells (reviewed in (Chin, Reference Chin2014)). The tRNAPyr/PyrRS pair from methanogenic bacteria that can incorporate pyrrolysine has also been a very useful tool, as it is orthogonal in E. coli, yeast and mammalian cell lines, and has allowed the incorporation of many lysine-based UAAs, including acetyllysine (Neumann et al. Reference Neumann, Peak-Chew and Chin2008). While these systems provide a useful starting point, it is usually necessary to use mutagenesis and rounds of negative and positive selection to improve on the selectivity and orthogonality of the pair. Directed evolution approaches can also be used for the generation of de novo tRNA/aaRS pairs, or to expand the function of other components of the translational machinery (reviewed in (Chin, Reference Chin2014)). Once an appropriate tRNA/aaRS pair is developed, however, the practical implementation of amber suppression for the UAA is relatively straightforward. E. coli cells, for example, can be transformed with two plasmids: (1) a plasmid encoding the protein of interest and an appropriate point mutation with the amber TAG codon, and (2) a plasmid carrying the appropriate DNA sequence to produce the optimized tRNA/aaRS pair. After addition of UAA to the media, gene expression is induced for both plasmids and the UAA is incorporated into the protein of interest by the bacterial translational machinery. To separate the full-length protein from prematurely truncated species, often a purification tag is added to the protein C-terminus – notably, these can involve ‘silent’ intein- or sortase-based purification tags (Batjargal et al. Reference Batjargal, Walters and Petersson2015; Warden-Rothman et al. Reference Warden-Rothman, Caturegli, Popik and Tsourkas2013). Well-established protocols for UAA incorporation are now available for yeast (Hancock et al. Reference Hancock, Uprety, Deiters and Chin2010), mammalian (Chen et al. Reference Chen, Groff, Guo, Ou, Cellitti, Geierstanger and Schultz2009) and insect cells (Koehler et al. Reference Koehler, Sauter, Wawryszyn, Girona, Gupta, Landry, Fritz, Radic, Hoffmann, Chen, Zou, Tan, Galik, Junttila, Stolt-Bergner, Pruneri, GYENESEI, Schultz, Biskup, Besir, Benes, Rappsilber, Jechlinger, Korbel, Berger, Braese and Lemke2016; Mukai et al. Reference Mukai, Wakiyama, Sakamoto and Yokoyama2010b) and amber suppression can even be performed in multicellular organisms including C. elegans (Greiss & Chin, Reference Greiss and Chin2011), D. melanogaster (Bianco et al. Reference Bianco, Townsley, Greiss, Lang and Chin2012), mice (Ernst et al. Reference Ernst, Krogager, Maywood, Zanchi, Beranek, Elliott, Barry, Hastings and Chin2016; Kang et al. Reference Kang, Kawaguchi, Coin, Xiang, O'leary, Slesinger and Wang2013) and plants (Li et al. Reference Li, Zhang, Sun, Pan, Zhou and Wang2013b).

Fig. 9. Unnatural amino acid (UAA) incorporation by amber suppression. (a) An orthogonal aminoacyl tRNA synthetase charges a matching tRNA with the UAA of interest. The ribosome incorporates the UAA into a growing polypeptide chain by decoding the amber stop codon (UAG) on the messenger RNA. The UAA toolbox includes UAAs that represent (b) protein post-translational modifications, (c) spectroscopic probes, (d) cross-linkers, (e) bio-orthogonal reactive handles.
3.6.2 The amber suppression toolbox
The amber suppression toolbox contains many UAAs designed with structural biology applications in mind (Fig. 9b–e ). For example, heavy atoms can be incorporated site-specifically for solving the phase problem in X-ray crystallography – appropriate UAAs include p-iodo-L-phenylalanine (Xie et al. Reference Xie, Wang, Wu, Brock, Spraggon and Schultz2004) and 3-iodo-L-tyrosine (Sakamoto et al. Reference Sakamoto, Murayama, Oki, Iraha, Kato-Murayama, Takahashi, Ohtake, Kobayashi, Kuramitsu, Shirouzu and Yokoyama2009), as well as metal-ion chelating amino acids (Lee et al. Reference Lee, Spraggon, Schultz and Wang2009b). For NMR spectroscopists, amber suppression offers a relatively cheap and efficient way to install site-specific isotopic labels in otherwise unlabeled proteins. Many of the NMR ‘friendly’ UAAs are fluorinated derivatives that exploit the unique spectroscopic properties of 19F as a reporter of global protein folding and dynamics (Jones et al. Reference Jones, Cellitti, Hao, Zhang, Jahnz, Summerer, Schultz, Uno and Geierstanger2010; Yang et al. Reference Yang, Yu, Liu, Qu, Gong, Liu, Li, Wang, He, Yi, Song, Tian, Xiao, Wang and Sun2015). Similarly, amber suppression has been used to install nitroxide spin labels for distance measurements by EPR, thus overcoming the problems often associated with cysteine-based approaches (Park et al. Reference Park, Wang, Radoicic, De Angelis, Berkamp and Opella2015; Schmidt et al. Reference Schmidt, Fedoseev, Bucker, Borbas, Peter, Drescher and Summerer2015). More importantly, however, amber suppression is a living cell protein engineering tool; thus, it is ideally suited for NMR or EPR studies designed to follow the structural fate of proteins in the cellular milieu.
Of particular interest to the structural biologist are UAAs carrying natural PTMs. Currently, amber suppression can directly incorporate the following modifications: phosphoserine (Rogerson et al. Reference Rogerson, Sachdeva, Wang, Haq, Kazlauskaite, Hancock, Huguenin-Dezot, Muqit, Fry, Bayliss and Chin2015), acetyllysine (Neumann et al. Reference Neumann, Peak-Chew and Chin2008), several lysine acylations (Gattner et al. Reference Gattner, Vrabel and Carell2013; Kim et al. Reference Kim, Kang, Kim, Chatterjee and Schultz2012), phosphotyrosine (Fan et al. Reference Fan, Ip and Soll2016) and sulfonated tyrosine (Liu et al. Reference Liu, Brustad, LIU and Schultz2007). To expand this toolbox, however, amber suppression can be used to install a reactive handle at the position of interest, and then the appropriate modification can be chemically generated after protein purification. Using this strategy, for example, the UAA δ-thiol-lysine can be incorporated, followed by traceless attachment of a ubiquitin moiety with NCL (Virdee et al. Reference Virdee, Kapadnis, Elliott, Lang, Madrzak, Nguyen, Riechmann and Chin2011). Similarly, methylated lysines can be generated by the incorporation of a suitable pre-cursor UAA (Nguyen et al. Reference Nguyen, Garcia Alai, Virdee and Chin2010; Wang et al. Reference Wang, Zeng, Kurra, Wang, Tharp, Vatansever, Hsu, Dai, Fang and Liu2017). Phosphoserine, on the other hand, can serve as a starting point for the generation of dehydroalanine, which in turn can be converted into a number of modified side-chains (Wright et al. Reference Wright, Bower, Chalker, Bernardes, Wiewiora, Ng, Raj, Faulkner, Vallee, Phanumartwiwath, Coleman, Thezenas, Khan, Galan, Lercher, Schombs, Gerstberger, Palm-Espling, Baldwin, Kessler, Claridge, Mohammed and Davis2016).
Amber suppression also allows the facile installation of site-specific cross-linkers, a valuable tool for the identification of protein–protein and protein–ligand interactions both in vitro and in vivo. There are several options for UV-activatable cross-linkers that exploit different cross-linking mechanisms, and afford temporal and spatial control of the reaction. The oldest members of this toolbox are p-benzophenylalanine (Chin et al. Reference Chin, Martin, King, Wang and Schultz2002a) and p-azido-L-phenylalanine (Chin et al. Reference Chin, Santoro, Martin, King, Wang and Schultz2002b), both available for bacterial and eukaryotic systems, and extensively used for cross-linking experiments of purified proteins or in the cellular environment. More recently, diazirine-modified lysine-based amber suppression systems have been developed (Ai et al. Reference Ai, Shen, Sagi, Chen and Schultz2011; Chou et al. Reference Chou, Uprety, Davis, Chin and Deiters2011; Zhang et al. Reference Zhang, Lin, Song, Liu, Fu, Ge, Fu, Chang and Chen2011), exhibiting superior cross-linking efficiency, smaller structural perturbation effects and ideally suited for cross-linking experiments of lysine-rich proteins such as histones. UAAs that can cross-link proteins to nucleic acids are also available, and these include p-benzophenylalanine (Winkelman et al. Reference Winkelman, Vvedenskaya, Zhang, Zhang, Bird, Taylor, Gourse, Ebright and Nickels2016), and a furan-based cross-linker activated upon red-light irradiation (Schmidt & Summerer, Reference Schmidt and Summerer2013). Interestingly, p-azido-L-phenylalanine can also serve as an infrared (IR) spectroscopy probe, and as such can be used to report on the structural transitions of a protein along its activation pathway (Ye et al. Reference Ye, Zaitseva, Caltabiano, Schertler, Sakmar, Deupi and Vogel2010). Similarly, para-cyanophenylalanine can be installed as a site-specific and sensitive vibrational probe of ligand binding (Schultz et al. Reference Schultz, Supekova, Ryu, Xie, Perera and Schultz2006).
A significant fraction of the amber suppression UAA library has been designed for imaging and fluorescence-based biophysical applications. Some fluorescent UAAs can be incorporated directly into polypeptide chains and these include coumarin- and dansyl-based modifications (Kuhn et al. Reference Kuhn, Rubini, Muller and Skerra2011; Summerer et al. Reference Summerer, Chen, Wu, Deiters, Chin and Schultz2006), as well as environmentally sensitive aminonaphthalene-based probes available for both yeast and mammalian applications (Chatterjee et al. Reference Chatterjee, Guo, Lee and Schultz2013a; Lee et al. Reference Lee, Guo, Lemke, Dimla and Schultz2009a). More commonly, however, optical probes are attached site-specifically using bio-orthogonal reactions. In this case, amber suppression is used to install a reactive handle at a site in the polypeptide chain, while the fluorescent probe is modified with a compatible reactive functionality. The conjugation reaction can be carried out in vitro with purified components, or the optical label can be added to the media and/or delivered into cells for bio-orthogonal reaction chemistry within the cellular milieu. While this places important UAA-fluorescent label design constraints with respect to cell permeability, chemical stability and reaction kinetics, this approach allows the incorporation of optical labels that work at a variety of wavelengths, amid reduced background fluorescence. Currently, UAAs with a diverse set of functionalities for bio-orthogonal reactions are available (reviewed in (Lang & Chin, Reference Lang and Chin2014)), and it is likely that this list will become much more expansive in the future as new fast and specific bio-compatible approaches are developed (Section 3.7).
3.6.3 Limitations and future directions
The remarkable plasticity of the natural and evolved cellular protein synthesis machinery has allowed chemical biologists to create a large and diverse set of UAAs that can be incorporated into biological polymers assembled in vivo. Yet, the incorporation of many of these UAAs is inefficient, context dependent and essentially limited to the inclusion of a single modification per protein. Since UAA incorporation relies on the reassignment of natural stop codons, the suppressor tRNAs compete for binding sites with the endogeneous release factors that terminate translation. Therefore, truncations of the desired protein are often produced, resulting in compromised yields, complicated purification protocols and potential toxicity for the recombinant cell. In E. coli, translation termination involves release factor protein 1 (RF1) responsible for recognition of ochre (UAA) and amber (UAG) stop codons, and release factor protein 2 (RF2) that identifies ochre (UAA) and opal (UGA) stop codons. Thus, it has been possible to engineer bacterial strains that lack RF1 to enhance amber codon translation efficiency, and in particular improve the incorporation of the same UAA at multiple positions in the protein sequence (Johnson et al. Reference Johnson, Xu, Shen, Takimoto, Schultz, Schmitz, Xiang, Ecker, Briggs and Wang2011; Mukai et al. Reference Mukai, Hayashi, Iraha, Sato, Ohtake, Yokoyama and Sakamoto2010a). These strains, however, can be problematic as misincorporation of glutamine and codon skipping have also been reported (George et al. Reference George, Aguirre, Spratt, Bi, Jeffery, Shaw and O'donoghue2016). More dramatically, organisms can be genomically recoded to replace the UAG codon completely with the synonomous UAA stop codons, and free this new unique codon for more efficient amber suppression (Lajoie et al. Reference Lajoie, Rovner, Goodman, Aerni, Haimovich, Kuznetsov, Mercer, Wang, Carr, Mosberg, Rohland, Schultz, Jacobson, Rinehart, Church and Isaacs2013).
Protein yields are also affected by the lower efficiency of the evolved synthetases and suboptimal interactions of the tRNA with the wild-type elongation factors and ribosomes. These problems can be partially alleviated by the design of more efficient plasmid systems that have optimized promoters and can produce higher copy numbers of the synthetase and the tRNA. For example, the pEVOL and the pUltra plasmids have significantly increased incorporation efficiency in E. coli (Chatterjee et al. Reference Chatterjee, Sun, Furman, Xiao and Schultz2013b; Young et al. Reference Young, Ahmad, Yin and Schultz2010). For mammalian cells, protein expression is further affected by the transfection efficiency of the delivered constructs, thus it is usually desirable to incorporate all of the necessary genetic components (multiple copies of the synthetase and tRNA, gene of interest, engineered release factor, etc.) on the same plasmid ((Cohen & Arbely, Reference Cohen and Arbely2016). Baculovirus vectors that can deliver a large cargo of genetic material (>30 kb) to a variety of mammalian cells have also been developed for more efficient UAA incorporation (Chatterjee et al. Reference Chatterjee, Xiao, Bollong, Ai and Schultz2013c; Zheng et al. Reference Zheng, Lewis, Igo, Polleux and Chatterjee2016). To avoid the heterogeneous expression levels associated with transient transfection and viral transduction altogether, the creation of stable mammalian cell lines capable of defined UAA incorporation is highly desirable and efforts have already been undertaken in this regard (Elsasser et al. Reference Elsasser, Ernst, Walker and Chin2016; Tian et al. Reference Tian, Lu, Manibusan, Sellers, Tran, Sun, Phuong, Barnett, Hehli, Song, Deguzman, Ensari, Pinkstaff, Sullivan, Biroc, Cho, Schultz, Dijoseph, Dougher, Ma, Dushin, Leal, Tchistiakova, Feyfant, Gerber and Sapra2014).
The incorporation of several different modifications into the same polypeptide chain not only requires optimized suppression of the most commonly reassigned amber stop codon, but also the availability of other unique or rare codons that can be used to develop orthogonal tRNA/aaRS pairs. Not surprisingly, the ochre and opal stop codons are often combined with the amber codon for dual incorporation of UAAs, primarily with fluorescence-type applications in mind (Chatterjee et al. Reference Chatterjee, Sun, Furman, Xiao and Schultz2013b; Wan et al. Reference Wan, Huang, Wang, Russell, Pai, Russell and Liu2010; Xiao et al. Reference Xiao, Chatterjee, Choi, Bajjuri, Sinha and Schultz2013). The use of two stop codons, however, suffers from and amplifies many of the drawbacks described above and, if successful, results in very low protein yields. To circumvent this problem, translation machinery engineered to recognize quadruplet codons has been developed (Neumann et al. Reference Neumann, Wang, Davis, Garcia-Alai and Chin2010; Wang et al. Reference Wang, Sachdeva, Cox, Wilf, Lang, Wallace, Mehl and Chin2014a). The use of quadruplet codons can in principle provide 256 blank codons, thus dramatically expanding the capabilities of the natural 64-codon based genomes. Such systems, however, require the engineering of orthogonal ribosomes that can efficiently recognize quadruplet messages (Neumann et al. Reference Neumann, Wang, Davis, Garcia-Alai and Chin2010). Alternatively, the genomes of organisms such as E. coli can be reprogrammed to create compressed codon schemes and free up unique codons for reassignment (Ostrov et al. Reference Ostrov, Landon, Guell, Kuznetsov, Teramoto, Cervantes, Zhou, Singh, Napolitano, Moosburner, Shrock, Pruitt, Conway, Goodman, Gardner, Tyree, Gonzales, Wanner, Norville, Lajoie and Church2016; Wang et al. Reference Wang, Fredens, Brunner, Kim, Chia and Chin2016). In the future, it may also be possible to create new coding schemes by expanding the genetic alphabet of living organisms and utilizing synthetic DNA bases in new synthetic organisms (Malyshev et al. Reference Malyshev, Dhami, Lavergne, Chen, Dai, Foster, Correa and Romesberg2014).
3.7 Chemical modification of unnatural amino acids
The ability to install unnatural amino acids in polypeptide chains with exquisite specificity has opened widely the doors to a post-cysteine world of bio-orthogonal reactions. There is now a rapidly expanding collection of reactions that work efficiently and selectively with low concentrations of reactants in physiological buffers or the cellular milieu. Here, we will review a small sample of these tools focusing primarily on reactions that might be used to install site-specific PTMs or their mimics, as well as probes for structural investigations. For a comprehensive discussion of the bio-orthogonal literature, we refer the interested reader to several excellent reviews on the subject (Lang & Chin, Reference Lang and Chin2014; Shieh & Bertozzi, Reference Shieh and Bertozzi2014; Spicer & Davis, Reference Spicer and Davis2014; Stephanopoulos & Francis, Reference Stephanopoulos and Francis2011).
The azido functional group is one of the most versatile modifications that can be installed on peptides and proteins by amber suppression, total chemical synthesis and/or peptide ligation. In addition to serving as a light-activatable cross-linker or infra-red probe, this small moiety is also at the heart of several important bio-orthogonal reactions. For example, in an extension of the classical Staudinger reduction reaction, azides react with triarylphosphines bearing an electrophilic trap (usually an ester) to form a stable amide bond (Saxon & Bertozzi, Reference Saxon and Bertozzi2000). While the first version of this methodology generated a residual phosphine oxide at the ligation junction, ‘traceless’ Staudinger ligation variants are now available (Nilsson et al. Reference Nilsson, Kiessling and Raines2000; Saxon et al. Reference Saxon, Armstrong and Bertozzi2000) (Fig. 10a ), as well as reactions based on azobenzene or caged phosphine reagents that can be activated with light (Shah et al. Reference Shah, Laughlin and Carrico2016; Szymanski et al. Reference Szymanski, Wu, Poloni, Janssen and Feringa2013). This reaction can be used to ligate a peptide fragment containing a C-terminal thioester to another fragment carrying an N-terminal azide resulting in an extended native polypeptide chain and bypassing the need for a cysteine residue at the ligation junction (Nilsson et al. Reference Nilsson, Kiessling and Raines2000). While displaying slower reaction kinetics compared with some other bio-orthogonal reactions, the Staudinger ligation can be impactful in protein engineering applications where versatility and orthogonality are desired. For example, it has been used to create native isopeptide linkages between ubiquitin and other proteins (Andersen & Raines, Reference Andersen and Raines2015), install multiple probes into proteins such as RNA polymerase or GPCRs (Chakraborty et al. Reference Chakraborty, Mazumder, Lin, Hasemeyer, Xu, Wang, Ebright and Ebright2015; Huber et al. Reference Huber, Naganathan, Tian, Ye and Sakmar2013), prepare glycoprotein or phosphoprotein mimics (Bernardes et al. Reference Bernardes, Linderoth, Doores, Boutureira and Davis2011; Serwa et al. Reference Serwa, Wilkening, Del Signore, Muhlberg, Claussnitzer, Weise, Gerrits and Hackenberger2009), and turn on the fluorescence of optical probes upon protein labeling (Lemieux et al. Reference Lemieux, De Graffenried and Bertozzi2003).

Fig. 10. Chemical modification of unnatural amino acids.
Another popular and versatile bio-orthogonal reaction is the azide-alkyne 1,3-dipolar cycloaddition (Rostovtsev et al. Reference Rostovtsev, Green, Fokin and Sharpless2002; Tornoe et al. Reference Tornoe, Christensen and Meldal2002), commonly referred to as ‘click chemistry’ (Kolb et al. Reference Kolb, Finn and Sharpless2001). The cycloaddition can be promoted either by the presence of Cu(I) ligands or the use of highly strained cyclooctyne systems (Agard et al. Reference Agard, Prescher and Bertozzi2004). Both versions are specific, exhibit relatively fast reaction kinetics and are easy to use, which has led to numerous applications in the protein conjugation area. Recent examples include the incorporation of dual EPR or FRET probes for distance measurements in proteins or RNA (Kucher et al. Reference Kucher, Korneev, Tyagi, Apfelbaum, Grohmann, Lemke, Klare, Steinhoff and Klose2016; Lavergne et al. Reference Lavergne, Lamichhane, Malyshev, Li, Li, Sperling, Williamson, Millar and Romesberg2016), and the attachment of rigid lanthanide tags to large proteins for paramagnetic relaxation experiments (Mallagaray et al. Reference Mallagaray, Dominguez, Peters and Perez-Castells2016). In vivo applications of the Cu(I) version have been limited by copper toxicity, although extracellular labeling of cells has been reported (Link et al. Reference Link, Vink and Tirrell2004; Uttamapinant et al. Reference Uttamapinant, Tangpeerachaikul, Grecian, Clarke, Singh, Slade, Gee and Ting2012). The strained cyclooctyne systems, on the other hand, are compatible with living systems and improved versions that exhibit faster labeling kinetics are available for genetic incorporation through amber suppression (Dommerholt et al. Reference Dommerholt, Schmidt, Temming, Hendriks, Rutjes, Van Hest, Lefeber, Friedl and Van Delft2010; Lang et al. Reference Lang, Davis, Wallace, Mahesh, Cox, Blackman, Fox and Chin2012b; Plass et al. Reference Plass, Milles, Koehler, Schultz and Lemke2011) (Fig. 10b ). These copper-free cycloadditions have thus been valuable in imaging and proteomic applications in living cells and organisms (Laughlin et al. Reference Laughlin, Baskin, Amacher and Bertozzi2008; Smits et al. Reference Smits, Borrmann, Roosjen, Van Hest and Vermeulen2016; Xie et al. Reference Xie, Dong, Huang, Hong, Lei and Chen2014), and can be useful for the structural biologist interested in understanding the dynamics of macromolecules in the cellular environment either by FRET or EPR (Kucher et al. Reference Kucher, Korneev, Tyagi, Apfelbaum, Grohmann, Lemke, Klare, Steinhoff and Klose2016).
The success of click chemistry bioconjugation has inspired the search for faster and more efficient reactions for labeling in the cellular milieu. An important development along this direction involves the use of inverse-electron demand Diels-Alder reactions between strained dienophiles and tetrazine dienes (Blackman et al. Reference Blackman, Royzen and Fox2008; Devaraj et al. Reference Devaraj, Weissleder and Hilderbrand2008). The dienophile system in this case can be a trans-cyclooctene or a norbornene, both exhibiting significantly faster reaction kinetics as compared to the 1,3-dipolar cycloadditions described above, and thus allowing labeling reactions of proteins at much lower concentrations (Fig. 10c ). These bio-orthogonal reactive handles are also available for genetic incorporation by amber suppression (Lang et al. Reference Lang, Davis, torres-Kolbus, Chou, Deiters and Chin2012a, Reference Lang, Davis, Wallace, Mahesh, Cox, Blackman, Fox and Chin2012b; Seitchik et al. Reference Seitchik, Peeler, Taylor, Blackman, Rhoads, Cooley, refakis, Fox and Mehl2012) and provide an expanded and rapidly evolving toolbox for bio-orthogonal protein manipulations and modifications in the cellular environment.
While most of the reactions described above are incredibly useful for the conjugation of small molecules and biophysical probes to proteins in vitro and in vivo, they generally leave a relatively large chemical ‘scar’ at the conjugation site to be practical in the generation of PTMs or their mimics. A versatile approach for the site-specific installation of PTMs involves the UAA dehydroalanine. This UAA can be generated from a cysteine residue installed by site-directed mutagenesis (Chalker et al. Reference Chalker, Lercher, Rose, Schofield and Davis2012) or from an O-phosphoserine precursor incorporated by amber suppression (Yang et al. Reference Yang, Ha, Ahn, Kim, Kim, Lee, Kim, Soll, Lee and Park2016). Under biocompatible conditions, dehydroalanine can then be reacted with a variety of alkyl halides, via a radical-mediated process, to produce an impressive list of more than 50 modified functionalities at the site of interest (Wright et al. Reference Wright, Bower, Chalker, Bernardes, Wiewiora, Ng, Raj, Faulkner, Vallee, Phanumartwiwath, Coleman, Thezenas, Khan, Galan, Lercher, Schombs, Gerstberger, Palm-Espling, Baldwin, Kessler, Claridge, Mohammed and Davis2016; Yang et al. Reference Yang, Ha, Ahn, Kim, Kim, Lee, Kim, Soll, Lee and Park2016). These include methylated lysine and arginine residues, as well as fluorinated, glycosylated, phosphorylated, alkylated or isotopically labeled natural and UAA side chains. It should be noted that this methodology currently leads to the generation of both D and L configurations at the mutation site (i.e. the reaction lacks stereochemical control), a potential limitation for some applications.
3.8 Enzymatic bioconjugation approaches
The site-specific incorporation of bio-orthogonal handles on proteins can also be achieved enzymatically. These enzymes choose their targets based on the recognition of a specific amino acid sequence and can perform various chemical modifications of cysteine, lysine, serine, glutamine or glycine residues within their target sequences. This toolbox, for example, includes the formylglycine generating enzyme (FGE) that recognizes the CXPXR sequence motif and converts the cysteine residue to formylglycine, thus introducing an aldehyde functional group (Wu et al. Reference Wu, Shui, Carlson, Hu, Rabuka, Lee and Bertozzi2009) (Fig. 11a ). The aldehyde handle can be subsequently elaborated with various probes via bio-orthogonal transformations such as oximation and Hydrazino-Pictet-Spengler reactions (Agarwal et al. Reference Agarwal, Kudirka, Albers, Barfield, DE Hart, Drake, Jones and Rabuka2013; Dirksen & Dawson, Reference Dirksen and Dawson2008). Another popular tool is lipoic acid ligase, an enzyme that modifies a lysine side-chain within the 13-residue target sequence (Uttamapinant et al. Reference Uttamapinant, White, Baruah, Thompson, Fernandez-Suarez, Puthenveetil and Ting2010). Engineered versions of this enzyme can accommodate lipoic acid analogs and have been used to introduce bio-orthogonal handles, including azides (Plaks et al. Reference Plaks, Falatach, Kastantin, Berberich and Kaar2015; Uttamapinant et al. Reference Uttamapinant, Tangpeerachaikul, Grecian, Clarke, Singh, Slade, Gee and Ting2012) (Fig. 11b ), aryl aldehydes and hydrazines (Cohen et al. Reference Cohen, Zou and Ting2012), p-iodophenyl derivatives (Hauke et al. Reference Hauke, Best, Schmidt, Baalmann, Krause and Wombacher2014), norbornenes (Best et al. Reference Best, Degen, Baalmann, Schmidt and Wombacher2015) an trans-cyclooctenes (Liu et al. Reference Liu, Tangpeerachaikul, Selvaraj, Taylor, Fox and Ting2012). It is also possible to engineer fluorescent lipoic acid analogs that can be installed directly on the protein of interest (Uttamapinant et al. Reference Uttamapinant, White, Baruah, Thompson, Fernandez-Suarez, Puthenveetil and Ting2010). Other members of the enzymatic toolbox include biotin ligase, farnesyltransferase, transglutaminase and N-myristoyltransferase (reviewed in (Rashidian et al. Reference Rashidian, Dozier and Distefano2013)).

Fig. 11. Examples of bioenzymatic conjugation approaches. (a) Site-specific modification of cysteine with formylglycine generating enzyme, followed by oxime ligation to attach a chemical or optical probe. (b) Introduction of ‘click’ handles into proteins using lipoic acid ligase and lipoic acid analogs.
Enzymatic bioconjugation is an increasingly useful tool for cell imaging applications, functionalization of therapeutic proteins, immobilization of proteins on solid supports or the preparation of protein—polymer or protein—nanoparticle conjugates (Hu et al. Reference Hu, Berti and Adamo2016; Slavoff et al. Reference Slavoff, Liu, Cohen and Ting2011; Walper et al. Reference Walper, Turner and Medintz2015). For the structural biologist and biophysicist, it offers an orthogonal strategy for protein modification that is based on genetically encodable peptide tags (Stephanopoulos & Francis, Reference Stephanopoulos and Francis2011). For example, it provides a conceptually straightforward way to introduce a second modification to a construct that already contains an UAA incorporated by amber suppression. Enzymatic bioconjugation can also be useful in the multiplex labeling of complex protein mixtures, and the modification of constructs that are large, difficult to purify or not easily amenable to the approaches outlined above. Since the recognition sequence is fused to the protein of interest, the modification site is usually limited in location to the N- and C-termini, or to a surface exposed and flexible loop of the protein. Other important considerations include the reaction kinetics, the stability and solubility of the enzymes, and the relatively large size of the modifications that are installed and that can perturb the function of the protein target.
4. Protein engineering approaches for tackling outstanding challenges in structural biology
4.1 X-ray crystallography
The molecular engineering toolbox presented here can aid crystallographers in all stages of the structure determination process. For example, when phase values cannot be determined by direct methods or molecular replacement, protein crystallographers can incorporate heavy atoms (e.g. Se, I or Br) into the protein crystal and use their anomalous diffraction patterns to solve the structure. Heavy atom incorporation can be accomplished in several ways (reviewed in (Pike et al. Reference Pike, Garman, Krojer, Von Delft and Carpenter2016). For example, cysteine residues can be derivatized with mercury by pre-labeling, co-crystallization or soaking of the protein crystals in mercury salts (Martinez et al. Reference Martinez, De Geus, Stanssens, Lauwereys and Cambillau1993). If necessary, cysteine accessibility can be pre-evaluated with Ellman's reagent (5,5′-dithiobis-2-nitrobenzoic acid) (Li et al. Reference Li, Pye and Caffrey2015). Another popular heavy atom labeling methodology relies on the incorporation of the modified amino acid selenomethionine (Hendrickson et al. Reference Hendrickson, Horton and Lemaster1990). In this case, selenomethionine is added to the culture of a methionine-auxotrophic bacterial strain resulting in substitution of all methionine residues in the protein. Selenomethionine incorporation is also possible in other non-bacterial expression systems, albeit with somewhat lower substitution efficiency (Cronin et al. Reference Cronin, Lim and Rogers2007; Nettleship et al. Reference Nettleship, Assenberg, Diprose, Rahman-Huq and Owens2010). Site-specific incorporation of appropriately modified unnatural amino acids (e.g. p-iodo-L-phenylalanine, p-bromo-L-phenylalanine and 3-iodo-L-tyrosine) can be performed with amber suppression or chemical synthesis (Kwon et al. Reference Kwon, Wang and Tirrell2006; Sakamoto et al. Reference Sakamoto, Murayama, Oki, Iraha, Kato-Murayama, Takahashi, Ohtake, Kobayashi, Kuramitsu, Shirouzu and Yokoyama2009; Xie et al. Reference Xie, Wang, Wu, Brock, Spraggon and Schultz2004; Yeung et al. Reference Yeung, Squire, Yosaatmadja, Panjikar, Lopez, Molina, Baker, Harris and Brimble2016). Solving the phase problem can also be aided by the development of metal-binding peptide tags (e.g. Tb3+) that can be genetically fused to the protein of interest (Silvaggi et al. Reference Silvaggi, Martin, Schwalbe, Imperiali and Allen2007).
Total chemical synthesis of proteins is an important tool for racemic and quasi-racemic crystallography (Yeates & Kent, Reference Yeates and Kent2012) (Fig. 12). These methods are based on the observation that mixing D- and L-forms of proteins can aid crystallization as macromolecules have access to a much larger set of crystallographic space groups, including those that contain mirror or center of inversion operations (Wukovitz & Yeates, Reference Wukovitz and Yeates1995). D-enantiomers of proteins can be made using solid-phase peptide synthesis and NCL, with synthesis efficiencies that have allowed the structure determination of constructs in the 200-amino acid range (Mandal et al. Reference Mandal, Uppalapati, Ault-Riche, Kenney, Lowitz, Sidhu and Kent2012; Pan et al. Reference Pan, Gao, Zheng, Tan, Lan, Tan, Sun, Lu, Wang, Zheng, Huang, Wang and LIU2016). The synthetic origin of such polypeptides also allows the facile co-incorporation of amino acids containing heavy atoms (Yeung et al. Reference Yeung, Squire, Yosaatmadja, Panjikar, Lopez, Molina, Baker, Harris and Brimble2016), or chemically well-defined post-translational modifications (Okamoto et al. Reference Okamoto, Mandal, Sawaya, Kajihara, Yeates and Kent2014).

Fig. 12. Principle of racemic crystallography. The L- and D- forms of the polypeptide chains are prepared separately by solid-phase peptide synthesis and native chemical ligation. The proteins are subsequently mixed and co-crystallized, thus gaining access to a much larger set of crystallographic space groups.
For structure characterization of larger biological macromolecules and complexes, it is often more practical to use mimics rather than the native PTM. For example, methylation mimics are accessible through the alkylation of cysteine residues, and produce constructs with high yields and chemical purity in all possible methylation states (Simon et al. Reference Simon, Chu, Racki, De La Cruz, Burlingame, Panning, Narlikar and Shokat2007). Such approaches have been invaluable in elucidating the impact of histone methylation on the nucleosome surface, a structural problem that requires the efficient crystallization of a biological assembly comprised of four different proteins and DNA (Lu et al. Reference Lu, Simon, Chodaparambil, Hansen, Shokat and Luger2008). Sometimes, non-native linkages and modifications are essential in trapping an important functional state of a protein complex, for example between an ubiquitylated protein and the corresponding deubiquitinating enzyme (Morgan et al. Reference Morgan, Haj-Yahya, Ringel, Bandi, Brik and Wolberger2016). The ongoing optimization of amber suppression systems for in-cell and cell-free protein synthesis has also made it easier to produce certain native modifications in high-enough yield for protein crystallization. There are, for example, several crystal structures of acetylated and phosphorylated proteins prepared by amber suppression in E. coli (Arbely et al. Reference Arbely, Natan, Brandt, Allen, Veprintsev, Robinson, Chin, Joerger and Fersht2011; Huguenin-Dezot et al. Reference Huguenin-Dezot, De Cesare, Peltier, Knebel, Kristaryianto, Rogerson, Kulathu, Trost and Chin2016; Kuhlmann et al. Reference Kuhlmann, Wroblowski, Knyphausen, DE Boor, Brenig, Zienert, Meyer-Teschendorf, Praefcke, Nolte, Kruger, SCHACHERL, Baumann, James, Chin and Lammers2016; Lammers et al. Reference Lammers, Neumann, Chin and James2010). This technology has also enabled the structural analysis of ubiquitin chains (Virdee et al. Reference Virdee, Ye, Nguyen, Komander and Chin2010). Crystallographic studies of proteins containing multiple modifications have been successfully addressed with cell-free protein synthesis (Wakamori et al. Reference Wakamori, Fujii, Suka, Shirouzu, Sakamoto, Umehara and Yokoyama2015) and EPL (Wang et al. Reference Wang, Kavran, Chen, Karukurichi, Leahy and Cole2014b).
Beyond the incorporation of PTMs, the modern protein engineering toolbox provides the means to precisely alter the covalent structure of proteins, importantly allowing access to both the amino acid side-chains and the polypeptide backbone. Crystallographic analysis of such modified proteins can be extremely powerful, yielding insights into processes as diverse as ion conduction (Grosse et al. Reference Grosse, Essen and Koert2011; Valiyaveetil et al. Reference Valiyaveetil, Leonetti, Muir and Mackinnon2006), enzyme catalysis (Torbeev et al. Reference Torbeev, Raghuraman, Hamelberg, Tonelli, Westler, Perozo and Kent2011; Wang et al. Reference Wang, Kavran, Chen, Karukurichi, Leahy and Cole2014b) and protein–protein interactions (Lu et al. Reference Lu, Randal, Kossiakoff and Kent1999; Morgan et al. Reference Morgan, Haj-Yahya, Ringel, Bandi, Brik and Wolberger2016).
4.2 Nuclear magnetic resonance
The site-specific incorporation of NMR probes into biological macromolecules presents two challenging requirements to the protein chemist. First, similarly to X-ray crystallography, large amounts of sample are required, and the incorporation methods should be robust and efficient. Second, the isotopic labeling precursors are often prohibitively expensive. It therefore comes as no surprise that most NMR labeling strategies exploit the metabolic pathways in bacteria to produce uniform or sparse isotopic labeling schemes, or incorporate isotopic labels in an amino acid specific fashion (for a comprehensive series of reviews on isotopic labeling of biomolecules, the interested reader is referred to volume 565 of Methods in Enzymology, 2015). Here, we will focus on labeling methodologies that allow more control over the placement of spectroscopic probes in a polypeptide chain, and that have historically been demonstrated to produce sufficient protein amounts for NMR analysis.
4.2.1 Segmental isotopic labeling
Segmental isotopic labeling is a powerful protein engineering approach that allows the generation of an intact and typically natively folded and functional protein where only a certain segment of the polypeptide chain is ‘visible’ by NMR (Xu et al. Reference Xu, Ayers, Cowburn and Muir1999; Yamazaki et al. Reference Yamazaki, Otomo, Oda, Kyogoku, Uegaki, Ito, Ishino and Nakamura1998). This strategy is particularly impactful if the protein of interest is large and/or has a degenerate amino acid sequence, thus simplifying assignment and interpretation of crowded and overlapped NMR spectra. Such constructs can be crucial in generating unambiguous structural constraints in large proteins and assemblies, and have been exploited in structural investigations both by solution and solid-state NMR (Mehler et al. Reference Mehler, Eckert, Busche, Kulhei, Michaelis, Becker-Baldus, Wachtveitl, Dotsch and Glaubitz2015; Schubeis et al. Reference Schubeis, Yuan, Ahmed, Nagaraj, Van Rossum and Ritter2015; Tremblay et al. Reference Tremblay, Xu, Lefevre, Sarker, Orrell, Leclerc, Meng, Pezolet, Auger, Liu and Rainey2015; Williams et al. Reference Williams, Milbradt, Embrey and Bobby2016). Several approaches can be used to achieve segmental labeling of polypeptide chains. For example, a protein domain can be fused to a contiguous intein and produced recombinantly in labeled media (e.g. supplemented with 15N and 13C-enriched nutrient sources). Following thiolysis of the intein, the thioester derivative of the labeled domain can then be ligated, via EPL, to a protein fragment that does not contain NMR isotopes (Xu et al. Reference Xu, Ayers, Cowburn and Muir1999). In principle, the NMR silent segment can be a synthetic peptide carrying a PTM or a paramagnetic tag, thus allowing the facile incorporation of C-terminal protein modifications. For example, this approach has been used to introduce a phosphorylated tyrosine residue at position 125 of the amyloid-related protein α-synuclein, while residues 1–106 were prepared recombinantly and isotopically labeled for NMR analysis (Hejjaoui et al. Reference Hejjaoui, Butterfield, Fauvet, Vercruysse, Cui, Dikiy, Prudent, Olschewski, ZHANG, Eliezer and Lashuel2012). In practice, however, the second fragment is usually produced recombinantly due to cost and yield considerations.
PTS offers an alternative intein-based approach for segmental isotopic labeling. The high affinity of naturally split intein fragments means that the splicing reactions can be conducted at very low concentrations of reactants (low μM), a capability that distinguishes the PTS strategy from EPL where reactant concentrations in the mM range are typically required. In practice, the target protein is divided into two segments, for example at domain boundaries, each fused to an appropriate intein fragment (Fig. 13a ). These constructs are expressed separately, allowing the differential incorporation of NMR-active isotopes. After purification, the two extein–intein fragments are mixed, protein trans-splicing takes place and the leftover intein components can be purified using an appropriate affinity tag or chromatographic separation. Notably, the splicing reaction can also be performed directly in the cellular environment. In this case, both constructs are transformed into the same cell, but with different promoters so that the expression of each construct can be controlled separately, and therefore timed with the addition of isotopically enriched nutrients to the media (Muona et al. Reference Muona, Aranko, Raulinaitis and Iwai2010) (Fig. 13b ). This strategy can be particularly helpful for labeling proteins that are hard to refold in vitro, e.g. membrane proteins (Mehler et al. Reference Mehler, Eckert, Busche, Kulhei, Michaelis, Becker-Baldus, Wachtveitl, Dotsch and Glaubitz2015). The first segmental labeling protocols relied on artificially split or natural inteins that were not very efficient, thus limiting the utility of the technology to few favorable cases (Yamazaki et al. Reference Yamazaki, Otomo, Oda, Kyogoku, Uegaki, Ito, Ishino and Nakamura1998). Today, however, the toolbox of useful split inteins has greatly expanded, and it is now possible to choose from inteins with diverse properties, e.g. those giving better expression yields for the protein of interest, capable of carrying out ligations at higher temperatures or denaturant concentrations, or more forgiving to the choice of extein junction site (see Section 3.4.3).

Fig. 13. Strategies for segmental isotopic labeling of proteins for NMR analysis. (a) Intein-based segmental labeling. (b) Strategy for segmental labeling of proteins in cells. (c) Labeling with sortases.
Another option for segmental isotopic labeling is provided by sortase-mediated ligation (Fig. 13c ). As discussed in Section 3.5, this method relies on the use of Sortase A, a transpeptidase that can ligate two constructs provided that one contains an LPXTG recognition sequence, and the other, a glycine repeat motif. Since it results in an LPXTG ‘scar’ at the ligation junction, sortase-mediated ligation is best suited for the segmental labeling of protein domains separated by a flexible, mutation-tolerant linker (Williams et al. Reference Williams, Milbradt, Embrey and Bobby2016). The reversibility of the sortase ligation also necessitates the removal of cleaved peptide byproducts during the reaction course for optimal yields (Freiburger et al. Reference Freiburger, Sonntag, Hennig, Li, Zou and Sattler2015). On the other hand, segmental labeling using sortase works well with low μM concentrations of reagents, and is a good alternative for constructs that do not express well as intein fusions.
The segmental labeling approaches described here have also been used to generate protein constructs with NMR silent solubility enhancement tags (Kobashigawa et al. Reference Kobashigawa, Kumeta, Ogura and Inagaki2009; Zuger & Iwai, Reference Zuger and Iwai2005). To keep proteins more stable in solution during a long multidimensional experiment, NMR spectroscopists can resort to fusing the protein of interest to a soluble domain such as SUMO, GB1, MBP or thioredoxin. If prepared as a single construct, however, the solubility tag will be isotopically labeled and will contribute to the NMR spectrum, a complication that can be avoided with segmental labeling.
4.2.2 Site-specific incorporation of magnetic resonance probes
The introduction of a single isotopic label at a well-defined position in a polypeptide chain provides NMR spectroscopists with a benign and unambiguous reporter of protein structure, dynamics and ligand binding. Strategically positioned isotopes can also be crucial in disentangling higher order structural interactions in complex biological assemblies such as amyloid fibrils (Debelouchina et al. Reference Debelouchina, Bayro, Fitzpatrick, Ladizhansky, Colvin, Caporini, Jaroniec, Bajaj, Rosay, Macphee, Vendruscolo, Maas, Dobson and Griffin2013; Petkova et al. Reference Petkova, Yau and Tycko2006). The incorporation of such probes is relatively straightforward in short polypeptides prepared by solid-phase peptide synthesis, and many protected isotopically labeled amino acids are available commercially at a reasonable cost. Longer polypeptide chains containing specific labels can be prepared by NCL as exemplified by the synthesis of the membrane protein M2 containing five 13C,15N-labeled amino acids dispersed throughout the sequence (Kwon et al. Reference Kwon, Tietze, White, Liao and Hong2015). A serendipitously positioned native cysteine residue in this case provided a convenient ligation site without the need for desulfurization.
In recent years, the biomolecular NMR field has seen a surge in the popularity of 19F as a tool to investigate large and complex systems both by solution and solid-state NMR spectroscopy. This nucleus is bio-orthogonal, has magnetic properties that ensure high sensitivity (~80% of the sensitivity of 1H nuclei) and a wide chemical shift range (~100-fold larger than the range of 1H). It can therefore serve as a highly sensitive probe of protein and peptide structure, folding and aggregation, dynamics and ligand binding (reviewed in (Chen et al. Reference Chen, Viel, Ziarelli and Peng2013; Sharaf & Gronenborn, Reference Sharaf and Gronenborn2015). Specific 19F labels can be installed by peptide synthesis, conjugation or fluorination of cysteine and lysine residues, or by supplementing the culture media with fluorinated amino acids (which results in amino acid specific labeling). Increasingly, amber suppression is becoming the site-specific incorporation method of choice ((Marsh & Suzuki, Reference Marsh and Suzuki2014; Sharaf & Gronenborn, Reference Sharaf and Gronenborn2015). There are several orthogonal tRNA/synthetase systems and fluorinated UAAs available, each imparting different spectroscopic signatures with respect to chemical shift anisotropy and relaxation. These include 3,5-difluorotyrosine (Li et al. Reference Li, Shi, Li, Yang, Wang, Zhang, Gao, Ding, Li, Li, Xiong, Sun, Gong, Tian and Wang2013a; Yang et al. Reference Yang, Yu, Liu, Qu, Gong, Liu, Li, Wang, He, Yi, Song, Tian, Xiao, Wang and Sun2015), p-trifluoromethoxyphenylalanine (Cellitti et al. Reference Cellitti, Jones, Lagpacan, Hao, Zhang, Hu, Brittain, Brinker, Caldwell, Bursulaya, Spraggon, Brock, Ryu, Uno, Schultz and Geierstanger2008) and p-trifluoromethylphenylalanine (Jackson et al. Reference Jackson, Hammill and Mehl2007; Loscha et al. Reference Loscha, Herlt, Qi, Huber, Ozawa and Otting2012). While most protein 19F NMR applications rely on amber suppression in E. coli, magnetic resonance imaging studies are already exploiting the unique spectroscopic properties of 19F in mammalian systems or whole organisms (Yu et al. Reference Yu, Hallac, Chiguru and Mason2013). Site-specific 13C and 15N probes can also be installed by amber suppression, including 13C/15N-labeled p-methoxyphenylalanine and 15N-labeled o-nitrobenzyl-tyrosine (Cellitti et al. Reference Cellitti, Jones, Lagpacan, Hao, Zhang, Hu, Brittain, Brinker, Caldwell, Bursulaya, Spraggon, Brock, Ryu, Uno, Schultz and Geierstanger2008). Ligand-binding studies of large molecular systems may also benefit from incorporation of o-tert-butyltyrosine, where rapid bond rotations of the tert-butyl group ensure a narrow linewidth of the characteristic peak in solution NMR spectra (Chen et al. Reference Chen, Kuppan, Lee, Jaudzems, Huber and Otting2015).
Another problem of particular relevance to the magnetic resonance spectroscopist concerns the site-directed installation of EPR and PRE probes. These probes carry stable nitroxide radicals or chelated metal ions such as Mn2+, Cu2+ or Gd3+, and can be used to measure distances by EPR (up to 80 Å), or change the relaxation properties of nuclear spins in their vicinity (up to 35 Å). Usually, paramagnetic tags are incorporated at available cysteine residues using alkylation with maleimides, and many appropriately functionalized reagents are available commercially. As discussed in Section 3.1, however, cysteine-based approaches often have limited selectivity, the generated maleimide-based linker can be unstable, and is generally not applicable to cellular investigations. The development of amber suppression and bio-orthogonal reactions has certainly provided more options for EPR and PRE label incorporation both in vitro and in vivo. For example, orthogonal tRNA/tRNA synthetase systems are available for direct incorporation of nitroxide-containing (Jones et al. Reference Jones, Cellitti, Hao, Zhang, Jahnz, Summerer, Schultz, Uno and Geierstanger2010; Schmidt et al. Reference Schmidt, Fedoseev, Bucker, Borbas, Peter, Drescher and Summerer2015) and metal chelating UAAs (Park et al. Reference Park, Wang, Radoicic, De Angelis, Berkamp and Opella2015). Another amber suppression incorporation route involves the UAA p-acetyl-L-phenylalanine that can react with paramagnetic tags functionalized with a hydroxylamine moiety (Fleissner et al. Reference Fleissner, Brustad, Kalai, Altenbach, Cascio, Peters, Hideg, Peuker, Schultz and hubbell2009). An NMR-specific and often prohibitive requirement for the incorporation of PRE tags with amber suppression, however, is the concurrent need to introduce isotopic labels in the protein of interest. Thus, general protocols that provide efficient UAA incorporation under suboptimal bacterial growth conditions (minimal 13C/15N-supplemented or perdeuterated media, for example) are desperately needed (Evans & Millhauser, Reference Evans and Millhauser2015; Venditti et al. Reference Venditti, Fawzi and Clore2012).
Another demanding problem concerns the site-specific installation of isotopically labeled PTMs such as different methylation states, glycosylation, ubiquitylation and various acylations (phosphorylation being somewhat of an exception as the naturally abundant 31P is the NMR active isotope). Coupled with the often problematic behavior of the modified proteins (e.g. aggregation prone intrinsically disordered and amyloidogeneic peptides and proteins, insoluble transmembrane domains or chromatin effectors), PTM installation in itself is a challenge even without the extra complication and associated cost of isotopic labeling. Not surprisingly, therefore, most NMR studies involving PTMs are performed with constructs that have been modified enzymatically. Enzymatic modification, however, is often incomplete and can result in a heterogeneous set of modifications, thus complicating the interpretation of NMR data. Well-established non-enzymatic site-directed protocols are available for ubiquitylation, taking advantage of the possibility to prepare and isotopically label ubiquitin recombinantly. The labeled ubiquitin moiety can then be installed at a specific position of the modified polypeptide using a variety of strategies including an asymmetric disulfide (Debelouchina et al. Reference Debelouchina, Gerecht and Muir2017), a condensation reaction of a C-terminal ubiquitin thioester and the ε-amine of a targeted lysine residue (Castaneda et al. Reference Castaneda, Liu, Chaturvedi, Nowicka, Cropp and Fushman2011a), or a combined recombinant expression–chemical synthesis approach (Castaneda et al. Reference Castaneda, Spasser, Bavikar, Brik and Fushman2011b). Site-specific phosphorylation can be incorporated by solid-phase peptide synthesis, followed by EPL to construct segmentally labeled and modified proteins (Hejjaoui et al. Reference Hejjaoui, Butterfield, Fauvet, Vercruysse, Cui, Dikiy, Prudent, Olschewski, ZHANG, Eliezer and Lashuel2012). Studies of glycosylated proteins also require the attachment of homogeneous glycans that are often isotopically labeled to facilitate resonance assignments and structure determination (Skrisovska et al. Reference Skrisovska, Schubert and Allain2010). Cysteine-based 13C-labeled methyllysine analogs are a convenient and affordable way to generate methylated constructs, and have been used in the NMR structural analysis of proteins such as HP1 and p53 (Cui et al. Reference Cui, Park, Badeaux, Kim, Lee, Thompson, Yan, Kaneko, Yuan, Botuyan, Bedford, Cheng and Mer2012; Munari et al. Reference Munari, Soeroes, Zenn, Schomburg, Kost, Schroder, Klingberg, Rezaei-Ghaleh, Stutzer, Gelato, Walla, Becker, Schwarzer, Zimmermann, Fischle and Zweckstetter2012).
4.3 Studies of dynamic interactions
4.3.1 Incorporation of optical probes
The advances in single-molecule optical spectroscopy and imaging have propelled the search for better fluorescent probes and the development of more efficient and targeted strategies for their incorporation in vitro and in cells. The information obtained from such experiments can complement high-resolution structural studies and provide data not easily accessible by other methods, including dynamic protein–protein interactions, conformational states and protein folding pathways. There are, of course, many excellent reviews on the subject (Dimura et al. Reference Dimura, Peulen, Hanke, Prakash, Gohlke and Seidel2016; Haney et al. Reference Haney, Wissner and Petersson2015; Minoshima & Kikuchi, Reference Minoshima and Kikuchi2017; Nikic & Lemke, Reference Nikic and Lemke2015). Therefore, we will limit our discussion to the problem of incorporating two small FRET probes for biophysical and structural investigations, still a surprisingly challenging task for the protein chemist.
A protein construct prepared for FRET experiments can be labeled with two different fluorophores (A and B) by cysteine mutagenesis at two appropriately selected sites. Incubation with the maleimide, iodoacetamide or methyl bromide derivatives of the two probes results in a statistical mixture of labeled constructs even if labeling is efficient (A–A, A–B, B–A, B–B). Since the mixture of products can complicate data analysis (Husada et al. Reference Husada, Gouridis, Vietrov, Schuurman-Wolters, Ploetz, De Boer, Poolman and Cordes2015), more selective strategies for labeling are highly desirable and have been explored in the literature. A popular approach, for example, combines cysteine labeling with amber suppression that can introduce a second fluorescent moiety either directly or through bio-orthogonal chemistries (Brustad et al. Reference Brustad, Lemke, Schultz and Deniz2008; Haney et al. Reference Haney, Wissner, Warner, Wang, Ferrie, Covell, Karpowicz, LEE and Petersson2016; Milles et al. Reference Milles, Tyagi, Banterle, Koehler, Vandelinder, Plass, Neal and Lemke2012; Ratzke et al. Reference Ratzke, Hellenkamp and Hugel2014). Suitable UAAs include p-acetylphenylalanine for labeling with commercially available hydroxylamine probe derivatives (Fig. 14a ), as well as azido- and alkyne-functionalized phenylalanine, tyrosine and lysine residues for click reactions. Cysteine labeling can also be combined with the site-specific incorporation of 1,2-aminothiols that react selectively with cyanobenzothiazole fluorescent-probe derivatives (Nguyen et al. Reference Nguyen, Elliott, Holt, Muir and Chin2011). A conceptually different cysteine-based labeling approach involves the FlAsH system that targets biarsenic reagents to the genetically encoded tetracysteine motif CCPGCC (Griffin et al. Reference Griffin, Adams and Tsien1998). This technology has the advantage that the small molecule ligands are cell permeable and virtually non-fluorescent until they bind their recognition motif, and are therefore well-suited for in-cell applications. In the context of dual labeling, FlAsH has been combined with amber suppression (Perdios et al. Reference Perdios, Lowe, Saladino, Bunney, Thiyagarajan, Alexandrov, Dunsby, French, Chin, Gervasio, Tate and Katan2017) (Fig. 14b ) or fluorescent proteins such as CFP (Hoffmann et al. Reference Hoffmann, Gaietta, Bunemann, Adams, Oberdorff-Maass, Behr, Vilardaga, Tsien, Ellisman and Lohse2005) for the investigation of protein conformational states in vitro or in the cellular environment.

Fig. 14. Dual labeling of proteins with fluorophores. (a) A labeling strategy based on the combination of cysteine chemistry and amber suppression (Brustad et al. Reference Brustad, Lemke, Schultz and Deniz2008). (b) The FlAsH labeling system can be used for the selective modification of a genetically encoded peptide tag, in combination with amber suppression (Perdios et al. Reference Perdios, Lowe, Saladino, Bunney, Thiyagarajan, Alexandrov, Dunsby, French, Chin, Gervasio, Tate and Katan2017). (c) A dual labeling strategy based on native chemical ligation and amber suppression (Wissner et al. Reference Wissner, Batjargal, Fadzen and Petersson2013). (d) Genetic incorporation of two UAAs using orthogonal ribosomes that can decode the AGTA quadruplet codon (Sachdeva et al. Reference Sachdeva, Wang, Elliott and Chin2014).
When the protein construct of interest contains functionally or structurally important cysteines, dual labeling can be attempted with NCL, EPL, the incorporation of genetically encodable UAAs and peptide tags for bioenzymatic conjugation, or a combination of these approaches. For example, a fluorescent probe and a thioamide quencher can be installed using a combination of amber suppression and protein ligation, where the quencher is a backbone modification that is only accessible through protein synthesis (Wissner et al. Reference Wissner, Batjargal, Fadzen and Petersson2013) (Fig. 14c ). As discussed in Section 3.6.3, incorporation of two UAAs in a polypeptide chain by genetic means is still difficult; however, the growing numbers of successful applications are almost exclusively developed for FRET-labeling. For example, using cells containing the orthogonal ribosome ribo-Q1, two fluorescent labels could be incorporated efficiently into calmodulin, one in response to the amber TAG codon, the other at the newly assigned AGTA quadruplet codon (Sachdeva et al. Reference Sachdeva, Wang, Elliott and Chin2014) (Fig. 14 d). Rapid, quantitative, one-pot labeling was achieved by using mutually orthogonal cycloadditions at UAAs bearing a terminal alkyne and a cyclopropene. Alternatively, amber–ochre or amber–opal dual incorporation schemes can be used (Chatterjee et al. Reference Chatterjee, Sun, Furman, Xiao and Schultz2013b; Wan et al. Reference Wan, Huang, Wang, Russell, Pai, Russell and Liu2010; Xiao et al. Reference Xiao, Chatterjee, Choi, Bajjuri, Sinha and Schultz2013).
4.3.2 Incorporation of vibrational probes
Two-dimensional infrared spectroscopy (2D IR) has emerged as a powerful technique to characterize the dynamic states and interactions of biological macromolecules (Baiz et al. Reference Baiz, Reppert and Tokmakoff2013; Le Sueur et al. Reference Le Sueur, Horness and Thielges2015). Dynamic and structural information is encoded in the vibrational modes of functional groups such as backbone amides and carbonyls, as well as side-chains with aromatic, carbonyl and guanidinium groups. Since even polypeptides of moderate length contain many overlapping signals, site-specific incorporation of vibrational reporters with distinct spectroscopic signatures is essential for data interpretation. The installation of such probes can take advantage of several of the protein engineering approaches described above (Zhang et al. Reference Zhang, Grechko, Moran and Zanni2016b). Segmental isotopic labeling, for example, can be used to distinguish the spectroscopic behavior of protein domains (Moran et al. Reference Moran, Woys, Buchanan, Bixby, Decatur and Zanni2012). In this case, one domain is labeled with 13C, while the other domain remains at natural abundance. The higher mass of the 13C isotope changes the vibrational modes of the backbone carbonyls, and results in a downward shift of the vibrational frequency, effectively decoupling the spectroscopic signatures of the two domains. Higher resolution in linear and multidimensional IR spectra can be achieved by the site-specific installation of isotopic labels such as 13C–18O pairs by solid-phase peptide synthesis, EPL, NCL or the incorporation of appropriately labeled methionine during recombinant protein expression (Courter et al. Reference Courter, Abdo, Brown, Tucker, Hochstrasser and Smith2014; Davis et al. Reference Davis, Cooper and Dyer2015; Dhayalan et al. Reference Dhayalan, Fitzpatrick, Mandal, Whittaker, Weiss, Tokmakoff and Kent2016; Marecek et al. Reference Marecek, Song, Brewer, Belyea, Dyer and Raleigh2007; Zhang et al. Reference Zhang, Grechko, Moran and Zanni2016b). Resolution and specificity can also be achieved by the introduction of orthogonal vibrational probes such as nitrile, cyano, azido and thiocyanate functional groups. To this end, several UAAs suitable for amber suppression are available, including 4-cyano-, 4-azido- and 4-azidomethyl-phenylalanine (Bazewicz et al. Reference Bazewicz, Liskov, Hines and Brewer2013; Schultz et al. Reference Schultz, Supekova, Ryu, Xie, Perera and Schultz2006; Ye et al. Reference Ye, Zaitseva, Caltabiano, Schertler, Sakmar, Deupi and Vogel2010). 13C, 15N-labeled thiocyanates, on the other hand, can be obtained by cyanylation of cysteine residues (van Wilderen et al. Reference Van Wilderen, Kern-Michler, Muller-Werkmeister and Bredenbeck2014).
5. Outlook
Our goal in this review has been to examine the contents of the modern protein engineering toolbox, with the particular needs of the structural biologist in mind. As is hopefully evident from the preceding sections, there are now many highly versatile strategies to manipulate and decorate protein constructs, allowing the creation of molecules and assemblies that faithfully represent the complexity of biological systems. We hope that this resource will serve as an inspiration to the structural biologist looking to improve on sample preparation protocols, construct interesting and relevant samples, or devise new strategies that push the boundaries of modern structural biology methods. Undoubtedly, these tools will continue to improve and evolve, expanding the range of options for both in vitro and in vivo applications. Yet, many challenges still lie ahead. For example, better tools are desperately needed to reign in obstinate systems such as membrane proteins, intrinsically disordered domains or amyloidogeneic polypeptides (Butterfield et al. Reference Butterfield, Hejjaoui, Fauvet, Awad and Lashuel2012; Uversky, Reference Uversky2015; Zuo et al. Reference Zuo, Tang and Zheng2015). These constructs, traditionally recalcitrant to protein engineering and structural biology analysis alike, beg for more efficient (semi)-synthetic, genetic or intein/sortase-based approaches that circumvent low expression yields and poor solubility issues. Next, the site-specific manipulation of large proteins and assemblies (especially those prepared in eukaryotic systems) is still an overwhelmingly difficult task, and new chemical tools and ideas are needed to supplement the existing genetic and enzymatic approaches. At the same time, the demand for constructs bearing multiple modifications will continue to increase. For example, dissecting the molecular basis of biological cross-talks requires access to homogeneous samples of proteins carrying multiple chemically diverse PTMs (Allis & Muir, Reference Allis and Muir2011; Bah & Forman-Kay, Reference Bah and Forman-Kay2016). Many biological molecules and assemblies are also too complex for structural analysis by one technique alone (Cramer, Reference Cramer2016; McGinty & Tan, Reference Mcginty and Tan2015; Tynan et al. Reference Tynan, Lo Schiavo, Zanetti-Domingues, Needham, Roberts, Hirsch, Rolfe, Korovesis, Clarke and Martin-Fernandez2016), and the increasing need for method integration will necessitate the construction of samples suitable for multi-modal studies.
Concurrently, structural biologists are devising new strategies to improve the sensitivity of their respective methodologies and reduce the amounts of precious biological materials required for structural analysis. New technological developments such as X-ray free-electron lasers (Neutze et al. Reference Neutze, Branden and Schertler2015), polarization enhancement strategies for NMR spectroscopy (Ardenkjaer-Larsen et al. Reference Ardenkjaer-Larsen, Boebinger, Comment, Duckett, Edison, Engelke, Griesinger, Griffin, Hilty, Maeda, Parigi, Prisner, Ravera, Van Bentum, Vega, Webb, Luchinat, Schwalbe and Frydman2015; Maly et al. Reference Maly, Debelouchina, Bajaj, Hu, Joo, Mak-Jurkauskas, Sirigiri, Van Der Wel, Herzfeld, Temkin and Griffin2008), and the enhanced capabilities of cryo-EM instrumentation (Nogales, Reference Nogales2016) will certainly be central to these efforts. The rise of mass spectrometry based structural approaches, including hydrogen–deuterium exchange, ion mobility-mass spectrometry or cross-linking based analysis, also holds the bright promise to reveal structural information from samples in the picomole regime (Lossl et al. Reference Lossl, Van De Waterbeemd and Heck2016). At the same time, structural biologists have started to turn to the cellular milieu as the future arena of structural endeavors (Beck & Baumeister, Reference Beck and Baumeister2016; Freedberg & Selenko, Reference Freedberg and Selenko2014). In this undoubtedly daunting task, structural biologists are not alone. Protein engineers, with their long track record of successes in the selective manipulation of complex systems both in vitro and in vivo, are ready to meet these exciting new challenges through innovations that will continue to push the boundaries of chemical biology.
Acknowledgements
We would like to thank Adam Stevens, Antony Burton, Felix Wojcik and Robert Thompson for many helpful discussions. We also acknowledge the contributions of many colleagues whose work could not be included in this review due to space limitations. This work was supported by US National Institutes of Health grants R37-GM086868, R01-GM107047 and P01-CA196539.















