We use cookies to distinguish you from other users and to provide you with a better experience on our websites. Close this message to accept cookies or find out how to manage your cookie settings.
This journal utilises an Online Peer Review Service (OPRS) for submissions. By clicking "Continue" you will be taken to our partner site
https://mc.manuscriptcentral.com/qrb-discovery.
Please be aware that your Cambridge account is not valid for this OPRS and registration is required. We strongly advise you to read all "Author instructions" in the "Journal information" area prior to submitting.
To save this undefined to your undefined account, please select one or more formats and confirm that you agree to abide by our usage policies. If this is the first time you used this feature, you will be asked to authorise Cambridge Core to connect with your undefined account.
Find out more about saving content to .
To send this article to your Kindle, first ensure [email protected] is added to your Approved Personal Document E-mail List under your Personal Document Settings on the Manage Your Content and Devices page of your Amazon account. Then enter the ‘name’ part of your Kindle email address below. Find out more about sending to your Kindle.
Find out more about saving to your Kindle.
Note you can select to save to either the @free.kindle.com or @kindle.com variations. ‘@free.kindle.com’ emails are free but can only be saved to your device when it is connected to wi-fi. ‘@kindle.com’ emails can be delivered even when you are not connected to wi-fi, but note that service fees apply.
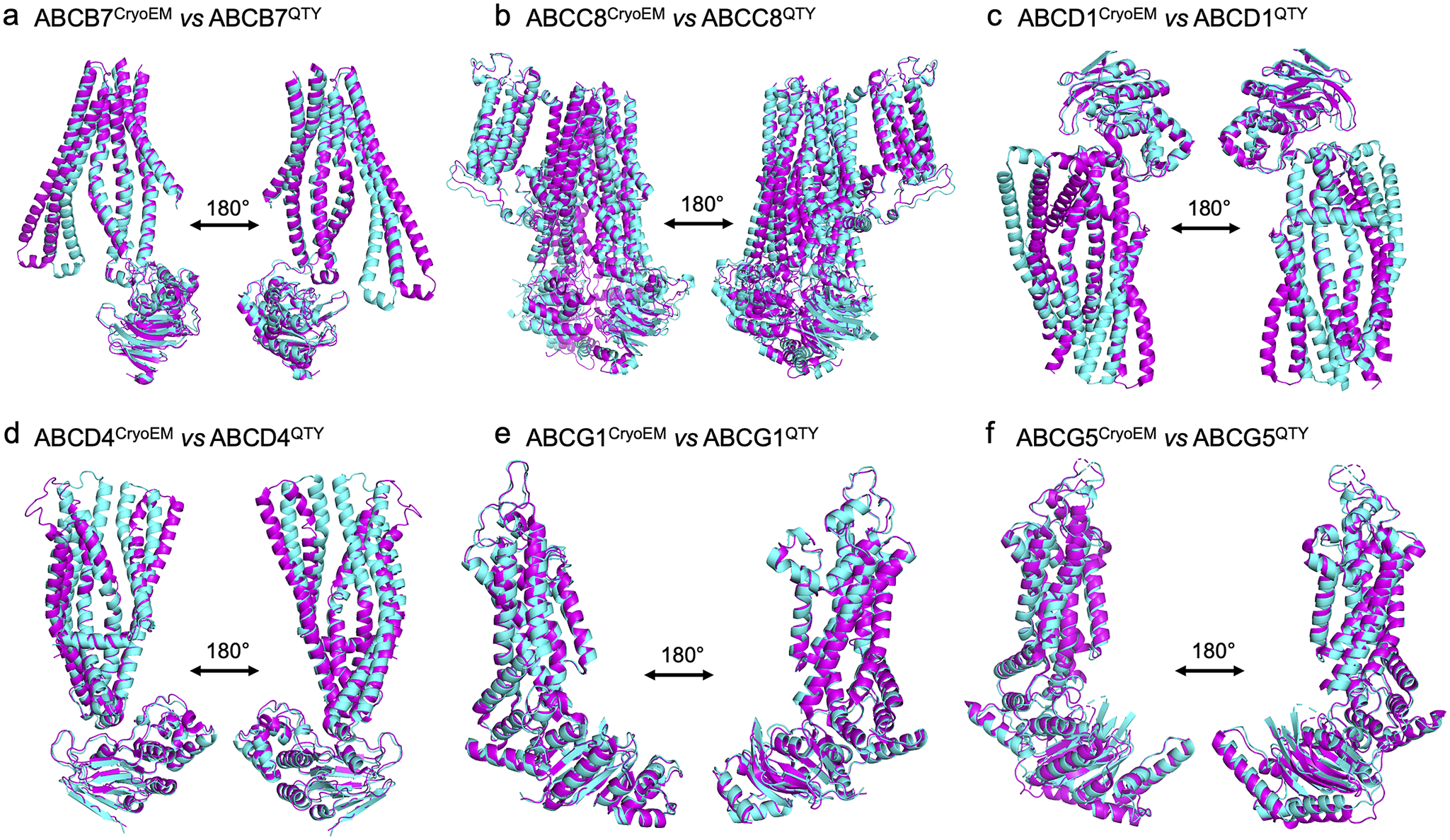
Human ATP-binding cassette (ABC) transporters are one of the largest families of membrane proteins and perform diverse functions. Many of them are associated with multidrug resistance that often results in cancer treatment with poor outcomes. Here, we present the structural bioinformatics study of six human ABC membrane transporters with experimentally determined cryo-electron microscopy (CryoEM) structures including ABCB7, ABCC8, ABCD1, ABCD4, ABCG1, ABCG5, and their AlphaFold2-predicted water-soluble QTY variants. In the native structures, there are hydrophobic amino acids such as leucine (L), isoleucine (I), valine (V), and phenylalanine (F) in the transmembrane alpha helices. These hydrophobic amino acids are systematically replaced by hydrophilic amino acids glutamine (Q), threonine (T), and tyrosine (Y). Therefore, these QTY variants become water soluble. We also present the superposed structures of native ABC transporters and their water-soluble QTY variants. The superposed structures show remarkable similarity with root mean square deviations between 1.064 and 3.413 Å despite significant (41.90–54.33%) changes to the protein sequence of the transmembrane domains. We also show the differences in hydrophobicity patches between the native ABC transporters and their QTY variants. We explain the rationale behind why the QTY membrane protein variants become water soluble. Our structural bioinformatics studies provide insight into the differences between the hydrophobic helices and hydrophilic helices and will likely further stimulate designs of water-soluble multispan transmembrane proteins and other aggregated proteins. The water-soluble ABC transporters may be useful as soluble antigens to generate therapeutic monoclonal antibodies for combating multidrug resistance in clinics.
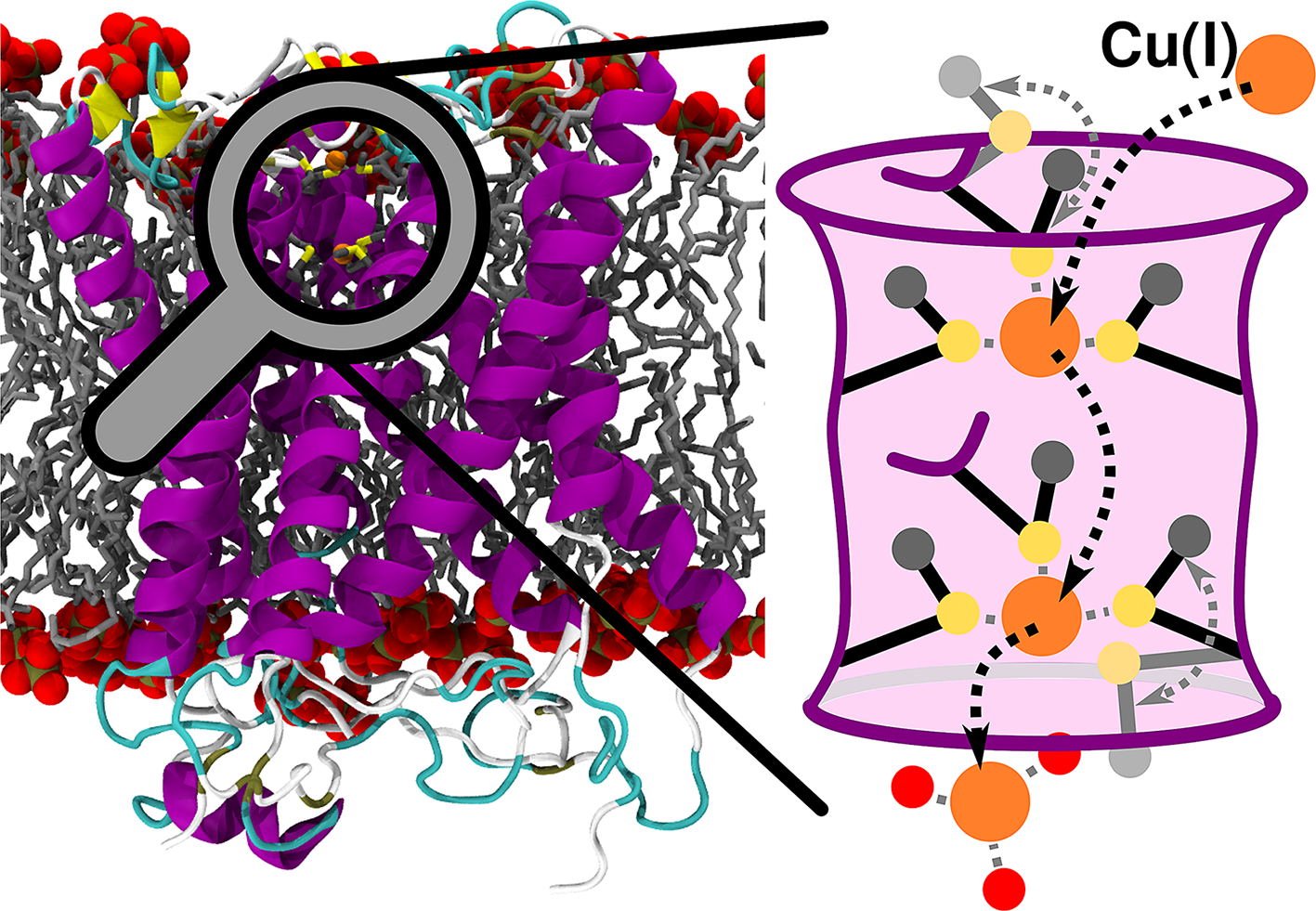
Copper is a trace element vital to many cellular functions. Yet its abnormal levels are toxic to cells, provoking a variety of severe diseases. The high affinity copper transporter 1 (CTR1), being the main in-cell copper [Cu(I)] entry route, tightly regulates its cellular uptake via a still elusive mechanism. Here, all-atoms simulations unlock the molecular terms of Cu(I) transport in eukaryotes disclosing that the two methionine (Met) triads, forming the selectivity filter, play an unprecedented dual role both enabling selective Cu(I) transport and regulating its uptake rate thanks to an intimate coupling between the conformational plasticity of their bulky side chains and the number of bound Cu(I) ions. Namely, the Met residues act as a gate reducing the Cu(I) import rate when two ions simultaneously bind to CTR1. This may represent an elegant autoregulatory mechanism through which CTR1 protects the cells from excessively high, and hence toxic, in-cell Cu(I) levels. Overall, our outcomes resolve fundamental questions in CTR1 biology and open new windows of opportunity to tackle diseases associated with an imbalanced copper uptake.
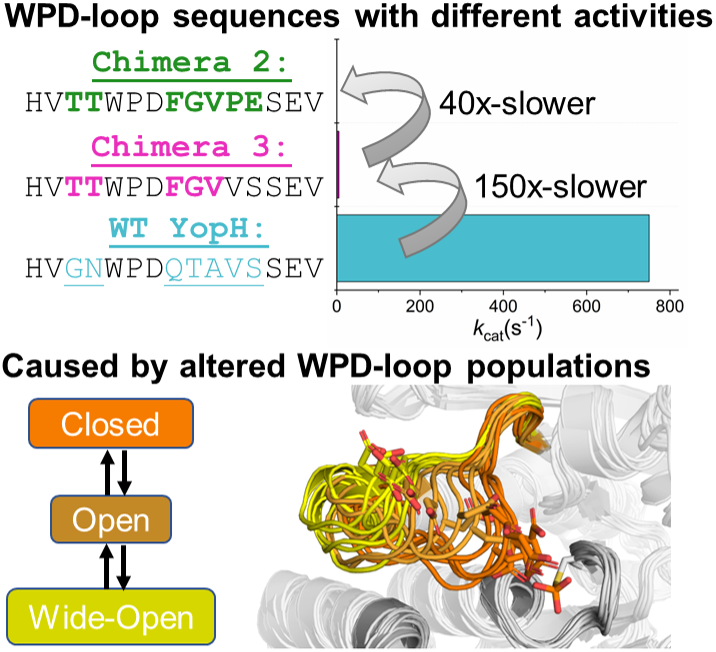
Protein tyrosine phosphatases (PTPs) are crucial regulators of cellular signaling. Their activity is regulated by the motion of a conserved loop, the WPD-loop, from a catalytically inactive open to a catalytically active closed conformation. WPD-loop motion optimally positions a catalytically critical residue into the active site, and is directly linked to the turnover number of these enzymes. Crystal structures of chimeric PTPs constructed by grafting parts of the WPD-loop sequence of PTP1B onto the scaffold of YopH showed WPD-loops in a wide-open conformation never previously observed in either parent enzyme. This wide-open conformation has, however, been observed upon binding of small molecule inhibitors to other PTPs, suggesting the potential of targeting it for drug discovery efforts. Here, we have performed simulations of both enzymes and show that there are negligible energetic differences in the chemical step of catalysis, but significant differences in the dynamical properties of the WPD-loop. Detailed interaction network analysis provides insight into the molecular basis for this population shift to a wide-open conformation. Taken together, our study provides insight into the links between loop dynamics and chemistry in these YopH variants specifically, and how WPD-loop dynamic can be engineered through modification of the internal protein interaction network.
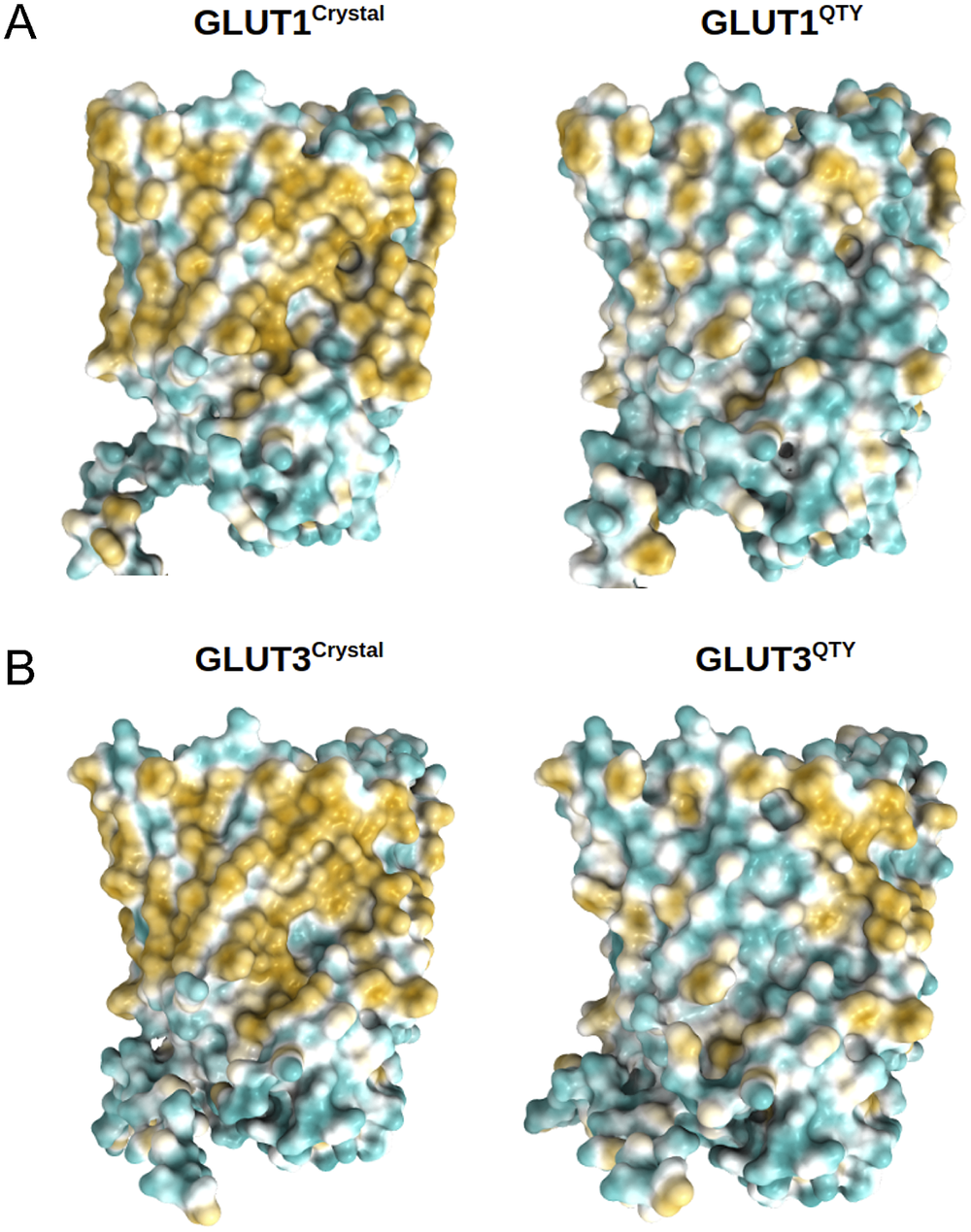
Membrane transporters including glucose transporters (GLUTs) are involved in cellular energy supplies, cell metabolism and other vital biological activities. They have also been implicated in cancer proliferation and metastasis, thus they represent an important target in combatting cancer. However, membrane transporters are very difficult to study due to their multispan transmembrane properties. The new computational tool, AlphaFold2, offers highly accurate predictions of three-dimensional protein structures. The glutamine, threonine and tyrosine (QTY) code provides a systematic method of rendering hydrophobic sequences into hydrophilic ones. Here, we present computational studies of native integral membrane GLUTs with 12 transmembrane helical segments determined by X-ray crystallography and CryoEM, comparing the AlphaFold2-predicted native structure to their water-soluble QTY variants predicted by AlphaFold2. In the native structures of the transmembrane helices, there are hydrophobic amino acids leucine (L), isoleucine (I), valine (V) and phenylalanine (F). Applying the QTY code, these hydrophobic amino acids are systematically replaced by hydrophilic amino acids, glutamine (Q), threonine (T) and tyrosine (Y) rendering them water-soluble. We present the superposed structures of native GLUTs and their water-soluble QTY variants. The superposed structures show remarkable similar residue mean square distance values between 0.47 and 3.6 Å (most about 1–2 Å) despite >44% transmembrane amino acid differences. We also show the differences of hydrophobicity patches between the native membrane transporters and their QTY variants. We explain the rationale why the membrane protein QTY variants become water-soluble. Our study provides insight into the differences between the hydrophobic helices and hydrophilic helices, and offers confirmation of the QTY method for studying multispan transmembrane proteins and other aggregated proteins through their water-soluble variants.
The resistance–nodulation–division efflux machinery confers antimicrobial resistance to Gram-negative bacteria by actively pumping antibiotics out of the cell. The protein complex is powered by proton motive force; however, the proton transfer mechanism itself and indeed even its stoichiometry is still unclear. Here we review computational studies from the last decade that focus on elucidating the number of protons transferred per conformational cycle of the pump. Given the difficulties in studying proton movement using even state-of-the-art structural biology methods, the contributions from computational studies have been invaluable from a mechanistic perspective.
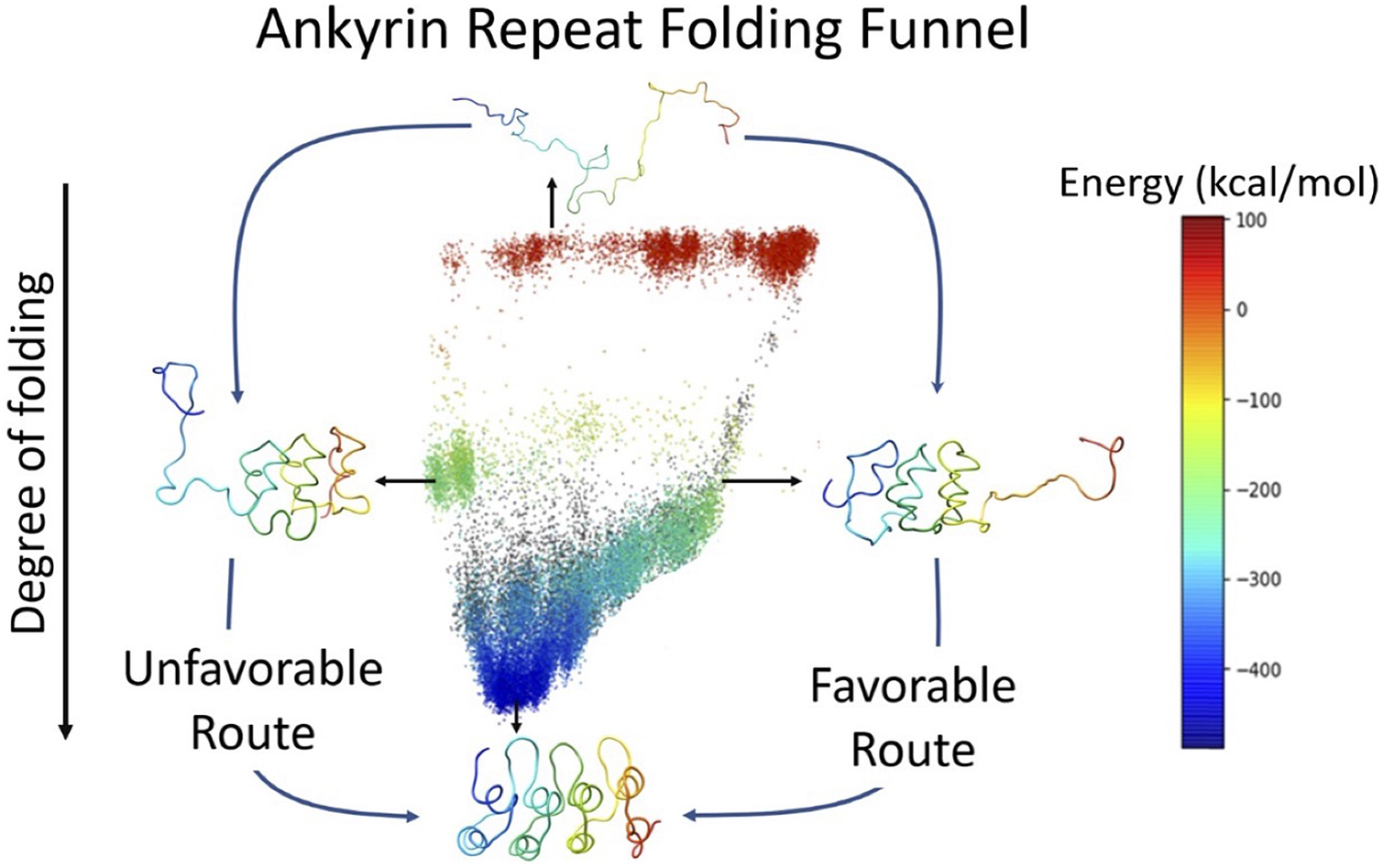
Ankyrin (ANK) repeat proteins are coded by tandem occurrences of patterns with around 33 amino acids. They often mediate protein–protein interactions in a diversity of biological systems. These proteins have an elongated non-globular shape and often display complex folding mechanisms. This work investigates the energy landscape of representative proteins of this class made up of 3, 4 and 6 ANK repeats using the energy-landscape visualisation method (ELViM). By combining biased and unbiased coarse-grained molecular dynamics AWSEM simulations that sample conformations along the folding trajectories with the ELViM structure-based phase space, one finds a three-dimensional representation of the globally funnelled energy surface. In this representation, it is possible to delineate distinct folding pathways. We show that ELViMs can project, in a natural way, the intricacies of the highly dimensional energy landscapes encoded by the highly symmetric ankyrin repeat proteins into useful low-dimensional representations. These projections can discriminate between multiplicities of specific parallel folding mechanisms that otherwise can be hidden in oversimplified depictions.
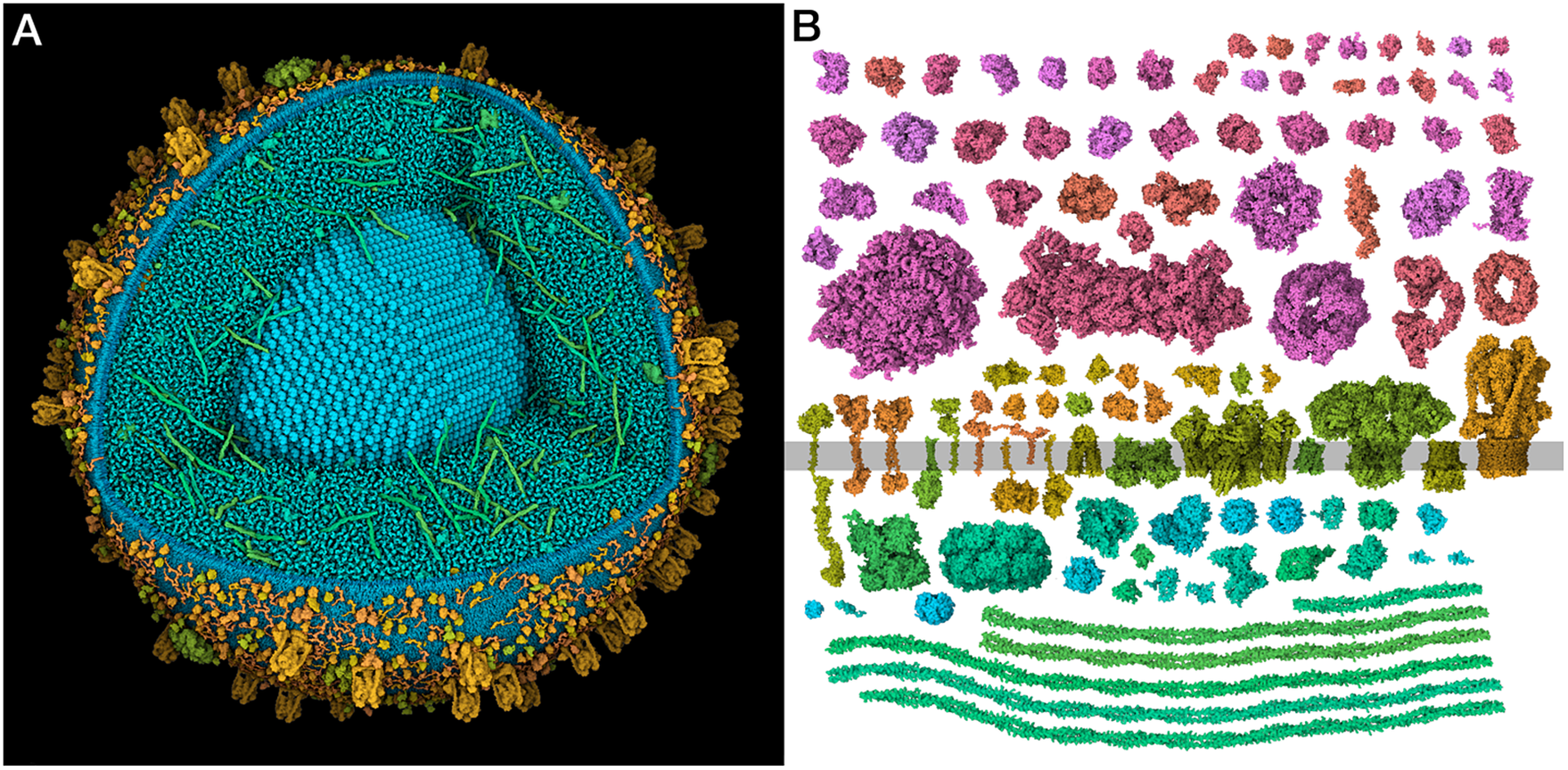
Models of insulin secretory vesicles from pancreatic beta cells have been created using the cellPACK suite of tools to research, curate, construct and visualise the current state of knowledge. The model integrates experimental information from proteomics, structural biology, cryoelectron microscopy and X-ray tomography, and is used to generate models of mature and immature vesicles. A new method was developed to generate a confidence score that reconciles inconsistencies between three available proteomes using expert annotations of cellular localisation. The models are used to simulate soft X-ray tomograms, allowing quantification of features that are observed in experimental tomograms, and in turn, allowing interpretation of X-ray tomograms at the molecular level.
Biomolecular recognition including binding of small molecules, peptides and proteins to their target receptors plays a key role in cellular function and has been targeted for therapeutic drug design. However, the high flexibility of biomolecules and slow binding and dissociation processes have presented challenges for computational modelling. Here, we review the challenges and computational approaches developed to characterise biomolecular binding, including molecular docking, molecular dynamics simulations (especially enhanced sampling) and machine learning. Further improvements are still needed in order to accurately and efficiently characterise binding structures, mechanisms, thermodynamics and kinetics of biomolecules in the future.
Machine learning (ML) has revolutionised the field of structure-based drug design (SBDD) in recent years. During the training stage, ML techniques typically analyse large amounts of experimentally determined data to create predictive models in order to inform the drug discovery process. Deep learning (DL) is a subfield of ML, that relies on multiple layers of a neural network to extract significantly more complex patterns from experimental data, and has recently become a popular choice in SBDD. This review provides a thorough summary of the recent DL trends in SBDD with a particular focus on de novo drug design, binding site prediction, and binding affinity prediction of small molecules.
Recent breakthroughs in deep learning-based protein structure prediction show that it is possible to obtain highly accurate models for a wide range of difficult protein targets for which only the amino acid sequence is known. The availability of accurately predicted models from sequences can potentially revolutionise many modelling approaches in structural biology, including the interpretation of cryo-EM density maps. Although atomic structures can be readily solved from cryo-EM maps of better than 4 Å resolution, it is still challenging to determine accurate models from lower-resolution density maps. Here, we report on the benefits of models predicted by AlphaFold2 (the best-performing structure prediction method at CASP14) on cryo-EM refinement using the Phenix refinement suite for AlphaFold2 models. To study the robustness of model refinement at a lower resolution of interest, we introduced hybrid maps (i.e. experimental cryo-EM maps) filtered to lower resolutions by real-space convolution. The AlphaFold2 models were refined to attain good accuracies above 0.8 TM scores for 9 of the 13 cryo-EM maps. TM scores improved for AlphaFold2 models refined against all 13 cryo-EM maps of better than 4.5 Å resolution, 8 hybrid maps of 6 Å resolution, and 3 hybrid maps of 8 Å resolution. The results show that it is possible (at least with the Phenix protocol) to extend the refinement success below 4.5 Å resolution. We even found isolated cases in which resolution lowering was slightly beneficial for refinement, suggesting that high-resolution cryo-EM maps might sometimes trap AlphaFold2 models in local optima.
Peptides mediate up to 40% of protein interactions, their high specificity and ability to bind in places where small molecules cannot make them potential drug candidates. However, predicting peptide–protein complexes remains more challenging than protein–protein or protein–small molecule interactions, in part due to the high flexibility peptides have. In this review, we look at the advances in docking, molecular simulations and machine learning to tackle problems related to peptides such as predicting structures, binding affinities or even kinetics. We specifically focus on explaining the number of docking programmes and force fields used in molecular simulations, so a prospective user can have an educated guess as to why choose one modelling tool or another to address their scientific questions.