



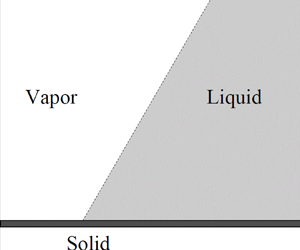
1. Introduction
The diffuse-interface model (DIM) was proposed by Korteweg (Reference Korteweg1901) as an attempt to avoid the abrupt change of parameters in the models of liquid/vapour interfaces existing at the time. It is based on the following two assumptions.
(i) The long-range attractive intermolecular force (the van der Waals force) can be modelled by a pairwise potential, so that the force affecting a molecule is the algebraic sum of those exerted on it by other molecules.
(ii) The characteristic distance over which the van der Waals force acts is much smaller than the interfacial thickness.
The resulting representation of the molecular interaction has been incorporated into models of various phenomena, such as phase transitions in ferroelectric materials (Ginzburg Reference Ginzburg1960), spinodal decomposition (Cahn Reference Cahn1961; Lowengrub & Truskinovsky Reference Lowengrub and Truskinovsky1998), growth of, and oscillations in, crystals (Collins & Levine Reference Collins and Levine1985; Tang, Carter & Cannon Reference Tang, Carter and Cannon2006; Heinonen et al. Reference Heinonen, Achim, Kosterlitz, Ying, Lowengrub and Ala-Nissila2016; Philippe, Henry & Plapp Reference Philippe, Henry and Plapp2020), solidification of alloys (Stinner, Nestler & Garcke Reference Stinner, Nestler and Garcke2004; Nestler, Garcke & Stinner Reference Nestler, Garcke and Stinner2005), phase separation in polymer blends (Thiele, Madruga & Frastia Reference Thiele, Madruga and Frastia2007; Madruga & Thiele Reference Madruga and Thiele2009), electrowetting (Lu et al. Reference Lu, Glasner, Bertozzi and Kim2007), contact lines (Jacqmin Reference Jacqmin2000; Pismen & Pomeau Reference Pismen and Pomeau2000; Ding & Spelt Reference Ding and Spelt2007; Yue, Zhou & Feng Reference Yue, Zhou and Feng2010; Yue & Feng Reference Yue and Feng2011; Sibley et al. Reference Sibley, Nold, Savva and Kalliadasis2014; Ding et al. Reference Ding, Zhu, Gao and Lu2017; Borcia et al. Reference Borcia, Borcia, Bestehorn, Varlamova, Hoefner and Reif2019), contact lines in liquids with surfactants (Zhu et al. Reference Zhu, Kou, Yao, Wu, Yao and Sun2019, Reference Zhu, Kou, Yao, Li and Sun2020), Faraday instability (Borcia & Bestehorn Reference Borcia and Bestehorn2014; Bestehorn et al. Reference Bestehorn, Sharma, Borcia and Amiroudine2021), Rayleigh–Taylor instability (Zanella et al. Reference Zanella, Tegze, Tellier and Henry2020, Reference Zanella, Le Tellier, Plapp, Tegze and Henry2021), cavitation (Petitpas et al. Reference Petitpas, Massoni, Saurel, Lapebie and Munier2009), nucleation and collapse of bubbles (Magaletti, Marino & Casciola Reference Magaletti, Marino and Casciola2015; Magaletti et al. Reference Magaletti, Gallo, Marino and Casciola2016; Gallo, Magaletti & Casciola Reference Gallo, Magaletti and Casciola2018; Gallo et al. Reference Gallo, Magaletti, Cocco and Casciola2020), capillary condensation (Pomeau Reference Pomeau1986; Benilov Reference Benilov2022a), liquid films (Benilov Reference Benilov2020c, Reference Benilov2022a), tumor growth (Frigeri, Grasselli & Rocca Reference Frigeri, Grasselli and Rocca2015; Rocca & Scala Reference Rocca and Scala2016; Dai et al. Reference Dai, Feireisl, Rocca, Schimperna and Schonbek2017), classification of high-dimensional data (Bertozzi & Flenner Reference Bertozzi and Flenner2012, Reference Bertozzi and Flenner2016), etc.
The present paper is mainly concerned with the application of the DIM to the evaporation of drops and the condensation of vapour on a solid. These phenomena have been examined using the single-component version of the DIM (Benilov Reference Benilov2022a,Reference Benilovb), where the fluid/gas interface is modelled by that between the liquid and vapour phases of the same fluid. The evaporation in this case was shown to be due to a flow caused by a weak imbalance of chemical potentials of the liquid and vapour phases.
It is unclear, however, how the results obtained via the single-component DIM are modified by the effect of air, whose density (at, say,  $25\,^{\circ }\mathrm {C}$) exceeds that of water vapour by a factor of more than
$25\,^{\circ }\mathrm {C}$) exceeds that of water vapour by a factor of more than  $50$. Furthermore, there are three additional physical effects in multicomponent fluids: diffusion (of water vapour in air), the Soret effect (thermodiffusion) and the Dufour effect (heat flux due to a density gradient). Only one of these, the diffusion, has been examined before (e.g. Deegan et al. Reference Deegan, Bakajin, Dupont, Huber, Nagel and Witten2000; Dunn et al. Reference Dunn, Wilson, Duffy, David and Sefiane2009; Eggers & Pismen Reference Eggers and Pismen2010; Colinet & Rednikov Reference Colinet and Rednikov2011; Rednikov & Colinet Reference Rednikov and Colinet2013; Morris Reference Morris2014; Stauber et al. Reference Stauber, Wilson, Duffy and Sefiane2014, Reference Stauber, Wilson, Duffy and Sefiane2015; Janeček et al. Reference Janeček, Doumenc, Guerrier and Nikolayev2015; Saxton et al. Reference Saxton, Whiteley, Vella and Oliver2016, Reference Saxton, Vella, Whiteley and Oliver2017; Rednikov & Colinet Reference Rednikov and Colinet2019; Wray, Duffy & Wilson Reference Wray, Duffy and Wilson2019), but the models employed in these papers do not include the flow-induced evaporative flux discovered via the DIM. Thus, the multicomponent DIM appears to be a tool describing all the mechanisms at work in evaporation/condensation of liquids into/from air.
$50$. Furthermore, there are three additional physical effects in multicomponent fluids: diffusion (of water vapour in air), the Soret effect (thermodiffusion) and the Dufour effect (heat flux due to a density gradient). Only one of these, the diffusion, has been examined before (e.g. Deegan et al. Reference Deegan, Bakajin, Dupont, Huber, Nagel and Witten2000; Dunn et al. Reference Dunn, Wilson, Duffy, David and Sefiane2009; Eggers & Pismen Reference Eggers and Pismen2010; Colinet & Rednikov Reference Colinet and Rednikov2011; Rednikov & Colinet Reference Rednikov and Colinet2013; Morris Reference Morris2014; Stauber et al. Reference Stauber, Wilson, Duffy and Sefiane2014, Reference Stauber, Wilson, Duffy and Sefiane2015; Janeček et al. Reference Janeček, Doumenc, Guerrier and Nikolayev2015; Saxton et al. Reference Saxton, Whiteley, Vella and Oliver2016, Reference Saxton, Vella, Whiteley and Oliver2017; Rednikov & Colinet Reference Rednikov and Colinet2019; Wray, Duffy & Wilson Reference Wray, Duffy and Wilson2019), but the models employed in these papers do not include the flow-induced evaporative flux discovered via the DIM. Thus, the multicomponent DIM appears to be a tool describing all the mechanisms at work in evaporation/condensation of liquids into/from air.
The same can hopefully be said about the dynamics of contact lines, as most of the existing models work for some fluids (including water) only if the so-called slip length – effectively, the interfacial thickness – is set to be unrealistically small (Podgorski, Flesselles & Limat Reference Podgorski, Flesselles and Limat2001; Winkels et al. Reference Winkels, Peters, Evangelista, Riepen, Daerr, Limat and Snoeijer2011; Puthenveettil, Senthilkumar & Hopfinger Reference Puthenveettil, Senthilkumar and Hopfinger2013; Limat Reference Limat2014; Benilov & Benilov Reference Benilov and Benilov2015).
Before using a new model, one generally needs to examine its basic properties, test it on problems with well-understood physics (to ensure that the mathematics captures it) and parameterise this model for the intended applications. These are the three aims of the present work in the context of the multicomponent DIM.
The following results are reported.
(i) Multicomponent flat interfaces with monotonically changing densities of the species (components) are all stable. This conclusion follows from the entropy principle and conservation laws.
(ii) Several new physical effects are described, the most interesting of which is evaporation of a flat liquid layer in contact with saturated vapour. This phenomenon appears to be similar to evaporation of drops surrounded by saturated vapour (Deegan et al. Reference Deegan, Bakajin, Dupont, Huber, Nagel and Witten2000; Eggers & Pismen Reference Eggers and Pismen2010; Benilov Reference Benilov2020d, Reference Benilov2021, Reference Benilov2022b), but with one important difference: the drops evaporate because the curvature of their boundary increases the effective saturation pressure (the so-called Kelvin effect), making the saturated vapour effectively under saturated and, thus, encouraging evaporation. This explanation is clearly inapplicable to layers with flat boundaries – yet they evaporate anyway. The actual mechanism is based on the long-range interaction of the liquid/vapour interface with the substrate, which implies that, for macroscopic liquid films, this effect is weak. It should be important, however, for nano-films whose thickness is comparable to the interfacial thickness.
(iii) The multicomponent DIM is parameterised for water/air interfaces at normal conditions, which can be used in the future for modelling evaporation and condensation of water in the Earth's atmosphere or the dynamics of contact lines of water drops.
The paper has the following structure. In § 2 the problem is formulated mathematically. Section 3 examines the entropy principle and conservation laws. In § 4 the governing equations are non-dimensionalised and the main non-dimensional parameters are identified. Sections 5–7 examine basic solutions of the DIM, and in § 8 the DIM is parameterised for water/air interfaces. Section 9 summarises the results obtained, including the effects partly mentioned in item (ii) of the above list.
2. Formulation
2.1. Thermodynamics
When studying hydrodynamics of a compressible fluid, one has to deal with its thermodynamic properties. In this section they are described briefly and in a self-contained form (for the benefit of readers specializing is incompressible hydrodynamics).
Consider an  $N$-component compressible non-ideal fluid, characterised by the temperature
$N$-component compressible non-ideal fluid, characterised by the temperature  $T$ and partial mass densities
$T$ and partial mass densities  $\rho _{i}$, where
$\rho _{i}$, where  $i=1\ldots N$. The fluid's thermodynamic properties are fully described by two functions: the internal energy
$i=1\ldots N$. The fluid's thermodynamic properties are fully described by two functions: the internal energy  $e(\rho _{1}\ldots \rho _{N},T)$ and entropy
$e(\rho _{1}\ldots \rho _{N},T)$ and entropy  $s(\rho _{1}\ldots \rho _{N},T)$, both specific, i.e. per unit mass. The dependence of the fluid pressure
$s(\rho _{1}\ldots \rho _{N},T)$, both specific, i.e. per unit mass. The dependence of the fluid pressure  $p$ on
$p$ on  $(\rho _{1}\ldots \rho _{N},T)$, or the equation of state, is defined by
$(\rho _{1}\ldots \rho _{N},T)$, or the equation of state, is defined by
 \begin{equation} p=\rho\sum_{i}\rho_{i}\left( \frac{\partial e}{\partial\rho_{i}} -T\frac{\partial s}{\partial\rho_{i}}\right) , \end{equation}
\begin{equation} p=\rho\sum_{i}\rho_{i}\left( \frac{\partial e}{\partial\rho_{i}} -T\frac{\partial s}{\partial\rho_{i}}\right) , \end{equation}(e.g. Giovangigli & Matuszewski Reference Giovangigli and Matuszewski2013), where
 \begin{equation} \rho=\sum_{i}\rho_{i} \end{equation}
\begin{equation} \rho=\sum_{i}\rho_{i} \end{equation}
is the total density (here and hereinafter, the summation is implied to be from  $1$ to
$1$ to  $N$ unless stated otherwise). The partial chemical potentials, in turn, are given by
$N$ unless stated otherwise). The partial chemical potentials, in turn, are given by
 \begin{equation} G_{i}=\frac{\partial( \rho e) }{\partial\rho_{i}}-T\frac {\partial( \rho s) }{\partial\rho_{i}}. \end{equation}
\begin{equation} G_{i}=\frac{\partial( \rho e) }{\partial\rho_{i}}-T\frac {\partial( \rho s) }{\partial\rho_{i}}. \end{equation}
Note that  $e(\rho _{1}\ldots \rho _{N},T)$ and
$e(\rho _{1}\ldots \rho _{N},T)$ and  $s(\rho _{1}\ldots \rho _{N},T)$ are not fully arbitrary, but should satisfy the fundamental thermodynamic relation, or the Gibbs relation, which can be written in the form
$s(\rho _{1}\ldots \rho _{N},T)$ are not fully arbitrary, but should satisfy the fundamental thermodynamic relation, or the Gibbs relation, which can be written in the form
 \begin{equation} \frac{\partial e}{\partial T}=T\frac{\partial s}{\partial T}. \end{equation}
\begin{equation} \frac{\partial e}{\partial T}=T\frac{\partial s}{\partial T}. \end{equation}The equivalence of this equality and the standard form of the Gibbs relation are shown in Appendix A.
Using (2.1)–(2.4), one can derive the identities
 $$\begin{gather} \frac{\partial p}{\partial T}=\sum_{i}\rho_{i}\frac{\partial G_{i}}{\partial T}+\rho s, \end{gather}$$
$$\begin{gather} \frac{\partial p}{\partial T}=\sum_{i}\rho_{i}\frac{\partial G_{i}}{\partial T}+\rho s, \end{gather}$$ $$\begin{gather}\frac{\partial p}{\partial\rho_{j}}=\sum_{i}\rho_{i}\frac{\partial G_{i} }{\partial\rho_{j}}, \end{gather}$$
$$\begin{gather}\frac{\partial p}{\partial\rho_{j}}=\sum_{i}\rho_{i}\frac{\partial G_{i} }{\partial\rho_{j}}, \end{gather}$$then rewrite (2.6) in the form
 \begin{equation} G_{i}=\frac{\partial}{\partial\rho_{i}}\left( \sum_{j}\rho_{j}G_{j}-p\right) . \end{equation}
\begin{equation} G_{i}=\frac{\partial}{\partial\rho_{i}}\left( \sum_{j}\rho_{j}G_{j}-p\right) . \end{equation}In what follows, one also needs the specific heat capacity at constant volume,
 \begin{equation} c=\frac{\partial e}{\partial T}, \end{equation}
\begin{equation} c=\frac{\partial e}{\partial T}, \end{equation}
where the traditional subscript  $_{V}$ is omitted. In this paper
$_{V}$ is omitted. In this paper  $c$ is assumed to be positive, which is indeed the case for neutral fluids (Lynden-Bell Reference Lynden-Bell1999).
$c$ is assumed to be positive, which is indeed the case for neutral fluids (Lynden-Bell Reference Lynden-Bell1999).
Define also
 \begin{equation} a_{i}={-}\frac{\partial e}{\partial\rho_{i}}, \end{equation}
\begin{equation} a_{i}={-}\frac{\partial e}{\partial\rho_{i}}, \end{equation}
which will be referred to as the generalized van der Waals parameter (of the  $i$th species), and
$i$th species), and
 \begin{equation} B={-}p-\rho\sum_{i}\rho_{i}a_{i}. \end{equation}
\begin{equation} B={-}p-\rho\sum_{i}\rho_{i}a_{i}. \end{equation}
Here  $B$ is not one of the standard thermodynamic functions, but is convenient to use when thermodynamics is coupled to hydrodynamics; as seen later,
$B$ is not one of the standard thermodynamic functions, but is convenient to use when thermodynamics is coupled to hydrodynamics; as seen later,  $B$ characterises the release (consumption) of heat due to the fluid's mechanical compression (expansion). Using equation of state (2.1) and definition (2.9) of the van der Waals parameter, one can rearrange expression (2.10) in the form
$B$ characterises the release (consumption) of heat due to the fluid's mechanical compression (expansion). Using equation of state (2.1) and definition (2.9) of the van der Waals parameter, one can rearrange expression (2.10) in the form
 \begin{equation} B=\rho T\sum_{i}\rho_{i}\frac{\partial s}{\partial\rho_{i}}. \end{equation}
\begin{equation} B=\rho T\sum_{i}\rho_{i}\frac{\partial s}{\partial\rho_{i}}. \end{equation}2.2. Examples: the Enskog–Vlasov and van der Waals fluids
In the low-density limit, the specific internal energy and entropy of any fluid should tend to those of an ideal gas, i.e.
 \begin{equation} e\sim\frac{T}{\rho}\sum_{i}c_{i}\rho_{i},\quad s\sim\frac{\ln T}{\rho} \sum_{i}c_{i}\rho_{i}-\frac{1}{\rho}\sum_{i}R_{i}\rho_{i}\ln\rho_{i} \quad \text{as}\quad \rho_{i}\rightarrow0, \end{equation}
\begin{equation} e\sim\frac{T}{\rho}\sum_{i}c_{i}\rho_{i},\quad s\sim\frac{\ln T}{\rho} \sum_{i}c_{i}\rho_{i}-\frac{1}{\rho}\sum_{i}R_{i}\rho_{i}\ln\rho_{i} \quad \text{as}\quad \rho_{i}\rightarrow0, \end{equation}
where  $c_{i}$ is the specific partial heat capacity and
$c_{i}$ is the specific partial heat capacity and  $R_{i}=k_{B}/m_{i}$ is the specific gas constant, with
$R_{i}=k_{B}/m_{i}$ is the specific gas constant, with  $m_{i}$ the molecular mass of the
$m_{i}$ the molecular mass of the  $i$th species and
$i$th species and
 \begin{equation} k_{B}=1.380649\times10^{{-}23}\ \mathrm{m}^{2}\ \mathrm{s}^{{-}2} \mathrm{kg\ K}^{{-}1} \end{equation}
\begin{equation} k_{B}=1.380649\times10^{{-}23}\ \mathrm{m}^{2}\ \mathrm{s}^{{-}2} \mathrm{kg\ K}^{{-}1} \end{equation}
the Boltzmann constant. Note also that one can replace  $\ln T\rightarrow \ln T/\bar {T}$ and
$\ln T\rightarrow \ln T/\bar {T}$ and  $\ln \rho \rightarrow \ln \rho /\bar {\rho }$, where
$\ln \rho \rightarrow \ln \rho /\bar {\rho }$, where  $\bar {T}$ and
$\bar {T}$ and  $\bar {\rho }$ are suitable dimensional scales. This would make the arguments of the logarithms non-dimensional (with none of physically measurable parameters depending on
$\bar {\rho }$ are suitable dimensional scales. This would make the arguments of the logarithms non-dimensional (with none of physically measurable parameters depending on  $\bar {T}$ and
$\bar {T}$ and  $\bar {\rho }$).
$\bar {\rho }$).
A simple description of non-ideal fluids is delivered by the Enskog–Vlasov (EV) model, according to which
 $$\begin{gather} e=\frac{T}{\rho}\sum_{i}c_{i}\rho_{i}-\frac{1}{\rho}\sum_{ij}a_{ij}\rho _{i}\rho_{j}, \end{gather}$$
$$\begin{gather} e=\frac{T}{\rho}\sum_{i}c_{i}\rho_{i}-\frac{1}{\rho}\sum_{ij}a_{ij}\rho _{i}\rho_{j}, \end{gather}$$ $$\begin{gather}s=\frac{\ln T}{\rho}\sum_{i}c_{i}\rho_{i}-\frac{1}{\rho}\sum_{i}R_{i}\rho _{i}\ln\rho_{i}-\varTheta(\rho_{1}\ldots \rho_{N}), \end{gather}$$
$$\begin{gather}s=\frac{\ln T}{\rho}\sum_{i}c_{i}\rho_{i}-\frac{1}{\rho}\sum_{i}R_{i}\rho _{i}\ln\rho_{i}-\varTheta(\rho_{1}\ldots \rho_{N}), \end{gather}$$
where  $\varTheta (\rho _{1}\ldots \rho _{N})$ is an arbitrary analytic function and
$\varTheta (\rho _{1}\ldots \rho _{N})$ is an arbitrary analytic function and  $a_{ij}$ characterises the long-range attraction between molecules of the
$a_{ij}$ characterises the long-range attraction between molecules of the  $i$th and
$i$th and  $j$th species (with the implication that
$j$th species (with the implication that  $a_{ij}=a_{ji}$). For a pure fluid,
$a_{ij}=a_{ji}$). For a pure fluid,  $a_{11}$ coincides with the generalized van der Waals parameter defined by (2.9) – hence, the notation.
$a_{11}$ coincides with the generalized van der Waals parameter defined by (2.9) – hence, the notation.
One can readily verify that expressions (2.14) and (2.15) satisfy the Gibbs relation (2.4). They can be viewed as two-term expansions in small  $T$, under an extra assumption that the heat capacity
$T$, under an extra assumption that the heat capacity  $c$ and the generalized van der Waals parameter
$c$ and the generalized van der Waals parameter  $a_{i}$ are independent of
$a_{i}$ are independent of  $T$ and are zeroth-degree homogeneous functions of
$T$ and are zeroth-degree homogeneous functions of  $\rho _{i}$ (for a pure fluid,
$\rho _{i}$ (for a pure fluid,  $c$ and
$c$ and  $a_{1}$ do not depend on
$a_{1}$ do not depend on  $\rho _{1}$ at all).
$\rho _{1}$ at all).
The EV fluid model originates from the EV kinetic theory (de Sobrino Reference de Sobrino1967; Grmela Reference Grmela1971) and, as such, is naturally suited for use with the DIM that can be viewed as a hydrodynamic approximation of the EV kinetic equation (Giovangigli Reference Giovangigli2020, Reference Giovangigli2021). Equations (2.14) and (2.15) work very well for inert gases (Benilov & Benilov Reference Benilov and Benilov2019), they will be shown to work reasonably well for water, nitrogen and oxygen (see § 8 of this paper).
The EV involves too many parameters to be used as an illustration of general results – so, in such cases, the simpler van der Waals model will be employed. It is a particular case of (2.14) and (2.15) with
 \begin{equation} \varTheta={-}\frac{1}{\rho}\sum_{i}R_{i}\rho_{i}\ln\left( 1- {\sum_{j}} b_{j}\rho_{j}\right) , \end{equation}
\begin{equation} \varTheta={-}\frac{1}{\rho}\sum_{i}R_{i}\rho_{i}\ln\left( 1- {\sum_{j}} b_{j}\rho_{j}\right) , \end{equation}
where  $b_{i}$ is the reciprocal of the maximum density of the
$b_{i}$ is the reciprocal of the maximum density of the  $i$th species. Physically,
$i$th species. Physically,  $1/b_{i}$ can be interpreted as density of the closest packing.
$1/b_{i}$ can be interpreted as density of the closest packing.
Given expressions (2.14)–(2.16), definition (2.1) of the pressure and definition (2.3) of the chemical potential yield
 $$\begin{gather} p=\frac{T}{1- {\displaystyle\sum_{j}} b_{j}\rho_{j}}\sum_{i}R_{i}\rho_{i}-\sum_{ij}a_{ij}\rho_{i}\rho_{j}, \end{gather}$$
$$\begin{gather} p=\frac{T}{1- {\displaystyle\sum_{j}} b_{j}\rho_{j}}\sum_{i}R_{i}\rho_{i}-\sum_{ij}a_{ij}\rho_{i}\rho_{j}, \end{gather}$$ $$\begin{gather}G_{i}=TR_{i}\ln\frac{\rho_{i}}{1- {\displaystyle\sum_{j}} b_{j}\rho_{j}}+\frac{Tb_{i} {\displaystyle\sum_{j}} R_{j}\rho_{j}}{1- {\displaystyle\sum_{j}} b_{j}\rho_{j}}-2\sum_{j}a_{ij}\rho_{j}+T( R_{i}+c_{i}-c_{i}\ln T) . \end{gather}$$
$$\begin{gather}G_{i}=TR_{i}\ln\frac{\rho_{i}}{1- {\displaystyle\sum_{j}} b_{j}\rho_{j}}+\frac{Tb_{i} {\displaystyle\sum_{j}} R_{j}\rho_{j}}{1- {\displaystyle\sum_{j}} b_{j}\rho_{j}}-2\sum_{j}a_{ij}\rho_{j}+T( R_{i}+c_{i}-c_{i}\ln T) . \end{gather}$$
For a pure fluid ( $N=1$), (2.17) reduces to the classical van der Waals equation of state (van der Waals Reference van der Waals1893).
$N=1$), (2.17) reduces to the classical van der Waals equation of state (van der Waals Reference van der Waals1893).
2.3. Hydrodynamics
Consider a fluid flow characterised by the species densities  $\rho _{i}(\boldsymbol {r},t)$, mass-averaged velocity
$\rho _{i}(\boldsymbol {r},t)$, mass-averaged velocity  $\boldsymbol {v}(\boldsymbol {r},t)$ and temperature
$\boldsymbol {v}(\boldsymbol {r},t)$ and temperature  $T(\boldsymbol {r},t)$, where
$T(\boldsymbol {r},t)$, where  $\boldsymbol {r}=[ x,y,z]$ is the position vector and
$\boldsymbol {r}=[ x,y,z]$ is the position vector and  $t$ the time. Let the species be affected by forces
$t$ the time. Let the species be affected by forces  $\boldsymbol {F}_{i}$ (which will be later identified with the van der Waals forces), and the fluid as a whole, affected by viscosity. The shear viscosity
$\boldsymbol {F}_{i}$ (which will be later identified with the van der Waals forces), and the fluid as a whole, affected by viscosity. The shear viscosity  $\mu _{s}$ and bulk viscosity
$\mu _{s}$ and bulk viscosity  $\mu _{b}$ depend generally on
$\mu _{b}$ depend generally on  $\rho _{i}$ and
$\rho _{i}$ and  $T$.
$T$.
Let the flow be governed by following equations:
 $$\begin{gather} \frac{\partial\rho_{i}}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho _{i}\boldsymbol{v}+\boldsymbol{J}_{i}) =0, \end{gather}$$
$$\begin{gather} \frac{\partial\rho_{i}}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho _{i}\boldsymbol{v}+\boldsymbol{J}_{i}) =0, \end{gather}$$ $$\begin{gather}\frac{\partial( \rho\boldsymbol{v}) }{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho\boldsymbol{vv}) =\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi }+\sum_{i}\rho_{i}\boldsymbol{F}_{i}-\boldsymbol{\nabla} p, \end{gather}$$
$$\begin{gather}\frac{\partial( \rho\boldsymbol{v}) }{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho\boldsymbol{vv}) =\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi }+\sum_{i}\rho_{i}\boldsymbol{F}_{i}-\boldsymbol{\nabla} p, \end{gather}$$ $$\begin{gather} \frac{\partial}{\partial t}\left( \rho e+\frac{1}{2}\rho\vert \boldsymbol{v}\vert^{2}\right) +\boldsymbol{\nabla} \boldsymbol{{\cdot}}\left[ \left( \rho e+\frac{1}{2}\rho\vert \boldsymbol{v}\vert^{2}+p\right) \boldsymbol{v}-\boldsymbol{\varPi}\boldsymbol{{\cdot}}\boldsymbol{v}+\boldsymbol{Q}\right] \nonumber\\ =\sum_{i}\boldsymbol{F}_{i}\boldsymbol{{\cdot}}(\rho_{i}\boldsymbol{v}+ \boldsymbol{J}_{i}). \end{gather}$$
$$\begin{gather} \frac{\partial}{\partial t}\left( \rho e+\frac{1}{2}\rho\vert \boldsymbol{v}\vert^{2}\right) +\boldsymbol{\nabla} \boldsymbol{{\cdot}}\left[ \left( \rho e+\frac{1}{2}\rho\vert \boldsymbol{v}\vert^{2}+p\right) \boldsymbol{v}-\boldsymbol{\varPi}\boldsymbol{{\cdot}}\boldsymbol{v}+\boldsymbol{Q}\right] \nonumber\\ =\sum_{i}\boldsymbol{F}_{i}\boldsymbol{{\cdot}}(\rho_{i}\boldsymbol{v}+ \boldsymbol{J}_{i}). \end{gather}$$Here, the viscous stress tensor is
 \begin{equation} \boldsymbol{\varPi}=\mu_{s}[ \boldsymbol{\boldsymbol{\nabla} } \boldsymbol{v}+( \boldsymbol{\boldsymbol{\nabla} }\boldsymbol{v})^{\text{tr}}-\tfrac{2}{3}\boldsymbol{I}( \boldsymbol{\boldsymbol{\nabla} } \boldsymbol{{\cdot}}\boldsymbol{v}) ] +\mu_{b}\boldsymbol{I}( \boldsymbol{\boldsymbol{\nabla} }\boldsymbol{{\cdot}}\boldsymbol{v}) , \end{equation}
\begin{equation} \boldsymbol{\varPi}=\mu_{s}[ \boldsymbol{\boldsymbol{\nabla} } \boldsymbol{v}+( \boldsymbol{\boldsymbol{\nabla} }\boldsymbol{v})^{\text{tr}}-\tfrac{2}{3}\boldsymbol{I}( \boldsymbol{\boldsymbol{\nabla} } \boldsymbol{{\cdot}}\boldsymbol{v}) ] +\mu_{b}\boldsymbol{I}( \boldsymbol{\boldsymbol{\nabla} }\boldsymbol{{\cdot}}\boldsymbol{v}) , \end{equation}
where the dotless product of two vectors (e.g.  $\boldsymbol {\boldsymbol {\nabla }} \boldsymbol {v}$) produces a second-order tensor and the superscript
$\boldsymbol {\boldsymbol {\nabla }} \boldsymbol {v}$) produces a second-order tensor and the superscript  $^{\text {tr}}$ denotes transposition. The diffusion fluxes
$^{\text {tr}}$ denotes transposition. The diffusion fluxes  $\boldsymbol {J}_{i}$ and the heat flux
$\boldsymbol {J}_{i}$ and the heat flux  $\boldsymbol {Q}$ are related to the forces
$\boldsymbol {Q}$ are related to the forces  $\boldsymbol {F}_{i}$, temperature
$\boldsymbol {F}_{i}$, temperature  $T$ and chemical potentials
$T$ and chemical potentials  $G_{i}$ by
$G_{i}$ by
 $$\begin{gather} \boldsymbol{J}_{i}=\sum_{j}D_{ij}\left[ \boldsymbol{F}_{j}-T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right] -\frac{\zeta_{i}}{T}\boldsymbol{\nabla} T, \end{gather}$$
$$\begin{gather} \boldsymbol{J}_{i}=\sum_{j}D_{ij}\left[ \boldsymbol{F}_{j}-T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right] -\frac{\zeta_{i}}{T}\boldsymbol{\nabla} T, \end{gather}$$ $$\begin{gather}\boldsymbol{Q}=\sum_{j}\zeta_{j}\left[ \boldsymbol{F}_{j}-T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right] -\frac{\kappa}{T}\boldsymbol{\nabla} T, \end{gather}$$
$$\begin{gather}\boldsymbol{Q}=\sum_{j}\zeta_{j}\left[ \boldsymbol{F}_{j}-T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right] -\frac{\kappa}{T}\boldsymbol{\nabla} T, \end{gather}$$
where  $D_{ij}$,
$D_{ij}$,  $\zeta _{i}$ and
$\zeta _{i}$ and  $\kappa$ are the transport coefficients (all three depend generally on
$\kappa$ are the transport coefficients (all three depend generally on 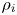 $\rho _{i}$ and
$\rho _{i}$ and  $T$).
$T$).
To understand the physical meaning of expressions (2.23) and (2.24), we rewrite them in the form
 $$\begin{gather} \boldsymbol{J}_{i}=\sum_{j}D_{ij}\boldsymbol{F}_{j}- \underset{\text{diffusion} }{\underbrace{\sum_{j}D_{ij}^{\prime}\boldsymbol{\nabla} \rho_{j}}} -\underset{\text{Soret effect}}{\underbrace{\zeta_{i}^{\prime} \rho\boldsymbol{\nabla} T}}, \end{gather}$$
$$\begin{gather} \boldsymbol{J}_{i}=\sum_{j}D_{ij}\boldsymbol{F}_{j}- \underset{\text{diffusion} }{\underbrace{\sum_{j}D_{ij}^{\prime}\boldsymbol{\nabla} \rho_{j}}} -\underset{\text{Soret effect}}{\underbrace{\zeta_{i}^{\prime} \rho\boldsymbol{\nabla} T}}, \end{gather}$$ $$\begin{gather}\boldsymbol{Q}=\sum_{j}\zeta_{j}\boldsymbol{F}_{j}- \underset{\text{Dufour effect}}{\underbrace{\sum_{j}\zeta_{j}\sum_{k}\frac{\partial G_{j}} {\partial\rho_{k}}\boldsymbol{\nabla} \rho_{k}}}-\underset{\text{heat conduction}}{\underbrace{\kappa^{\prime}\boldsymbol{\nabla} T}}, \end{gather}$$
$$\begin{gather}\boldsymbol{Q}=\sum_{j}\zeta_{j}\boldsymbol{F}_{j}- \underset{\text{Dufour effect}}{\underbrace{\sum_{j}\zeta_{j}\sum_{k}\frac{\partial G_{j}} {\partial\rho_{k}}\boldsymbol{\nabla} \rho_{k}}}-\underset{\text{heat conduction}}{\underbrace{\kappa^{\prime}\boldsymbol{\nabla} T}}, \end{gather}$$where
 $$\begin{gather} D_{ik}^{\prime}=\sum_{j}D_{ij}\frac{\partial G_{j}}{\partial\rho_{k}} ,\quad \zeta_{i}^{\prime}=\frac{\zeta_{i}}{T\rho}+\sum_{j}\frac{D_{ij}}{\rho} \left( \frac{\partial G_{j}}{\partial T}-\frac{G_{j}}{T}\right) , \end{gather}$$
$$\begin{gather} D_{ik}^{\prime}=\sum_{j}D_{ij}\frac{\partial G_{j}}{\partial\rho_{k}} ,\quad \zeta_{i}^{\prime}=\frac{\zeta_{i}}{T\rho}+\sum_{j}\frac{D_{ij}}{\rho} \left( \frac{\partial G_{j}}{\partial T}-\frac{G_{j}}{T}\right) , \end{gather}$$ $$\begin{gather} \kappa^{\prime}=\frac{\kappa}{T}+\sum_{j}\zeta_{j}\left( \frac{\partial G_{j}}{\partial T}-\frac{G_{j}}{T}\right) \end{gather}$$
$$\begin{gather} \kappa^{\prime}=\frac{\kappa}{T}+\sum_{j}\zeta_{j}\left( \frac{\partial G_{j}}{\partial T}-\frac{G_{j}}{T}\right) \end{gather}$$
are the standard diffusivities, thermodiffusivities and thermal conductivity, respectively. The second term in expression (2.25) corresponds to the classical Fick law (the fluxes depend linearly on the density gradients), and the last term in expression (2.26) characterises heat conduction described by the Fourier law. The last term in (2.25) describes the Soret effect ( $\boldsymbol {\nabla } T$ gives rise to diffusion) and the second term in (2.26), the Dufour effect (
$\boldsymbol {\nabla } T$ gives rise to diffusion) and the second term in (2.26), the Dufour effect ( $\boldsymbol {\nabla } \rho _{j}$ gives rise to heat conduction).
$\boldsymbol {\nabla } \rho _{j}$ gives rise to heat conduction).
The same four effects – diffusion, heat conduction, the Soret and Dufour effects – are described, obviously, by the original expressions (2.23) and (2.24), albeit in a form where the terms cannot be matched to a single effect each.
One might think that representing the fluxes in terms of  $\boldsymbol {\nabla } \rho _{j}$ would be more natural than using
$\boldsymbol {\nabla } \rho _{j}$ would be more natural than using  $\boldsymbol {\nabla } ( G_{j}/T)$. Observe, however, that the coefficient of
$\boldsymbol {\nabla } ( G_{j}/T)$. Observe, however, that the coefficient of  $( \boldsymbol {\nabla } T) /T$ in expression (2.23) coincides with the coefficient of
$( \boldsymbol {\nabla } T) /T$ in expression (2.23) coincides with the coefficient of  $[ \boldsymbol {F}_{j}-T\boldsymbol {\nabla } ( G_{j}/T) ]$ in (2.24). This symmetry reflects the so-called Onsager reciprocal relations (Ferziger & Kaper Reference Ferziger and Kaper1972), which also imply that
$[ \boldsymbol {F}_{j}-T\boldsymbol {\nabla } ( G_{j}/T) ]$ in (2.24). This symmetry reflects the so-called Onsager reciprocal relations (Ferziger & Kaper Reference Ferziger and Kaper1972), which also imply that
 \begin{equation} D_{ij}=D_{ji}, \end{equation}
\begin{equation} D_{ij}=D_{ji}, \end{equation}
i.e. the diffusion of an  $i$th species in a
$i$th species in a  $j$th species occurs the same way as that of the
$j$th species occurs the same way as that of the  $j$th species in the
$j$th species in the  $i$th species.
$i$th species.
It should also be assumed that the extended transport matrix,
 \begin{equation} D_{ij}^{(ext)}= \begin{bmatrix} & & & \zeta_{1}\\ & D_{ij} & & \vdots\\ & & & \zeta_{N}\\ \zeta_{1} & \ldots & \zeta_{N} & \kappa \end{bmatrix}, \end{equation}
\begin{equation} D_{ij}^{(ext)}= \begin{bmatrix} & & & \zeta_{1}\\ & D_{ij} & & \vdots\\ & & & \zeta_{N}\\ \zeta_{1} & \ldots & \zeta_{N} & \kappa \end{bmatrix}, \end{equation}
is positive semidefinite ( $D_{ij}^{(ext)}\succeq 0$), i.e.
$D_{ij}^{(ext)}\succeq 0$), i.e.
 \begin{equation} \sum_{i=1}^{N+1}\sum_{j=1}^{N+1}d_{i}\,D_{ji}^{(ext)}d_{j}\geq0 \end{equation}
\begin{equation} \sum_{i=1}^{N+1}\sum_{j=1}^{N+1}d_{i}\,D_{ji}^{(ext)}d_{j}\geq0 \end{equation}
for all  $( N+1)$-dimensional arrays
$( N+1)$-dimensional arrays  $d_{i}$. As seen later, this property is essential for the entropy principle to hold.
$d_{i}$. As seen later, this property is essential for the entropy principle to hold.
Furthermore, the transport coefficients should be such that
 \begin{equation} \sum_{i}D_{ij}=0,\quad \sum_{i}\zeta_{i}=0. \end{equation}
\begin{equation} \sum_{i}D_{ij}=0,\quad \sum_{i}\zeta_{i}=0. \end{equation}As a result, the density (2.19) and expressions (2.23) for the diffusion fluxes imply that
 \begin{equation} \frac{\partial\rho}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho \boldsymbol{v}) =0, \end{equation}
\begin{equation} \frac{\partial\rho}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho \boldsymbol{v}) =0, \end{equation}
where  $\rho$ is the total density given by (2.2). Observe that, for a pure fluid, restrictions (2.32a,b) can be satisfied only if
$\rho$ is the total density given by (2.2). Observe that, for a pure fluid, restrictions (2.32a,b) can be satisfied only if  $D_{11}=0$ and
$D_{11}=0$ and  $\zeta _{1}=0$, which means that pure fluids neither diffuse nor thermodiffuse.
$\zeta _{1}=0$, which means that pure fluids neither diffuse nor thermodiffuse.
Equations (2.19)–(2.24) have been first derived from the thermodynamics of irreversible processes (Meixner Reference Meixner1941). They were also derived from statistical mechanics (Bearman & Kirkwood Reference Bearman and Kirkwood1958; Mori Reference Mori1958) and non-equilibrium statistical thermodynamics (Keizer Reference Keizer1987); see (Giovangigli Reference Giovangigli1999) for more references. A derivation of the small-density version of (2.19)–(2.24) from the Boltzmann kinetic equation can be found in any textbook on kinetic theory (e.g. Ferziger & Kaper Reference Ferziger and Kaper1972).
In all these cases, the derived expressions for the transport coefficients automatically satisfy the Onsager relations and the rest of the properties listed above.
Note also that a reduction of the above equations for a binary fluid with no Soret and Dufour effects ( $N=2$,
$N=2$,  $\zeta _{i}=0$) was used by Liu, Amberg & Do-Quang (Reference Liu, Amberg and Do-Quang2016) to show that such a model is able to describe the phase equilibrium for a real binary mixture of
$\zeta _{i}=0$) was used by Liu, Amberg & Do-Quang (Reference Liu, Amberg and Do-Quang2016) to show that such a model is able to describe the phase equilibrium for a real binary mixture of  $\mathrm {CO}_{2}$ and ethanol.
$\mathrm {CO}_{2}$ and ethanol.
2.4. Alternative forms of the momentum and energy equations
Since the transport fluxes (2.23) and (2.24) are expressed in terms of  $G_{i}$ and
$G_{i}$ and  $T$ (not
$T$ (not  $\rho _{i}$ and
$\rho _{i}$ and  $T$), it is convenient to do the same for the pressure gradient in the momentum equation (2.20). Recalling identities (2.6)–(2.5) which imply that
$T$), it is convenient to do the same for the pressure gradient in the momentum equation (2.20). Recalling identities (2.6)–(2.5) which imply that
 \begin{equation} \boldsymbol{\nabla} p=\sum_{i}\rho_{i}\boldsymbol{\nabla} G_{i}+\rho s\boldsymbol{\nabla} T, \end{equation}
\begin{equation} \boldsymbol{\nabla} p=\sum_{i}\rho_{i}\boldsymbol{\nabla} G_{i}+\rho s\boldsymbol{\nabla} T, \end{equation}and using (2.33) to simply the left-hand side of the momentum equation (2.20), one reduces it to
 \begin{equation} \rho\left[ \frac{\partial\boldsymbol{v}}{\partial t}+( \boldsymbol{v} \boldsymbol{{\cdot}}\boldsymbol{\nabla} ) \boldsymbol{v}\right] =\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi}+\sum_{i}\rho_{i} ( \boldsymbol{F}_{i}-\boldsymbol{\nabla} G_{i}) -\rho s\boldsymbol{\nabla} T. \end{equation}
\begin{equation} \rho\left[ \frac{\partial\boldsymbol{v}}{\partial t}+( \boldsymbol{v} \boldsymbol{{\cdot}}\boldsymbol{\nabla} ) \boldsymbol{v}\right] =\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi}+\sum_{i}\rho_{i} ( \boldsymbol{F}_{i}-\boldsymbol{\nabla} G_{i}) -\rho s\boldsymbol{\nabla} T. \end{equation}
The energy equation, in turn, can be conveniently rewritten in terms of the temperature (which is a measurable quantity, unlike the internal energy  $e$). Replacing, thus, in (2.21),
$e$). Replacing, thus, in (2.21),
 \begin{equation} \frac{\partial e}{\partial t}=\sum_{i}\frac{\partial e}{\partial\rho_{i}} \frac{\partial\rho_{i}}{\partial t}+\frac{\partial e}{\partial T} \frac{\partial T}{\partial t}, \end{equation}
\begin{equation} \frac{\partial e}{\partial t}=\sum_{i}\frac{\partial e}{\partial\rho_{i}} \frac{\partial\rho_{i}}{\partial t}+\frac{\partial e}{\partial T} \frac{\partial T}{\partial t}, \end{equation}
one should use the density equation to eliminate  $\partial \rho _{i}/\partial t$. Then using (2.19) and (2.35), and recalling identities (2.8), (2.9) and (2.11), one obtains
$\partial \rho _{i}/\partial t$. Then using (2.19) and (2.35), and recalling identities (2.8), (2.9) and (2.11), one obtains
 \begin{equation} \rho c\left( \frac{\partial T}{\partial t}+\boldsymbol{v}\boldsymbol{{\cdot}}\boldsymbol{\nabla} T\right) +\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{Q}=\boldsymbol{\varPi}:( \boldsymbol{\nabla} \boldsymbol{v}) +B\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v}+\sum_{i}( \boldsymbol{F}_{i}-\rho a_{i}\boldsymbol{\nabla} ) \boldsymbol{{\cdot}}\boldsymbol{J}_{i},\end{equation}
\begin{equation} \rho c\left( \frac{\partial T}{\partial t}+\boldsymbol{v}\boldsymbol{{\cdot}}\boldsymbol{\nabla} T\right) +\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{Q}=\boldsymbol{\varPi}:( \boldsymbol{\nabla} \boldsymbol{v}) +B\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v}+\sum_{i}( \boldsymbol{F}_{i}-\rho a_{i}\boldsymbol{\nabla} ) \boldsymbol{{\cdot}}\boldsymbol{J}_{i},\end{equation}
where the symbol ‘ $:$’ denotes the double scalar product of two tensors.
$:$’ denotes the double scalar product of two tensors.
The first term on the right-hand side of (2.37) describes heat production by viscosity and the second term that by fluid compression or expansion.
2.5. The van der Waals force
Assume that a molecule of a  $j$th species exerts on a molecule of an
$j$th species exerts on a molecule of an  $i$th species an isotropic force with a potential
$i$th species an isotropic force with a potential  $\varPhi _{ij}(r)$, where
$\varPhi _{ij}(r)$, where  $r=( x^{2}+y^{2}+z^{2})^{1/2}$. Assuming for simplicity that the fluid is unbounded, one can write the mass-averaged force affecting the
$r=( x^{2}+y^{2}+z^{2})^{1/2}$. Assuming for simplicity that the fluid is unbounded, one can write the mass-averaged force affecting the  $i$th species in the hydrodynamic equations (2.20) and (2.21) in the form
$i$th species in the hydrodynamic equations (2.20) and (2.21) in the form
 \begin{equation} \boldsymbol{F}_{i}(\boldsymbol{r},t)=\boldsymbol{\nabla} \sum_{j}\int\frac{\rho _{j}(\boldsymbol{r}^{\prime},t)}{m_{i}m_{j}} \varPhi_{ij}(|\boldsymbol{r}^{\prime}- \boldsymbol{r|})\,\mathrm{d}^{3}\boldsymbol{r}^{\prime}, \end{equation}
\begin{equation} \boldsymbol{F}_{i}(\boldsymbol{r},t)=\boldsymbol{\nabla} \sum_{j}\int\frac{\rho _{j}(\boldsymbol{r}^{\prime},t)}{m_{i}m_{j}} \varPhi_{ij}(|\boldsymbol{r}^{\prime}- \boldsymbol{r|})\,\mathrm{d}^{3}\boldsymbol{r}^{\prime}, \end{equation}
where  $m_{i}$ is the molecular mass and the integration is implied to be over the whole space. To guarantee the convergence of the integral in (2.38) and those arising later, the potential
$m_{i}$ is the molecular mass and the integration is implied to be over the whole space. To guarantee the convergence of the integral in (2.38) and those arising later, the potential  $\varPhi _{ij}(r)$ is assumed to decay exponentially as
$\varPhi _{ij}(r)$ is assumed to decay exponentially as  $r\rightarrow \infty$.
$r\rightarrow \infty$.
Next, let the spatial scale of  $\rho (\boldsymbol {r},t)$ be much larger than that of
$\rho (\boldsymbol {r},t)$ be much larger than that of  $\varPhi _{ij}(r)$, in which case expression (2.38) can be simplified asymptotically. To do so, change in it
$\varPhi _{ij}(r)$, in which case expression (2.38) can be simplified asymptotically. To do so, change in it  $\boldsymbol {r}^{\prime }\rightarrow \boldsymbol {r}^{\prime }+\boldsymbol {r}$ and then expand
$\boldsymbol {r}^{\prime }\rightarrow \boldsymbol {r}^{\prime }+\boldsymbol {r}$ and then expand  $\rho _{j}(\boldsymbol {r}^{\prime }+\boldsymbol {r},t)$ about
$\rho _{j}(\boldsymbol {r}^{\prime }+\boldsymbol {r},t)$ about  $\boldsymbol {r}^{\prime }$, which yields
$\boldsymbol {r}^{\prime }$, which yields
 \begin{align} \boldsymbol{F}_{i}(\boldsymbol{r},t)=\boldsymbol{\nabla} \sum_{j}\int\left[ \rho _{j}(\boldsymbol{r},t)+\boldsymbol{r}^{\prime}\boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{j} (\boldsymbol{r},t)+\frac{1}{2}\boldsymbol{r}^{\prime}\boldsymbol{r}^{\prime} :\boldsymbol{\nabla}\boldsymbol{\nabla} \rho_{j}(\boldsymbol{r},t)+\cdots \right] \frac{\varPhi _{ij}(r^{\prime})}{m_{i}m_{j}}\,\mathrm{d}^{3}\boldsymbol{r}^{\prime}. \end{align}
\begin{align} \boldsymbol{F}_{i}(\boldsymbol{r},t)=\boldsymbol{\nabla} \sum_{j}\int\left[ \rho _{j}(\boldsymbol{r},t)+\boldsymbol{r}^{\prime}\boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{j} (\boldsymbol{r},t)+\frac{1}{2}\boldsymbol{r}^{\prime}\boldsymbol{r}^{\prime} :\boldsymbol{\nabla}\boldsymbol{\nabla} \rho_{j}(\boldsymbol{r},t)+\cdots \right] \frac{\varPhi _{ij}(r^{\prime})}{m_{i}m_{j}}\,\mathrm{d}^{3}\boldsymbol{r}^{\prime}. \end{align}
Given the isotropy of  $\varPhi _{ij}(r^{\prime })$, the second integral in the above expansion vanishes, and one obtains
$\varPhi _{ij}(r^{\prime })$, the second integral in the above expansion vanishes, and one obtains
 \begin{equation} \boldsymbol{F}_{i}=\sum_{j}W_{ij}\boldsymbol{\nabla} \rho_{j}+\sum_{j}K_{ij} \boldsymbol{\nabla} \nabla^{2}\rho_{j}+\cdots , \end{equation}
\begin{equation} \boldsymbol{F}_{i}=\sum_{j}W_{ij}\boldsymbol{\nabla} \rho_{j}+\sum_{j}K_{ij} \boldsymbol{\nabla} \nabla^{2}\rho_{j}+\cdots , \end{equation}where
 \begin{equation} W_{ij}=\int\frac{\varPhi_{ij}(r^{\prime})}{m_{i}m_{j}}\,\mathrm{d}^{3} \boldsymbol{r}^{\prime},\quad K_{ij}=\int\frac{r^{\prime2}}{2}\frac{\varPhi _{ij}(r^{\prime})}{m_{i}m_{j}}\,\mathrm{d}^{3}\boldsymbol{r}^{\prime}. \end{equation}
\begin{equation} W_{ij}=\int\frac{\varPhi_{ij}(r^{\prime})}{m_{i}m_{j}}\,\mathrm{d}^{3} \boldsymbol{r}^{\prime},\quad K_{ij}=\int\frac{r^{\prime2}}{2}\frac{\varPhi _{ij}(r^{\prime})}{m_{i}m_{j}}\,\mathrm{d}^{3}\boldsymbol{r}^{\prime}. \end{equation}
Since Newton's third law implies that  $\varPhi _{ij}=\varPhi _{ji}$, the matrices
$\varPhi _{ij}=\varPhi _{ji}$, the matrices  $W_{ij}$ and
$W_{ij}$ and  $K_{ij}$ are symmetric.
$K_{ij}$ are symmetric.
Once expansion (2.40) is substituted into the hydrodynamic equations (2.20) and (2.21), its first term can be absorbed into the internal energy, i.e. eliminated by the change
 \begin{equation} e\rightarrow e+\frac{1}{2\rho}\sum_{ij}W_{ij}\rho_{i}\rho_{j},\quad G_{i}\rightarrow G_{i}+\sum_{j}W_{ij}\rho_{j}. \end{equation}
\begin{equation} e\rightarrow e+\frac{1}{2\rho}\sum_{ij}W_{ij}\rho_{i}\rho_{j},\quad G_{i}\rightarrow G_{i}+\sum_{j}W_{ij}\rho_{j}. \end{equation}
This does not come as a surprise, as the energy associated with potential interactions of molecules can be viewed as a kind of internal energy; in fact, the second term of expansion (2.40) could also be (and sometimes is) absorbed into  $e$. This is not done in this paper, however, as it would make
$e$. This is not done in this paper, however, as it would make  $e$ a functional (instead of a function), with the implication that all the thermodynamic definitions and identities in § 2.1 would need to be rewritten in terms of functional derivatives.
$e$ a functional (instead of a function), with the implication that all the thermodynamic definitions and identities in § 2.1 would need to be rewritten in terms of functional derivatives.
Thus, without loss of generality, one can set in expression (2.40),  $W_{ij}=0$. Omitting also the small terms hidden in ‘
$W_{ij}=0$. Omitting also the small terms hidden in ‘ $\ldots$’, one obtains
$\ldots$’, one obtains
 \begin{equation} \boldsymbol{F}_{i}=\sum_{j}K_{ij}\boldsymbol{\nabla} \nabla^{2}\rho_{j}, \end{equation}
\begin{equation} \boldsymbol{F}_{i}=\sum_{j}K_{ij}\boldsymbol{\nabla} \nabla^{2}\rho_{j}, \end{equation}
which is the multicomponent extension of the standard DIM formula for the van der Waals force (e.g. Pismen & Pomeau Reference Pismen and Pomeau2000). The matrix  $K_{ij}$ is the extension of the so-called Korteweg parameter for pure fluids, and it will be referred to as the Korteweg matrix. It should be positive definite,
$K_{ij}$ is the extension of the so-called Korteweg parameter for pure fluids, and it will be referred to as the Korteweg matrix. It should be positive definite,  $K_{ij}\succ 0$, as the van der Waals force should be attractive, not repulsive.
$K_{ij}\succ 0$, as the van der Waals force should be attractive, not repulsive.
Since the original representation (2.38) was a phenomenological model and, thus, the pairwise potential  $\varPhi _{ij}$ cannot be measured, the Korteweg matrix should be viewed as a set of adjustable parameters. As seen later, they can be deduced from the measurements of the equation of state and surface tension. One should keep in mind, however, that the Korteweg matrix should not depend on the temperature. Such a dependence would be physically unjustified, as the intermolecular attraction (characterised by
$\varPhi _{ij}$ cannot be measured, the Korteweg matrix should be viewed as a set of adjustable parameters. As seen later, they can be deduced from the measurements of the equation of state and surface tension. One should keep in mind, however, that the Korteweg matrix should not depend on the temperature. Such a dependence would be physically unjustified, as the intermolecular attraction (characterised by  $K_{ij}$) should not depend on the molecules’ velocities (characterised by
$K_{ij}$) should not depend on the molecules’ velocities (characterised by  $T$).
$T$).
2.6. Boundary conditions at a solid wall
Let the fluid occupy a domain  $\mathcal {D}$, bounded by a smooth surface
$\mathcal {D}$, bounded by a smooth surface  $\partial \mathcal {D}$. For simplicity, the so-called Navier slip is disallowed in this work, so that the boundary condition for the velocity is
$\partial \mathcal {D}$. For simplicity, the so-called Navier slip is disallowed in this work, so that the boundary condition for the velocity is
 \begin{equation} \boldsymbol{v}=\boldsymbol{0}\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D}. \end{equation}
\begin{equation} \boldsymbol{v}=\boldsymbol{0}\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D}. \end{equation}To ensure mass conservation, one should require
 \begin{equation} \boldsymbol{n}\boldsymbol{{\cdot}} \boldsymbol{J}_{i}=0\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D}, \end{equation}
\begin{equation} \boldsymbol{n}\boldsymbol{{\cdot}} \boldsymbol{J}_{i}=0\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D}, \end{equation}
where  $\boldsymbol {n}$ is the outward-pointing unit normal to
$\boldsymbol {n}$ is the outward-pointing unit normal to  $\partial \mathcal {D}$.
$\partial \mathcal {D}$.
The boundary condition for the temperature, in turn, depends on the problem at hand. Since this paper is concerned, inter alia, with the entropy principle and energy conservation, it will be assumed that no heat escapes through the boundary,
 \begin{equation} \boldsymbol{n}\boldsymbol{{\cdot}} \boldsymbol{Q}=0\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D}. \end{equation}
\begin{equation} \boldsymbol{n}\boldsymbol{{\cdot}} \boldsymbol{Q}=0\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D}. \end{equation}Boundary conditions (2.44)–(2.46) would be sufficient for the standard compressible multicomponent hydrodynamics, but the DIM requires an extra condition (due to the presence of higher-order derivatives of the density field in expression (2.43)).
The most common version of such a condition – prescribing a linear combination of the boundary value of the density and its normal gradient – ascends to the paper by Cahn & Hilliard (Reference Cahn and Hilliard1958). In application to pure fluids, the Neumann reduction of this condition was proposed by Seppecher (Reference Seppecher1996) and the Dirichlet reduction by Pismen & Pomeau (Reference Pismen and Pomeau2000). As shown by Benilov (Reference Benilov2020b), the latter follows from the assumptions under which the whole DIM is derived (pairwise intermolecular interactions, slowly varying density field), and so it is used in the present paper. Thus, we require that
 \begin{equation} \rho_{i}=\rho_{0,i}\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D},\end{equation}
\begin{equation} \rho_{i}=\rho_{0,i}\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D},\end{equation}
where the constant  $\rho _{0,i}$ is specific to the fluid/solid combination under consideration. The general version of the boundary condition for
$\rho _{0,i}$ is specific to the fluid/solid combination under consideration. The general version of the boundary condition for  $\rho _{i}$ is discussed briefly in Appendix B.
$\rho _{i}$ is discussed briefly in Appendix B.
To clarify the physical meaning of condition (2.47), consider the van der Waals forces acting on a fluid molecule located infinitesimally close to the wall: the solid attracts it towards the wall, while the other fluid molecules pull it away. The former force is fixed, whereas the latter grows with the near-wall density – so that the balance is achieved when the density assumes a certain value – namely, the parameter  $\rho _{0,i}$ in condition (2.47). This argument suggests that a smaller value of
$\rho _{0,i}$ in condition (2.47). This argument suggests that a smaller value of  $\rho _{0,i}$ corresponds to a hydrophobic wall (characterised by a large contact angle) and larger
$\rho _{0,i}$ corresponds to a hydrophobic wall (characterised by a large contact angle) and larger  $\rho _{0,i}$ to a hydrophilic one.
$\rho _{0,i}$ to a hydrophilic one.
According to its physical meaning,  $\rho _{0,i}$ does not depend on the temperature. As shown later, its value can be deduced from a measurement of the contact angle.
$\rho _{0,i}$ does not depend on the temperature. As shown later, its value can be deduced from a measurement of the contact angle.
3. The entropy principle
3.1. Conservation laws and the H-theorem
It can be verified that the governing equations and boundary conditions introduced above conserve the mass of each species
 \begin{equation} M_{i}=\int_{\mathcal{D}}\rho_{i}\,\mathrm{d}^{3}\boldsymbol{r}, \end{equation}
\begin{equation} M_{i}=\int_{\mathcal{D}}\rho_{i}\,\mathrm{d}^{3}\boldsymbol{r}, \end{equation}and the total energy
 \begin{equation} E=\int_{\mathcal{D}+ }\left[ \rho e+\frac{1}{2}\rho\vert \boldsymbol{v} \vert^{2}+\frac{1}{2}\sum_{ij}K_{ij}( \boldsymbol{\nabla} \rho _{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \rho_{j}) \right]\, \mathrm{d}^{3}\boldsymbol{r}.\end{equation}
\begin{equation} E=\int_{\mathcal{D}+ }\left[ \rho e+\frac{1}{2}\rho\vert \boldsymbol{v} \vert^{2}+\frac{1}{2}\sum_{ij}K_{ij}( \boldsymbol{\nabla} \rho _{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \rho_{j}) \right]\, \mathrm{d}^{3}\boldsymbol{r}.\end{equation}The three terms in expression (3.2) represent the internal, kinetic and van der Waals energies.
The governing equations and boundary conditions also satisfy an H-theorem, reflecting the fact that the net entropy of a fluid in a thermally insulated container cannot decrease. To prove this, consider the following combination of the governing equations:
 \begin{equation}
(2.37)+T\rho\sum_{i}\frac{\partial
s}{\partial\rho_{i}} \times(2.19)+Ts\times
(2.26). \end{equation}
\begin{equation}
(2.37)+T\rho\sum_{i}\frac{\partial
s}{\partial\rho_{i}} \times(2.19)+Ts\times
(2.26). \end{equation}
After straightforward algebra involving the use of the thermodynamic identities presented in § 2.1 and expressions (2.23) and (2.24) for the fluxes, one obtains
 \begin{align} &\frac{\partial( \rho s)}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}\left( \rho s\boldsymbol{v}-\sum_{i}\frac{G_{i}}{T}\boldsymbol{J}_{i}+ \frac{\boldsymbol{Q}}{T}\right) =\frac{\boldsymbol{\varPi}: ( \boldsymbol{\nabla} \boldsymbol{v}) }{T}\nonumber\\ &\quad +\left\{ \sum_{ij}D_{ij}\left\vert \boldsymbol{F}_{j}-T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right\vert^{2}+2\sum_{i}\zeta_{i}\left[ \boldsymbol{F}_{i}-T\boldsymbol{\nabla} \left( \frac{G_{i}}{T}\right) \right] \boldsymbol{{\cdot}}\left( -\frac{\boldsymbol{\nabla} T}{T}\right) \phantom{\left\vert -\frac{\boldsymbol{\nabla} T}{T}\right\vert^{2}}\right. \nonumber\\ &\quad +\left.\vphantom{\sum_{ij}\left\vert \left( \frac{G_{j}}{T}\right) \right\vert^{2}}\kappa T\left\vert -\frac{\boldsymbol{\nabla} T}{T} \right\vert^{2}\right\} . \end{align}
\begin{align} &\frac{\partial( \rho s)}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}\left( \rho s\boldsymbol{v}-\sum_{i}\frac{G_{i}}{T}\boldsymbol{J}_{i}+ \frac{\boldsymbol{Q}}{T}\right) =\frac{\boldsymbol{\varPi}: ( \boldsymbol{\nabla} \boldsymbol{v}) }{T}\nonumber\\ &\quad +\left\{ \sum_{ij}D_{ij}\left\vert \boldsymbol{F}_{j}-T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right\vert^{2}+2\sum_{i}\zeta_{i}\left[ \boldsymbol{F}_{i}-T\boldsymbol{\nabla} \left( \frac{G_{i}}{T}\right) \right] \boldsymbol{{\cdot}}\left( -\frac{\boldsymbol{\nabla} T}{T}\right) \phantom{\left\vert -\frac{\boldsymbol{\nabla} T}{T}\right\vert^{2}}\right. \nonumber\\ &\quad +\left.\vphantom{\sum_{ij}\left\vert \left( \frac{G_{j}}{T}\right) \right\vert^{2}}\kappa T\left\vert -\frac{\boldsymbol{\nabla} T}{T} \right\vert^{2}\right\} . \end{align}The first term on the right-hand side of this equation is non-negative due to the (easily verifiable) identity
 \begin{equation} \boldsymbol{\varPi}:( \boldsymbol{\nabla} \boldsymbol{v}) =\tfrac{1}{2}\mu _{s}\vert ( \boldsymbol{\nabla} \boldsymbol{v}) +( \boldsymbol{\nabla} \boldsymbol{v})^{\text{tr}}-\tfrac{2}{3}\boldsymbol{I} \boldsymbol{\nabla}\boldsymbol{{\cdot}}\boldsymbol{v}\vert^{2}+\mu_{b}( \boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v})^{2}, \end{equation}
\begin{equation} \boldsymbol{\varPi}:( \boldsymbol{\nabla} \boldsymbol{v}) =\tfrac{1}{2}\mu _{s}\vert ( \boldsymbol{\nabla} \boldsymbol{v}) +( \boldsymbol{\nabla} \boldsymbol{v})^{\text{tr}}-\tfrac{2}{3}\boldsymbol{I} \boldsymbol{\nabla}\boldsymbol{{\cdot}}\boldsymbol{v}\vert^{2}+\mu_{b}( \boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v})^{2}, \end{equation}
whereas the expression in curly brackets is non-negative because the extended transport matrix is positive semidefinite (see § 2.3). Thus, integrating (3.4) over  $\mathcal {D}$, and using boundary conditions (2.44)–(2.46), one obtains
$\mathcal {D}$, and using boundary conditions (2.44)–(2.46), one obtains
 \begin{equation} \frac{\mathrm{d}S}{\mathrm{d}t}\geq0, \end{equation}
\begin{equation} \frac{\mathrm{d}S}{\mathrm{d}t}\geq0, \end{equation}where
 \begin{equation} S=\int_{\mathcal{D}}\rho s\,\mathrm{d}^{3}\boldsymbol{r}. \end{equation}
\begin{equation} S=\int_{\mathcal{D}}\rho s\,\mathrm{d}^{3}\boldsymbol{r}. \end{equation}Inequality (3.6) is the desired H-theorem.
It follows from (3.4) and (3.5) that the exact equality in (3.6) can only hold if the velocity field is spatially uniform; together with the no-flow boundary conditions, this requirement amounts to  $\boldsymbol {v}=\boldsymbol {0}$ (i.e. the fluid is static).
$\boldsymbol {v}=\boldsymbol {0}$ (i.e. the fluid is static).
3.2. Stability via the entropy principle
The most common way to examine the stability of a steady solution of a set of equations consist in linearising these equations with respect to a small perturbation, assuming the harmonic dependence of the perturbation on  $t$, and solving the resulting eigenvalue problem. In the problem at hand, however, it is much simpler to examine stability using the entropy principle.
$t$, and solving the resulting eigenvalue problem. In the problem at hand, however, it is much simpler to examine stability using the entropy principle.
If, at a certain steady state, the total entropy  $S$ has a local maximum constrained by the conditions of fixed energy
$S$ has a local maximum constrained by the conditions of fixed energy  $E$ and mass
$E$ and mass  $M_{i}$, this state is stable. The inverse is also true: if
$M_{i}$, this state is stable. The inverse is also true: if  $S$ does not have a maximum, the corresponding state is unstable, because a perturbed solution with a higher entropy cannot evolve ‘back’. Neutrally stable oscillations are also prohibited by the entropy principle – hence, the system can only evolve further away, towards a steady state where the entropy does have a maximum.
$S$ does not have a maximum, the corresponding state is unstable, because a perturbed solution with a higher entropy cannot evolve ‘back’. Neutrally stable oscillations are also prohibited by the entropy principle – hence, the system can only evolve further away, towards a steady state where the entropy does have a maximum.
Let a fluid be enclosed in a container (which can be later assumed to be infinitely large, if need be) and seek a maximum of  $S$, constrained by the conditions of fixed
$S$, constrained by the conditions of fixed  $M_{i}$ and
$M_{i}$ and  $E$. This problem amounts to finding the stationary points of the functional
$E$. This problem amounts to finding the stationary points of the functional
 \begin{equation} H[\rho_{1}\ldots \rho_{N},T,\boldsymbol{v}]=S+\sum_{i}\eta_{i}M_{i}+\lambda E, \end{equation}
\begin{equation} H[\rho_{1}\ldots \rho_{N},T,\boldsymbol{v}]=S+\sum_{i}\eta_{i}M_{i}+\lambda E, \end{equation}
where  $\lambda$ and
$\lambda$ and  $\mu _{i}$ are the Lagrange multipliers, and
$\mu _{i}$ are the Lagrange multipliers, and  $S$,
$S$,  $E$ and
$E$ and  $M_{i}$ are given by (3.7), (3.2) and (3.1), respectively. Varying
$M_{i}$ are given by (3.7), (3.2) and (3.1), respectively. Varying  $H$ with respect to
$H$ with respect to  $\boldsymbol {v}$ and equating the variation to zero, one obtains
$\boldsymbol {v}$ and equating the variation to zero, one obtains
 \begin{equation} \boldsymbol{v}=\boldsymbol{0}, \end{equation}
\begin{equation} \boldsymbol{v}=\boldsymbol{0}, \end{equation}
i.e. a steady state must be (unsurprisingly) static. Next, varying  $H$ with respect to
$H$ with respect to  $T$, one obtains
$T$, one obtains
 \begin{equation} \frac{\partial s}{\partial T}+\lambda\frac{\partial e}{\partial T}=0. \end{equation}
\begin{equation} \frac{\partial s}{\partial T}+\lambda\frac{\partial e}{\partial T}=0. \end{equation}Comparison of this equality with the Gibbs relation (2.4) yields
 \begin{equation} \lambda={-}\frac{1}{T}. \end{equation}
\begin{equation} \lambda={-}\frac{1}{T}. \end{equation}
Since  $\lambda$ is a constant, (3.11) implies that
$\lambda$ is a constant, (3.11) implies that  $T$ is spatially uniform, i.e. a steady state must be isothermal.
$T$ is spatially uniform, i.e. a steady state must be isothermal.
Finally, varying  $H$ with respect to
$H$ with respect to  $\rho _{i}$, recalling expressions (3.9)–(3.11) for
$\rho _{i}$, recalling expressions (3.9)–(3.11) for  $\boldsymbol {v}$ and
$\boldsymbol {v}$ and  $\lambda$, and keeping in mind definition (2.3) of the chemical potential
$\lambda$, and keeping in mind definition (2.3) of the chemical potential  $G_{i}$, one obtains
$G_{i}$, one obtains
 \begin{equation} \frac{1}{T}\left( G_{i}-\sum_{j}K_{ij}\nabla^{2}\rho_{j}\right) +\eta_{i}=0. \end{equation}
\begin{equation} \frac{1}{T}\left( G_{i}-\sum_{j}K_{ij}\nabla^{2}\rho_{j}\right) +\eta_{i}=0. \end{equation}
This equation describes all steady states of the governing equations, and it will be extensively used in the rest of this paper. The temperature  $T$ in (3.12) should be treated as a known parameter, whereas the Lagrange multiplier
$T$ in (3.12) should be treated as a known parameter, whereas the Lagrange multiplier  $\eta _{i}$ is to be deduced from the boundary conditions. The latter will be illustrated for the case of an infinite domain, plus an assumption that the fluid at infinity be spatially uniform and characterised by a coordinate-independent chemical potential
$\eta _{i}$ is to be deduced from the boundary conditions. The latter will be illustrated for the case of an infinite domain, plus an assumption that the fluid at infinity be spatially uniform and characterised by a coordinate-independent chemical potential  $G_{i}=G_{\infty,i}$. In this case, (3.12) yields
$G_{i}=G_{\infty,i}$. In this case, (3.12) yields
 \begin{equation} \eta_{i}={-}\frac{G_{\infty,i}}{T}, \end{equation}
\begin{equation} \eta_{i}={-}\frac{G_{\infty,i}}{T}, \end{equation}as required.
Note also that (3.12) can also be recovered by adapting the governing equations for the state of equilibrium. To do so, one should set  $\boldsymbol {v}=\boldsymbol {0}$,
$\boldsymbol {v}=\boldsymbol {0}$,  $T=\text {const.}$ and
$T=\text {const.}$ and  $\partial \rho _{i}/\partial t=0$ in (2.19) and (2.20) and, recalling expression (2.23) for the diffusion fluxes, obtain (3.12), as required.
$\partial \rho _{i}/\partial t=0$ in (2.19) and (2.20) and, recalling expression (2.23) for the diffusion fluxes, obtain (3.12), as required.
To examine a solution of (3.12) for stability, one needs to calculate the second variation of  $H$. Omitting the algebra (involving the use of identities (2.3) and (2.4), definition (2.8) of the heat capacity
$H$. Omitting the algebra (involving the use of identities (2.3) and (2.4), definition (2.8) of the heat capacity  $c$ and expression (3.11) for
$c$ and expression (3.11) for  $\lambda$), one obtains
$\lambda$), one obtains
 \begin{align} \delta^{2}H=\frac{1}{T}\int_{\mathcal{D}}\left\{ \sum_{ij}\left[ -\frac{\partial G_{i}}{\partial\rho_{j}}( \delta\rho_{i}) ( \delta\rho_{j}) -K_{ij}( \boldsymbol{\nabla} \delta\rho_{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \delta\rho_{j}) \right] -\frac{\rho c} {T}( \delta T)^{2}-\rho\vert \delta\boldsymbol{v}\vert^{2}\right\}\, \mathrm{d}^{3}\boldsymbol{r}. \end{align}
\begin{align} \delta^{2}H=\frac{1}{T}\int_{\mathcal{D}}\left\{ \sum_{ij}\left[ -\frac{\partial G_{i}}{\partial\rho_{j}}( \delta\rho_{i}) ( \delta\rho_{j}) -K_{ij}( \boldsymbol{\nabla} \delta\rho_{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \delta\rho_{j}) \right] -\frac{\rho c} {T}( \delta T)^{2}-\rho\vert \delta\boldsymbol{v}\vert^{2}\right\}\, \mathrm{d}^{3}\boldsymbol{r}. \end{align}
Evidently, perturbations of the temperature and velocity are negative and can only lower the total entropy – hence, the type of stationary point (maximum versus saddle) is fully determined by the variation of the density. Thus, setting  $\delta T=0$ and
$\delta T=0$ and  $\delta \boldsymbol {v}=\boldsymbol {0}$, one obtains
$\delta \boldsymbol {v}=\boldsymbol {0}$, one obtains
 \begin{equation} \delta^{2}H=\frac{1}{T}\int_{\mathcal{D}}\sum_{ij}\left[ -\frac{\partial G_{i}}{\partial\rho_{j}}( \delta\rho_{i}) ( \delta\rho _{j}) -K_{ij}( \boldsymbol{\nabla} \delta\rho_{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \delta\rho_{j}) \right] \,\mathrm{d}^{3}\boldsymbol{r}. \end{equation}
\begin{equation} \delta^{2}H=\frac{1}{T}\int_{\mathcal{D}}\sum_{ij}\left[ -\frac{\partial G_{i}}{\partial\rho_{j}}( \delta\rho_{i}) ( \delta\rho _{j}) -K_{ij}( \boldsymbol{\nabla} \delta\rho_{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \delta\rho_{j}) \right] \,\mathrm{d}^{3}\boldsymbol{r}. \end{equation}Expression (3.15) is the main tool used in this paper for studying the stability properties of steady states described by (3.12).
4. Non-dimensionalisation and the governing parameters
Introduce a characteristic density scale  $\varrho$, a pressure scale
$\varrho$, a pressure scale  $P$, a temperature scale
$P$, a temperature scale  $T_{0}$ and a typical value
$T_{0}$ and a typical value  $R$ of the specific gas constant
$R$ of the specific gas constant  $R_{i}$ introduced in § 2.2. These scales allow one to non-dimensionalise all thermodynamics variables and functions introduced in § 2.1,
$R_{i}$ introduced in § 2.2. These scales allow one to non-dimensionalise all thermodynamics variables and functions introduced in § 2.1,
 \begin{equation} \left.\begin{array}{c@{}}
\displaystyle(
\rho_{i})_{nd}=\dfrac{\rho_{i}}{\varrho},\quad \rho_{nd}
=\dfrac{\rho}{\varrho},\quad T_{nd}=\dfrac{T}{T_{0}},\quad
e_{nd} =\dfrac{\varrho}{P}e,\quad s_{nd}=\dfrac{s}{R},\\
\displaystyle p_{nd}=\dfrac{p}{P},\quad ( G_{i})
_{nd}=\dfrac{\rho}{P} G_{i},\quad c_{nd}=\dfrac{c}{R},\quad
a_{nd}=\dfrac{\varrho^{2}}{P}a,\quad B_{nd}=\dfrac{B}{P}.
\end{array}\right\} \end{equation}
\begin{equation} \left.\begin{array}{c@{}}
\displaystyle(
\rho_{i})_{nd}=\dfrac{\rho_{i}}{\varrho},\quad \rho_{nd}
=\dfrac{\rho}{\varrho},\quad T_{nd}=\dfrac{T}{T_{0}},\quad
e_{nd} =\dfrac{\varrho}{P}e,\quad s_{nd}=\dfrac{s}{R},\\
\displaystyle p_{nd}=\dfrac{p}{P},\quad ( G_{i})
_{nd}=\dfrac{\rho}{P} G_{i},\quad c_{nd}=\dfrac{c}{R},\quad
a_{nd}=\dfrac{\varrho^{2}}{P}a,\quad B_{nd}=\dfrac{B}{P}.
\end{array}\right\} \end{equation}
It is also convenient to non-dimensionalise the coefficients that appear in the governing equations. Using their respective scales, one obtains
 \begin{equation} \left.\begin{array}{c@{}}
\displaystyle( K_{ij})_{nd}=\dfrac{K_{ij}}{K},\quad (
\mu_{s})_{nd}= \dfrac{\mu_{s}}{\mu},\quad (
\mu_{b})_{nd}=\dfrac{\mu_{b} }{\mu},\\ \displaystyle(
R_{i})_{nd}=\dfrac{R_{i}}{R},\quad
\kappa_{nd}=\dfrac{\kappa }{\varkappa},\quad (
D_{ij})_{nd}=\dfrac{D_{ij}}{D},\quad (
\zeta_{i})_{nd}=\dfrac{\zeta_{i}}{\zeta}.
\end{array}\right\} \end{equation}
\begin{equation} \left.\begin{array}{c@{}}
\displaystyle( K_{ij})_{nd}=\dfrac{K_{ij}}{K},\quad (
\mu_{s})_{nd}= \dfrac{\mu_{s}}{\mu},\quad (
\mu_{b})_{nd}=\dfrac{\mu_{b} }{\mu},\\ \displaystyle(
R_{i})_{nd}=\dfrac{R_{i}}{R},\quad
\kappa_{nd}=\dfrac{\kappa }{\varkappa},\quad (
D_{ij})_{nd}=\dfrac{D_{ij}}{D},\quad (
\zeta_{i})_{nd}=\dfrac{\zeta_{i}}{\zeta}.
\end{array}\right\} \end{equation}
Finally, we introduce
 \begin{equation} \left.\begin{array}{c@{}}
\displaystyle\boldsymbol{r}_{nd}=\dfrac{\boldsymbol{r}}{l},\quad
t_{nd}=\dfrac{Vt}{l},\quad
\boldsymbol{v}_{nd}=\dfrac{\boldsymbol{v}}{V},\\
\displaystyle\boldsymbol{\varPi}_{nd}=\dfrac{l}{\mu
V}\boldsymbol{\varPi},\quad (
\boldsymbol{F}_{i})_{nd}=\dfrac{l^{3}}{K\varrho}\boldsymbol{F}_{i}
,\quad (
\boldsymbol{J}_{i})_{nd}=\dfrac{\boldsymbol{J}_{i}}{\varrho
V},\quad \boldsymbol{Q}_{nd}=\dfrac{\boldsymbol{Q}}{PV},
\end{array}\right\} \end{equation}
\begin{equation} \left.\begin{array}{c@{}}
\displaystyle\boldsymbol{r}_{nd}=\dfrac{\boldsymbol{r}}{l},\quad
t_{nd}=\dfrac{Vt}{l},\quad
\boldsymbol{v}_{nd}=\dfrac{\boldsymbol{v}}{V},\\
\displaystyle\boldsymbol{\varPi}_{nd}=\dfrac{l}{\mu
V}\boldsymbol{\varPi},\quad (
\boldsymbol{F}_{i})_{nd}=\dfrac{l^{3}}{K\varrho}\boldsymbol{F}_{i}
,\quad (
\boldsymbol{J}_{i})_{nd}=\dfrac{\boldsymbol{J}_{i}}{\varrho
V},\quad \boldsymbol{Q}_{nd}=\dfrac{\boldsymbol{Q}}{PV},
\end{array}\right\} \end{equation}
where
 \begin{equation} l=\sqrt{\frac{\varrho^{2}K}{P}} \end{equation}
\begin{equation} l=\sqrt{\frac{\varrho^{2}K}{P}} \end{equation}is the characteristic interfacial thickness and
 \begin{equation} V=\frac{Pl}{\mu} \end{equation}
\begin{equation} V=\frac{Pl}{\mu} \end{equation}
is the velocity scale reflecting the three-way balance of the pressure gradient, viscous stress and van der Waals force (Benilov Reference Benilov2020a). Note also that the characteristic interfacial thickness  $l$ is on a nanoscale (Magaletti et al. Reference Magaletti, Gallo, Marino and Casciola2016; Benilov Reference Benilov2020b; Gallo et al. Reference Gallo, Magaletti, Cocco and Casciola2020).
$l$ is on a nanoscale (Magaletti et al. Reference Magaletti, Gallo, Marino and Casciola2016; Benilov Reference Benilov2020b; Gallo et al. Reference Gallo, Magaletti, Cocco and Casciola2020).
Rewriting (2.19), (2.35)–(2.37), (2.23), (2.24) and (2.43), and omitting the subscript  $_{nd}$, one obtains
$_{nd}$, one obtains
 $$\begin{gather} \frac{\partial\rho_{i}}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho _{i}\boldsymbol{v}+\boldsymbol{J}_{i}) =0, \end{gather}$$
$$\begin{gather} \frac{\partial\rho_{i}}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho _{i}\boldsymbol{v}+\boldsymbol{J}_{i}) =0, \end{gather}$$ $$\begin{gather}\fbox{$\alpha$}\rho\left[ \frac{\partial\boldsymbol{v}}{\partial t}+( \boldsymbol{v}\boldsymbol{{\cdot}}\boldsymbol{\nabla} ) \boldsymbol{v}\right] =\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi}+\sum_{i}\rho_{i}( \boldsymbol{F}_{i}-\boldsymbol{\nabla} G_{i}) -\rho s\boldsymbol{\nabla} T, \end{gather}$$
$$\begin{gather}\fbox{$\alpha$}\rho\left[ \frac{\partial\boldsymbol{v}}{\partial t}+( \boldsymbol{v}\boldsymbol{{\cdot}}\boldsymbol{\nabla} ) \boldsymbol{v}\right] =\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi}+\sum_{i}\rho_{i}( \boldsymbol{F}_{i}-\boldsymbol{\nabla} G_{i}) -\rho s\boldsymbol{\nabla} T, \end{gather}$$ $$\begin{gather}\fbox{$\tau$}\rho c\left( \frac{\partial T}{\partial t}+\boldsymbol{v} \boldsymbol{{\cdot}}\boldsymbol{\nabla} T\right) +\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{Q}=\boldsymbol{\varPi }:(\boldsymbol{\nabla} \boldsymbol{v}) -B\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v}+\sum _{i}(\boldsymbol{F}_{i}+\rho a_{i}\boldsymbol{\nabla} ) \boldsymbol{{\cdot}} \boldsymbol{J}_{i}, \end{gather}$$
$$\begin{gather}\fbox{$\tau$}\rho c\left( \frac{\partial T}{\partial t}+\boldsymbol{v} \boldsymbol{{\cdot}}\boldsymbol{\nabla} T\right) +\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{Q}=\boldsymbol{\varPi }:(\boldsymbol{\nabla} \boldsymbol{v}) -B\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v}+\sum _{i}(\boldsymbol{F}_{i}+\rho a_{i}\boldsymbol{\nabla} ) \boldsymbol{{\cdot}} \boldsymbol{J}_{i}, \end{gather}$$ $$\begin{gather}\boldsymbol{\varPi}=\mu_{s}\left[ \boldsymbol{\nabla} \boldsymbol{v}+( \boldsymbol{\nabla} \boldsymbol{v})^{\text{tr}}-\frac{2}{3}(\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v}) \boldsymbol{I}\right] +\mu_{b}(\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v}) \boldsymbol{I}, \end{gather}$$
$$\begin{gather}\boldsymbol{\varPi}=\mu_{s}\left[ \boldsymbol{\nabla} \boldsymbol{v}+( \boldsymbol{\nabla} \boldsymbol{v})^{\text{tr}}-\frac{2}{3}(\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v}) \boldsymbol{I}\right] +\mu_{b}(\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{v}) \boldsymbol{I}, \end{gather}$$ $$\begin{gather}\fbox{$\delta$}\boldsymbol{J}_{i}=\sum_{j}D_{ij}\left[ \boldsymbol{F}_{j} -T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right] -\fbox{$\dfrac {\nu\delta}{\beta}$}\frac{\zeta_{i}}{T}\boldsymbol{\nabla} T, \end{gather}$$
$$\begin{gather}\fbox{$\delta$}\boldsymbol{J}_{i}=\sum_{j}D_{ij}\left[ \boldsymbol{F}_{j} -T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right] -\fbox{$\dfrac {\nu\delta}{\beta}$}\frac{\zeta_{i}}{T}\boldsymbol{\nabla} T, \end{gather}$$ $$\begin{gather}\fbox{$\beta$}\boldsymbol{Q}=\fbox{$\nu$}\sum_{j}\zeta_{j}\left[ \boldsymbol{F} _{j}-T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right] -\kappa \boldsymbol{\nabla} T, \end{gather}$$
$$\begin{gather}\fbox{$\beta$}\boldsymbol{Q}=\fbox{$\nu$}\sum_{j}\zeta_{j}\left[ \boldsymbol{F} _{j}-T\boldsymbol{\nabla} \left( \frac{G_{j}}{T}\right) \right] -\kappa \boldsymbol{\nabla} T, \end{gather}$$ $$\begin{gather}\boldsymbol{F}_{i}=\boldsymbol{\nabla} \sum_{j}K_{ij}\nabla^{2}\rho_{j}, \end{gather}$$
$$\begin{gather}\boldsymbol{F}_{i}=\boldsymbol{\nabla} \sum_{j}K_{ij}\nabla^{2}\rho_{j}, \end{gather}$$where the non-dimensional parameters
 \begin{equation} \alpha=\frac{K\varrho^{3}}{\mu^{2}},\quad \tau=\frac{\varrho RT_{0}}{P} ,\quad \beta=\frac{KP\varrho^{2}}{\mu\varkappa T_{0}},\quad \nu=\frac{\zeta P}{\varkappa T_{0}\varrho},\quad \delta=\frac{K\varrho^{4}}{D\mu P}, \end{equation}
\begin{equation} \alpha=\frac{K\varrho^{3}}{\mu^{2}},\quad \tau=\frac{\varrho RT_{0}}{P} ,\quad \beta=\frac{KP\varrho^{2}}{\mu\varkappa T_{0}},\quad \nu=\frac{\zeta P}{\varkappa T_{0}\varrho},\quad \delta=\frac{K\varrho^{4}}{D\mu P}, \end{equation}are placed for better ‘visibility’ in boxes.
Since  $\alpha$ appears in front of the material derivative in (4.6), it should be interpreted as the microscopic Reynolds number (associated with the flow near the interface, not the global flow). Here
$\alpha$ appears in front of the material derivative in (4.6), it should be interpreted as the microscopic Reynolds number (associated with the flow near the interface, not the global flow). Here  $\tau$ is the non-dimensional temperature;
$\tau$ is the non-dimensional temperature;  $\beta$ is the isothermality parameter (Benilov Reference Benilov2020a): if it is small, the temperature field is close to being spatially uniform (isothermal). The Nusselt number
$\beta$ is the isothermality parameter (Benilov Reference Benilov2020a): if it is small, the temperature field is close to being spatially uniform (isothermal). The Nusselt number  $\nu$ characterises the importance of heat diffusion relative to heat conduction; see expression (4.11) for the heat flux. Finally, the position of
$\nu$ characterises the importance of heat diffusion relative to heat conduction; see expression (4.11) for the heat flux. Finally, the position of  $\delta$ in (4.10) suggests that this parameter characterises advection relative to diffusion (recall that the flux
$\delta$ in (4.10) suggests that this parameter characterises advection relative to diffusion (recall that the flux  $\boldsymbol {J}_{i}$ was non-dimensionalised using the advection scale
$\boldsymbol {J}_{i}$ was non-dimensionalised using the advection scale  $\varrho V$).
$\varrho V$).
One can also introduce the Prandtl and Schmidt numbers,
 \begin{equation} Pr=\frac{\beta\tau}{\alpha}=\frac{\mu R}{\varkappa},\quad Sc=\frac{\delta }{\alpha}=\frac{\mu\varrho}{DP}, \end{equation}
\begin{equation} Pr=\frac{\beta\tau}{\alpha}=\frac{\mu R}{\varkappa},\quad Sc=\frac{\delta }{\alpha}=\frac{\mu\varrho}{DP}, \end{equation}characterizing viscosity relative to heat conduction and diffusion, respectively.
The non-dimensional versions of the boundary conditions look exactly the same as their dimensional counterparts, (2.44)–(2.47); as do the non-dimensional versions of the thermodynamic identities of § 2.1 except definition (2.8) of the heat capacity, which becomes
 \begin{equation} c=\frac{1}{\tau}\frac{\partial e}{\partial T}. \end{equation}
\begin{equation} c=\frac{1}{\tau}\frac{\partial e}{\partial T}. \end{equation}This paper does not aim to present a comprehensive classification of asymptotic regimes of the multicomponent DIM (for the pure-fluid DIM, see Benilov Reference Benilov2020a). Only the simplest regime will be described and used later as a qualitative illustration of theoretically predicted behaviours. It corresponds to the following assumptions:
 \begin{equation} \alpha\ll1,\quad \beta\ll1,\quad \nu\ll1. \end{equation}
\begin{equation} \alpha\ll1,\quad \beta\ll1,\quad \nu\ll1. \end{equation}
The smallness of  $\alpha$ allows one to take advantage of the slow-flow approximation; whereas the other two assumptions and (4.8) and (4.11) imply that
$\alpha$ allows one to take advantage of the slow-flow approximation; whereas the other two assumptions and (4.8) and (4.11) imply that
 \begin{equation} T=1+{O}(\beta), \end{equation}
\begin{equation} T=1+{O}(\beta), \end{equation}
i.e. the fluid is almost isothermal. Thus, setting  $T=\textrm {const.}$ in expression (4.10) for the diffusion flux and substituting it into the density equation (4.6), one obtains
$T=\textrm {const.}$ in expression (4.10) for the diffusion flux and substituting it into the density equation (4.6), one obtains
 \begin{equation} \frac{\partial\rho_{i}}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}\left[ \rho _{i}\boldsymbol{v}+\sum_{j}D_{ij}\boldsymbol{\nabla} \left( \sum_{n}K_{jn}\nabla^{2}\rho_{n}-G_{j}\right) \right] =0, \end{equation}
\begin{equation} \frac{\partial\rho_{i}}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}\left[ \rho _{i}\boldsymbol{v}+\sum_{j}D_{ij}\boldsymbol{\nabla} \left( \sum_{n}K_{jn}\nabla^{2}\rho_{n}-G_{j}\right) \right] =0, \end{equation}
where it was assumed, without loss of generality, that  $\delta =1$. Similarly, simplifying (4.7) and substituting into it expression (4.12) for
$\delta =1$. Similarly, simplifying (4.7) and substituting into it expression (4.12) for  $\boldsymbol {F}_{i}$, one obtains
$\boldsymbol {F}_{i}$, one obtains
 \begin{equation} \boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi}+\sum_{i}\rho_{i}\boldsymbol{\nabla} \left( \sum_{j}K_{ij}\nabla^{2}\rho_{j}-G_{i}\right) =0. \end{equation}
\begin{equation} \boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi}+\sum_{i}\rho_{i}\boldsymbol{\nabla} \left( \sum_{j}K_{ij}\nabla^{2}\rho_{j}-G_{i}\right) =0. \end{equation}Observe that (4.18) and (4.19) do not involve the (small) temperature variations – hence, the temperature (4.8) can be simply omitted.
Equations (4.18) and (4.19) and expression (4.9) for the viscous stress form a closed set of approximate equations for the unknowns  $\rho _{i}$ and
$\rho _{i}$ and  $\boldsymbol {v}$. The chemical potential
$\boldsymbol {v}$. The chemical potential  $G_{i}$ in these equations should be treated as a known function of
$G_{i}$ in these equations should be treated as a known function of  $\rho _{1}\ldots \rho _{N}$, and the temperature
$\rho _{1}\ldots \rho _{N}$, and the temperature  $T$ as a known parameter.
$T$ as a known parameter.
Unlike the exact set – which describes fast acoustic waves and slow interfacial flow – the approximate equations describe only the latter. This is a clear advantage: in a numerical simulation, for example, waves necessitate a small time step and, thus, dramatically slow down the computation. At the same time, the two sets of equations have very similar properties: they share the same steady solutions, both conserve mass and energy, and satisfy the H-theorem.
5. Basic solutions and their stability
5.1. Spatially uniform states
Consider a uniform fluid where there is no flow and all species are in vapour phase. If the temperature drops, one of the species may become overcooled, giving rise to rapid condensation. A similar instability may occur when all or some of the species are in liquid phase and the temperature increases, giving rise to rapid evaporation.
To determine exactly which states are unstable (thus, do not occur in real world), one could perform the usual linear analysis. For a pure fluid ( $N=1$), this is a straightforward task yielding the following stability criterion:
$N=1$), this is a straightforward task yielding the following stability criterion:
 \begin{equation} \frac{\partial p}{\partial\rho}>0. \end{equation}
\begin{equation} \frac{\partial p}{\partial\rho}>0. \end{equation}Thus, instability occurs if an increase in density lowers the pressure, so that the flow generated by the pressure gradient brings even more fluid to this region.
For an arbitrary  $N$, however, the analysis of linearised equations is extremely cumbersome. Instead, it will be examined via the entropy principle, i.e. using expression (3.15). Taking the limit
$N$, however, the analysis of linearised equations is extremely cumbersome. Instead, it will be examined via the entropy principle, i.e. using expression (3.15). Taking the limit  $\mathcal {D}\rightarrow \mathbb {R} ^{3}$ (unbounded fluid), assuming that
$\mathcal {D}\rightarrow \mathbb {R} ^{3}$ (unbounded fluid), assuming that  $\rho _{i}$ is spatially uniform and recalling that
$\rho _{i}$ is spatially uniform and recalling that  $K_{ij}\succ 0$ and
$K_{ij}\succ 0$ and  $c>0$, one can deduce from (3.15) that
$c>0$, one can deduce from (3.15) that  $H$ has a maximum if
$H$ has a maximum if
 \begin{equation} \frac{\partial G_{i}}{\partial\rho_{j}}\succ0. \end{equation}
\begin{equation} \frac{\partial G_{i}}{\partial\rho_{j}}\succ0. \end{equation}This is the standard stability criterion for a spatially uniform state of a multicomponent fluid (Glansdorff & Prigogine Reference Glansdorff and Prigogine1971).
The following four comments are in order.
(i) For a physically meaningful
 $G_{i}(\rho _{1}\ldots \rho _{N},T)$, condition (5.2) holds for a sufficiently rarefied vapour or a sufficiently dense liquid.
$G_{i}(\rho _{1}\ldots \rho _{N},T)$, condition (5.2) holds for a sufficiently rarefied vapour or a sufficiently dense liquid.(ii) The states with marginal stability are sometimes referred to as ‘spinodal points’ and stable vapour as ‘subspinodal vapour’.
(iii) Interestingly, the viscosity and transport coefficients do not appear in criterion (5.2). The corresponding effects can only slow the instability down, but not eliminate it.
(iv) To reconcile criterion (5.2) with its pure-fluid counterpart (5.1), note that, for
$N=1$, $\partial G/\partial \rho$ and $\partial p/\partial \rho$ have the same sign (as implied by identity (2.6)).




5.2. Two-phase saturated states
Consider an interface separating liquid and vapour of the same pure fluid. If they are in equilibrium, their temperatures are equal, and the rest of the parameters satisfy the so-called Maxwell construction (Maxwell Reference Maxwell1875),
 \begin{equation} G(\rho^{(l)},T)=G(\rho^{(v)},T),\quad p(\rho^{(l)},T)=p(\rho^{(v)},T), \end{equation}
\begin{equation} G(\rho^{(l)},T)=G(\rho^{(v)},T),\quad p(\rho^{(l)},T)=p(\rho^{(v)},T), \end{equation}
where the superscripts  ${(l)}$ and
${(l)}$ and  ${(v)}$ mark the parameters of the liquid and vapour, respectively. The former and latter equalities in (5.3a,b) guarantee thermodynamic and mechanical equilibria of the interface, respectively.
${(v)}$ mark the parameters of the liquid and vapour, respectively. The former and latter equalities in (5.3a,b) guarantee thermodynamic and mechanical equilibria of the interface, respectively.
Assume also that both phases are stable,
 \begin{equation} \left( \frac{\partial G}{\partial\rho}\right) _{\rho=\rho^{(l)}} >0,\quad \left( \frac{\partial G}{\partial\rho}\right) _{\rho=\rho^{(v)} }>0, \end{equation}
\begin{equation} \left( \frac{\partial G}{\partial\rho}\right) _{\rho=\rho^{(l)}} >0,\quad \left( \frac{\partial G}{\partial\rho}\right) _{\rho=\rho^{(v)} }>0, \end{equation}and the density of the liquid exceeds that of the vapour,
 \begin{equation} \rho^{(l)}>\rho^{(v)}. \end{equation}
\begin{equation} \rho^{(l)}>\rho^{(v)}. \end{equation}
Subject to these conditions, the Maxwell construction (5.3a,b) uniquely determines the saturated densities  $\rho ^{(l)}$ and
$\rho ^{(l)}$ and  $\rho ^{(v)}$ as functions of
$\rho ^{(v)}$ as functions of  $T$.
$T$.
In what follows, the multicomponent version of the Maxwell construction will be shown to follow from the DIM's entropy principle.
Consider an insulated container with fluid subdivided between two states, liquid and vapour. If the liquid/vapour and fluid/wall interfaces are sufficiently thin, the corresponding full entropy, mass and energy can be approximately written in the form
 $$\begin{gather} S=V^{(l)}\rho^{(l)} s(\rho_{1}^{(l)}\ldots \rho_{N}^{(l)},T^{(l)})+V^{(v)} \rho^{(v)} s(\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T^{(v)}), \end{gather}$$
$$\begin{gather} S=V^{(l)}\rho^{(l)} s(\rho_{1}^{(l)}\ldots \rho_{N}^{(l)},T^{(l)})+V^{(v)} \rho^{(v)} s(\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T^{(v)}), \end{gather}$$ $$\begin{gather}M_{i}=V^{(l)}\rho_{i}^{(l)}+V^{(v)}\rho_{i}^{(v)}, \end{gather}$$
$$\begin{gather}M_{i}=V^{(l)}\rho_{i}^{(l)}+V^{(v)}\rho_{i}^{(v)}, \end{gather}$$ $$\begin{gather}E=V^{(l)}\rho^{(l)} e(\rho_{1}^{(l)}\ldots \rho_{N}^{(l)},T^{(l)})+V^{(v)} \rho^{(v)} e(\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T^{(v)}), \end{gather}$$
$$\begin{gather}E=V^{(l)}\rho^{(l)} e(\rho_{1}^{(l)}\ldots \rho_{N}^{(l)},T^{(l)})+V^{(v)} \rho^{(v)} e(\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T^{(v)}), \end{gather}$$
where  $V^{(l)}$ and
$V^{(l)}$ and  $V^{(v)}$ are the volumes of the liquid and vapour phases, respectively. We also introduce the full volume of the container,
$V^{(v)}$ are the volumes of the liquid and vapour phases, respectively. We also introduce the full volume of the container,
 \begin{equation} V=V^{(l)}+V^{(v)}. \end{equation}
\begin{equation} V=V^{(l)}+V^{(v)}. \end{equation}
The Maxwell construction can be derived by maximizing  $S$ subject to the constraints of fixed
$S$ subject to the constraints of fixed  $M_{i}$,
$M_{i}$,  $E$ and
$E$ and  $V$ (which are now functions, as opposed to being functionals in the previous subsection). Straightforward calculations show that the maximum of entropy is achieved if
$V$ (which are now functions, as opposed to being functionals in the previous subsection). Straightforward calculations show that the maximum of entropy is achieved if  $T^{(l)}=T^{(v)}$ (isothermality) and
$T^{(l)}=T^{(v)}$ (isothermality) and
 $$\begin{gather} G_{i}(\rho_{1}^{(l)}\ldots \rho_{N}^{(l)},T)=G_{i}(\rho_{1}^{(v)}\ldots \rho_{N} ^{(v)},T), \end{gather}$$
$$\begin{gather} G_{i}(\rho_{1}^{(l)}\ldots \rho_{N}^{(l)},T)=G_{i}(\rho_{1}^{(v)}\ldots \rho_{N} ^{(v)},T), \end{gather}$$ $$\begin{gather}p(\rho_{1}^{(l)}\ldots \rho_{N}^{(l)},T)=p(\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T). \end{gather}$$
$$\begin{gather}p(\rho_{1}^{(l)}\ldots \rho_{N}^{(l)},T)=p(\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T). \end{gather}$$The following four comments are in order.
(i) Since the solution describing coexistence of two phases satisfies the entropy principle, it is automatically stable.
(ii) The Maxwell construction can sometimes yield a solution with negative
$V^{(l)}$ or $V^{(v)}$. In such cases, the two-phase equilibrium is irrelevant, and the fluid evolves towards the one-phase state with the same masses of the species and total energy.(iii) The multicomponent Maxwell construction (5.10) and (5.11) comprises
$N+1$ equations for $2N$ unknowns – hence, does not uniquely fix the liquid and vapour densities.To close set (5.10) and (5.11), one should assume the masses
$M_{i}$ and the total energy $E$ to be known and view equalities (5.7)–(5.9) as additional equations. They bring the total number of equations to $2N+3$ (making the ‘expanded’ Maxwell construction appear overdetermined), but in this formulation $V^{(l)}$, $V^{(v)}$ and $T$ should also be viewed as unknowns.Physically, if a certain amount of fluid, with a certain amount of energy, is placed in a box, the entropy principle uniquely determines the final temperature and the proportion in which the box is subdivided between the liquid and vapour phases.
(iv) The closure of the Maxwell construction described in the previous bullet is, obviously, inapplicable to containers of infinite volume. To understand how conditions (5.10) and (5.11) should be closed in this case, consider the interface between the Earth's atmosphere and ocean. For this setting, one should prescribe the (atmospheric) pressure and composition of dry air above the ocean's surface. With these parameters given, (5.10) and (5.11) yield the saturated moisture content of the air and the saturated amounts of nitrogen, oxygen, etc. dissolved in the oceanic water.










To illustrate the use of the Maxwell construction, consider a van der Waals fluid, whose pressure and chemical potential are described by expressions (2.17) and (2.18). Assume for simplicity that the fluid is pure ( $N=1$) and monatomic (
$N=1$) and monatomic ( $c_{i}=3R_{i}/2$), and let the scales
$c_{i}=3R_{i}/2$), and let the scales  $\varrho$,
$\varrho$,  $P$ and
$P$ and  $R$ used for non-dimensionalisation be such that
$R$ used for non-dimensionalisation be such that
 \begin{equation} a_{11}=1,\quad b_{1}=1,\quad R_{1}=1. \end{equation}
\begin{equation} a_{11}=1,\quad b_{1}=1,\quad R_{1}=1. \end{equation}
If  $T<8/27$ (which is a subcritical temperature of the van der Waals fluid), two states exist representing the liquid and vapour phases, with some (but not all) of the states in between being unstable (see figure 1). If the temperature is supercritical, only one phase exists and interfaces do not.
$T<8/27$ (which is a subcritical temperature of the van der Waals fluid), two states exist representing the liquid and vapour phases, with some (but not all) of the states in between being unstable (see figure 1). If the temperature is supercritical, only one phase exists and interfaces do not.

Figure 1. The pressure (a) and chemical potential (b) of a pure van der Waals fluid as functions of the density, for three values of the temperature: (1)  $T=0.26$ (subcritical), (2)
$T=0.26$ (subcritical), (2)  $T=8/27$ (critical), (3)
$T=8/27$ (critical), (3)  $T=0.33$ (supercritical). The critical point on curve (2) is marked with a filled circle; the saturation and spinodal points on curve (1) are shown by filled and empty squares, respectively. The labels ‘undersat(urated) vapour’, ‘oversat(urated)v(apour)’, etc. apply only to curve (1).
$T=0.33$ (supercritical). The critical point on curve (2) is marked with a filled circle; the saturation and spinodal points on curve (1) are shown by filled and empty squares, respectively. The labels ‘undersat(urated) vapour’, ‘oversat(urated)v(apour)’, etc. apply only to curve (1).
The simplest model describing a water/air interface is that with  $N=2$. The Maxwell construction in this case should be complemented with one extra condition, setting the pressure above the interface equal to its atmospheric value,
$N=2$. The Maxwell construction in this case should be complemented with one extra condition, setting the pressure above the interface equal to its atmospheric value,
 \begin{equation} p(\rho_{1}^{(v)},\rho_{2}^{(v)},T)=p_{A}. \end{equation}
\begin{equation} p(\rho_{1}^{(v)},\rho_{2}^{(v)},T)=p_{A}. \end{equation}The difference between a pure and a multicomponent fluid is illustrated in figure 2 for parameters (5.12a–c) and
 \begin{equation} a_{22}=0.2,\quad a_{12}=0,\quad b_{2}=1,\quad R_{2}=0.6,\quad p_{A}=0.03. \end{equation}
\begin{equation} a_{22}=0.2,\quad a_{12}=0,\quad b_{2}=1,\quad R_{2}=0.6,\quad p_{A}=0.03. \end{equation}
These values reflect a compromise between simplicity, illustrative purposes (the curves with  $N=1$ and
$N=1$ and  $N=2$ should be visibly different) and an attempt to loosely match the parameters of water and air (loosely, because the van der Waals model includes few adjustable constants). In particular, parameters (5.14a–e) make the critical temperature of the second species noticeably smaller than that of the first species. As a result, a temperature range exists where the former is definitely vapour, whereas the latter can be either vapour or liquid (as is indeed the case with water under normal conditions).
$N=2$ should be visibly different) and an attempt to loosely match the parameters of water and air (loosely, because the van der Waals model includes few adjustable constants). In particular, parameters (5.14a–e) make the critical temperature of the second species noticeably smaller than that of the first species. As a result, a temperature range exists where the former is definitely vapour, whereas the latter can be either vapour or liquid (as is indeed the case with water under normal conditions).

Figure 2. The saturated densities (a) and pressure (b) versus the temperature, for a pure (solid line) and two-component (dotted line) van der Waals fluid, with the parameters (5.12a–c)–(5.14a–e). The empty circles mark the boiling point, the filled circle marks the critical point. The saturated densities of the second species (of the two-component fluid) are not shown.
The most important difference between a multicomponent fluid at fixed pressure and a pure fluid is that the former exhibits a boiling point. It occurs when the saturated pressure of the liquid-phase species matches the applied (atmospheric) pressure: as a result, the second species is completely replaced by the vapour of the first species, and so the fluid becomes pure. If it is heated beyond the boiling point, the pressure can no longer be kept fixed, but ought to increase (to match the saturated pressure of the first species, which grows with  $T$).
$T$).
One should keep in mind that the small- $T$ part of figure 2 is physically meaningless due to freezing (which is not described by the DIM).
$T$ part of figure 2 is physically meaningless due to freezing (which is not described by the DIM).
6. One-dimensional steady states
6.1. Flat liquid/vapour interfaces
Consider an unbounded fluid involving liquid and vapour phases in equilibrium, separated by a static flat interface. Its spatial structure is described by (3.12) derived from the entropy principle. To adapt it specifically for a flat interface, let  $\rho _{i}$ depend on a single coordinate – say,
$\rho _{i}$ depend on a single coordinate – say,  $z$ – so that the interface is horizontal. Let the fluid below the interface be liquid and above vapour,
$z$ – so that the interface is horizontal. Let the fluid below the interface be liquid and above vapour,
 $$\begin{gather} \rho_{i}\rightarrow\rho_{i}^{(l)}\quad \text{as}\ z\rightarrow-\infty, \end{gather}$$
$$\begin{gather} \rho_{i}\rightarrow\rho_{i}^{(l)}\quad \text{as}\ z\rightarrow-\infty, \end{gather}$$ $$\begin{gather}\rho_{i}\rightarrow\rho_{i}^{(v)}\quad \text{as}\ z\rightarrow+\infty. \end{gather}$$
$$\begin{gather}\rho_{i}\rightarrow\rho_{i}^{(v)}\quad \text{as}\ z\rightarrow+\infty. \end{gather}$$
Taking in (3.12) the limit  $z\rightarrow +\infty$, one can determine the constant
$z\rightarrow +\infty$, one can determine the constant  $\eta _{i}$ and rewrite (3.12) in the form
$\eta _{i}$ and rewrite (3.12) in the form
 \begin{equation} \sum_{j}K_{ij}\frac{\mathrm{d}^{2}\rho_{j}}{\mathrm{d}z^{2}}=G_{i}-G_{i} (\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T). \end{equation}
\begin{equation} \sum_{j}K_{ij}\frac{\mathrm{d}^{2}\rho_{j}}{\mathrm{d}z^{2}}=G_{i}-G_{i} (\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T). \end{equation}
The boundary-value problem (6.1)–(6.3) is invariant with respect to the change  $z\rightarrow z+\textrm {const.}$ – hence, its solution is not unique. To make it such, an extra condition is needed, say,
$z\rightarrow z+\textrm {const.}$ – hence, its solution is not unique. To make it such, an extra condition is needed, say,
 \begin{equation} \rho_{1}=\tfrac{1}{2}( \rho_{1}^{(l)}+\rho_{1}^{(v)}) \quad \text{at}\ z=0. \end{equation}
\begin{equation} \rho_{1}=\tfrac{1}{2}( \rho_{1}^{(l)}+\rho_{1}^{(v)}) \quad \text{at}\ z=0. \end{equation}
As shown before, the parameters  $\rho _{i}^{(l)}$ and
$\rho _{i}^{(l)}$ and  $\rho _{i}^{(v)}$ are not arbitrary but have to satisfy the Maxwell construction, i.e. conditions (5.10) and (5.11). Thus, they must be intrinsic to the boundary-value problem (6.1)–(6.4) and can be derived directly from it, as a condition for the existence of solution.
$\rho _{i}^{(v)}$ are not arbitrary but have to satisfy the Maxwell construction, i.e. conditions (5.10) and (5.11). Thus, they must be intrinsic to the boundary-value problem (6.1)–(6.4) and can be derived directly from it, as a condition for the existence of solution.
Indeed, consider (6.3) in the limit  $z\rightarrow -\infty$ and recall (6.1), which immediately yields the first half of the Maxwell construction, (5.10). To derive the second half, consider
$z\rightarrow -\infty$ and recall (6.1), which immediately yields the first half of the Maxwell construction, (5.10). To derive the second half, consider
 \begin{equation} \sum_{i}\int_{-\infty}^{\infty}(6.3) \times \frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\,\mathrm{d}z. \end{equation}
\begin{equation} \sum_{i}\int_{-\infty}^{\infty}(6.3) \times \frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\,\mathrm{d}z. \end{equation}The integral in this equation can be evaluated via identity (2.7), and the constant of integration can be fixed via boundary condition (6.2). After straightforward algebra, one obtains
 \begin{equation} \frac{1}{2}\sum_{ij}K_{ij}\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z} \frac{\mathrm{d}\rho_{j}}{\mathrm{d}z}=\sum_{i}\rho_{i}[ G_{i} -G_{i}(\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T)] -p+p(\rho_{1}^{(v)} \ldots \rho_{N}^{(v)},T). \end{equation}
\begin{equation} \frac{1}{2}\sum_{ij}K_{ij}\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z} \frac{\mathrm{d}\rho_{j}}{\mathrm{d}z}=\sum_{i}\rho_{i}[ G_{i} -G_{i}(\rho_{1}^{(v)}\ldots \rho_{N}^{(v)},T)] -p+p(\rho_{1}^{(v)} \ldots \rho_{N}^{(v)},T). \end{equation}
Taking in this equation the limit  $z\rightarrow -\infty$ and recalling boundary condition (6.1), one recovers the second part of the Maxwell construction, (5.11), as required.
$z\rightarrow -\infty$ and recalling boundary condition (6.1), one recovers the second part of the Maxwell construction, (5.11), as required.
The boundary-value problem (6.1)–(6.4) was solved numerically for a two-component van der Waals fluid, using the MATLAB function BVP4c based on the three-stage Lobatto IIIa formula (Kierzenka & Shampine Reference Kierzenka and Shampine2001). Examples of numerical solutions with parameters (5.12a–c)–(5.14a–e) and
 \begin{equation} K_{11}=K_{22}=1,\quad K_{12}=0 \end{equation}
\begin{equation} K_{11}=K_{22}=1,\quad K_{12}=0 \end{equation}are shown in figure 3. Observe that an increase in temperature makes the interface thicker, and it becomes infinitely thick when the critical temperature is reached.

Figure 3. Comparison between liquid/vapour interfaces in pure (solid line) and two-component (dotted line) van der Waals fluids with parameters (5.12a–c)–(5.14a–e) and (6.7a,b). Curves (1)–(3) correspond to  $T=0.17,0.23,0.28132$. The last of these values is the (approximate) boiling point, so that the solid and dotted curves virtually coincide and
$T=0.17,0.23,0.28132$. The last of these values is the (approximate) boiling point, so that the solid and dotted curves virtually coincide and  $\rho _{2}$ is virtually zero.
$\rho _{2}$ is virtually zero.
As shown in Appendix C.1, all solutions describing liquid/vapour interfaces are stable as long as  $\mathrm {d}\rho _{i} /\mathrm {d}z$ never vanishes (
$\mathrm {d}\rho _{i} /\mathrm {d}z$ never vanishes ( $\rho _{i}(z)$ is strictly monotonic) for all
$\rho _{i}(z)$ is strictly monotonic) for all  $i$.
$i$.
6.2. One-dimensional drops and bubbles
Apart from liquid/vapour interfaces, (3.12) admits spatially localised solutions, describing drops or bubbles floating in vapour or liquid, respectively. In this subsection the simplest – one-dimensional (1-D) – solutions of this kind are discussed, describing a flat layer of increased or decreased density. They both correspond to the following boundary condition:
 \begin{equation} \rho_{i}\rightarrow\rho_{\infty,i}\ \text{as}\ z\rightarrow\pm\infty. \end{equation}
\begin{equation} \rho_{i}\rightarrow\rho_{\infty,i}\ \text{as}\ z\rightarrow\pm\infty. \end{equation}
Here  $\rho _{\infty,i}$ is the density of the vapour (liquid). Unlike the interface solutions examined above, the fluid outside the drop (bubble) does not have to be saturated vapour (liquid); it can actually be any stable or metastable state.
$\rho _{\infty,i}$ is the density of the vapour (liquid). Unlike the interface solutions examined above, the fluid outside the drop (bubble) does not have to be saturated vapour (liquid); it can actually be any stable or metastable state.
The 1-D reduction of the steady state (3.12) and expression (3.13) for  $\eta _{i}$ can be written in the form
$\eta _{i}$ can be written in the form
 \begin{equation} \sum_{j}K_{ij}\frac{\mathrm{d}^{2}\rho_{j}}{\mathrm{d}z^{2}}=G_{i} -G_{\infty,i}, \end{equation}
\begin{equation} \sum_{j}K_{ij}\frac{\mathrm{d}^{2}\rho_{j}}{\mathrm{d}z^{2}}=G_{i} -G_{\infty,i}, \end{equation}
where  $G_{\infty,i}=G_{i}(\rho _{\infty,1}\ldots \rho _{\infty,N},T)$ is the chemical potential at infinity.
$G_{\infty,i}=G_{i}(\rho _{\infty,1}\ldots \rho _{\infty,N},T)$ is the chemical potential at infinity.
Figure 4 illustrates typical solutions of the boundary-value problem (6.8) and (6.9) computed for a pure fluid. The following features can be observed in panel (a) depicting some 1-D drop solutions.
(i) Drop solutions exist only if
$\rho _{\infty,1}$ corresponds to oversaturated vapour.(ii) As
$\rho _{\infty,1}\rightarrow \rho _{1}^{(v)}+0$, the drop becomes increasingly thick.(iii) Once
$\rho _{\infty,1}$ passes $\rho _{1}^{(v)}$, drop solutions cease to exist. Physically, this is because liquid drops surrounded by undersaturated vapour evaporate.





Figure 4. Solutions of boundary-value problem (6.8) and (6.9) for a pure van der Waals fluid with  $T=0.2$. (a) Drop solutions with
$T=0.2$. (a) Drop solutions with  $\rho _{\infty,1}=0.1, 0.05, 0.03587, 0.0358485$ (curves (1)–(4), respectively). (b) Bubble solutions with
$\rho _{\infty,1}=0.1, 0.05, 0.03587, 0.0358485$ (curves (1)–(4), respectively). (b) Bubble solutions with  $\rho _{\infty,1} =0.63, 0.68, 0.7286, 0.728671$ (curves (1)–(4), respectively). The labels ‘u(ndersaturated) v(apour)’, ‘o(versaturated) v(apour)’, etc. characterise
$\rho _{\infty,1} =0.63, 0.68, 0.7286, 0.728671$ (curves (1)–(4), respectively). The labels ‘u(ndersaturated) v(apour)’, ‘o(versaturated) v(apour)’, etc. characterise  $\rho _{\infty,1}$.
$\rho _{\infty,1}$.
Similar tendencies have been observed for 1-D bubble solutions, illustrated in figure 4(b).
Most importantly, all drops (bubbles) in oversaturated vapour (undersaturated liquid) are likely to be unstable. For pure fluids ( $N=1$), the instability can be proven rigorously; as shown in Appendix C.2, the entropy has a saddle (not maximum) on these solutions.
$N=1$), the instability can be proven rigorously; as shown in Appendix C.2, the entropy has a saddle (not maximum) on these solutions.
To understand how slightly perturbed drops and bubbles evolve, they were simulated numerically using the simplest asymptotic version of the DIM, the one based on (4.18), (4.19) and (4.9). It is adapted for  $N=1$ and a single spatial coordinate in Appendix D, which also outlines the numerical method used.
$N=1$ and a single spatial coordinate in Appendix D, which also outlines the numerical method used.
Various initial conditions have been simulated for the van der Waals pure fluid, with only two patterns of evolution observed. If the initial condition includes a steady-drop solution  $\rho _{1}^{(sd)}(z)$ plus some extra fluid, spontaneous condensation is typically triggered off, giving rise to two shock waves propagating away from the drop's centre. This behaviour is illustrated in figure 5(a) for
$\rho _{1}^{(sd)}(z)$ plus some extra fluid, spontaneous condensation is typically triggered off, giving rise to two shock waves propagating away from the drop's centre. This behaviour is illustrated in figure 5(a) for
 \begin{equation} T=0.2,\quad \rho_{\infty,1}=0.05, \end{equation}
\begin{equation} T=0.2,\quad \rho_{\infty,1}=0.05, \end{equation}and the initial condition
 \begin{equation} \rho_{1}=\rho_{1}^{(sd)}(1.01\,z),\quad w=0\ \text{at} t=0,\end{equation}
\begin{equation} \rho_{1}=\rho_{1}^{(sd)}(1.01\,z),\quad w=0\ \text{at} t=0,\end{equation}
where  $w$ is the vertical velocity (the other two velocity components are zero due to the 1-D nature of the flow).
$w$ is the vertical velocity (the other two velocity components are zero due to the 1-D nature of the flow).

Figure 5. Evolution of a perturbed drop, described by (D3) and (D4) and boundary conditions (D5a,b) and (D6a,b), for a pure van der Waals fluid and parameters (6.10a,b). (a) Initial condition (6.11) (the steady solution plus extra fluid); the time  $t$ of a snapshot and the corresponding curve number
$t$ of a snapshot and the corresponding curve number  $n$ are inter-related via
$n$ are inter-related via  $t=5000\,n$. (b) Initial condition (6.12) (the steady solution minus extra fluid);
$t=5000\,n$. (b) Initial condition (6.12) (the steady solution minus extra fluid);  $t=500\,n$.
$t=500\,n$.
If, however, the initial condition includes less fluid than  $\rho _{1}^{(nm)}(z)$, the drop would typically evaporate. This pattern is illustrated in figure 5(b) for parameters (6.10a,b) and the following initial condition:
$\rho _{1}^{(nm)}(z)$, the drop would typically evaporate. This pattern is illustrated in figure 5(b) for parameters (6.10a,b) and the following initial condition:
 \begin{equation} \rho_{1}=\rho_{1}^{(sd)}(0.99\,z),\quad w=0\quad \text{at}\ t=0. \end{equation}
\begin{equation} \rho_{1}=\rho_{1}^{(sd)}(0.99\,z),\quad w=0\quad \text{at}\ t=0. \end{equation}The two patterns can be understood physically on the basis of the fact that the steady-drop solutions exist only for drops surrounded by oversaturated metastable vapour, which is stable with respect to infinitesimally small perturbations, but may be unstable with respect to finite ones. One can thus assume that the drop solution provides a lower bound for the mass of perturbations capable of triggering off instability of the surrounding vapour.
It is also interesting to see how a drop would evolve if it is surrounded by saturated (not oversaturated) vapour.
The fact that the thickness of a steady drop becomes infinite as  $\rho _{\infty,1}\rightarrow \rho _{1}^{(v)}$ suggests that a finite-size liquid layer, surrounded by saturated vapour, evaporates. The mechanism of this evaporation, however, is not immediately clear. Two-dimensional and three-dimensional drops, for example, evaporate because the curvature of their boundary gives rise to a liquid-to-vapour mass flux (Benilov Reference Benilov2020d, Reference Benilov2021, Reference Benilov2022b), but the boundaries of 1-D drops are flat. One can only assume that they evaporate due to a long-range interaction of the drop's upper and lower interfaces.
$\rho _{\infty,1}\rightarrow \rho _{1}^{(v)}$ suggests that a finite-size liquid layer, surrounded by saturated vapour, evaporates. The mechanism of this evaporation, however, is not immediately clear. Two-dimensional and three-dimensional drops, for example, evaporate because the curvature of their boundary gives rise to a liquid-to-vapour mass flux (Benilov Reference Benilov2020d, Reference Benilov2021, Reference Benilov2022b), but the boundaries of 1-D drops are flat. One can only assume that they evaporate due to a long-range interaction of the drop's upper and lower interfaces.
To explore this hypothesis, 1-D drops floating in saturated vapour were simulated numerically (using the same model and numerical method as before). It turned out that these drops do evaporate, and their evolution can be subdivided into the following three distinct stages.
(i) The boundaries of the drop rapidly assume the profile of a steady liquid/vapour interface, of the kind examined in the previous subsection.
(ii) The drop begins to get thinner, but the density of the drop's core remains close to
$\rho _{1}^{(l)}$.(iii) Once the thickness of the drop becomes comparable to the interfacial thickness, the density at the drop's centre begins to rapidly decrease, and the drop disappears.

The main characteristic of such behaviour is the evaporation time  $t_{e}$, which can be defined as the interval over which the density at the drop's centre falls by a factor of, say,
$t_{e}$, which can be defined as the interval over which the density at the drop's centre falls by a factor of, say,  $10$, i.e.
$10$, i.e.
 \begin{equation} \frac{\rho_{1}(0,t_{e})}{\rho_{1}(0,0)}=0.1. \end{equation}
\begin{equation} \frac{\rho_{1}(0,t_{e})}{\rho_{1}(0,0)}=0.1. \end{equation}
Here  $t_{e}$ was computed as a function of the drop's initial size
$t_{e}$ was computed as a function of the drop's initial size  $W$ for the following initial condition:
$W$ for the following initial condition:
 \begin{equation} \rho_{1}=\rho_{1}^{(v)}+\frac{\rho_{1}^{(l)}-\rho_{1}^{(v)}}{1+\left( \dfrac{2z}{W}\right)^{6}},\quad w=0\quad \text{at}\ t=0. \end{equation}
\begin{equation} \rho_{1}=\rho_{1}^{(v)}+\frac{\rho_{1}^{(l)}-\rho_{1}^{(v)}}{1+\left( \dfrac{2z}{W}\right)^{6}},\quad w=0\quad \text{at}\ t=0. \end{equation}
It turned out that, for  $W$ changing from
$W$ changing from  $0.2$ to
$0.2$ to  $10$, the evaporation time
$10$, the evaporation time  $t_{e}$ grows from
$t_{e}$ grows from  $48.5$ to
$48.5$ to  $10^{6}$, i.e. exponentially. This agrees with the hypothesis that the drop evaporation is caused by long-range interaction of the drop's boundaries and the fact that the density in liquid/vapour interfaces tends to
$10^{6}$, i.e. exponentially. This agrees with the hypothesis that the drop evaporation is caused by long-range interaction of the drop's boundaries and the fact that the density in liquid/vapour interfaces tends to  $\rho _{1}^{(v)}$ and
$\rho _{1}^{(v)}$ and  $\rho _{1}^{(l)}$ (as
$\rho _{1}^{(l)}$ (as  $z\rightarrow \pm \infty$) exponentially quickly.
$z\rightarrow \pm \infty$) exponentially quickly.
6.3. Solid/fluid interfaces
Let the fluid be bounded below by a flat horizontal wall (substrate) located at  $z=0$, so that the boundary condition (2.47) reduces to
$z=0$, so that the boundary condition (2.47) reduces to
 \begin{equation} \rho_{i}=\rho_{0,i}\quad \text{at}\ z=0. \end{equation}
\begin{equation} \rho_{i}=\rho_{0,i}\quad \text{at}\ z=0. \end{equation}Far above the substrate, the fluid is homogeneous,
 \begin{equation} \rho_{i}\rightarrow\rho_{\infty,i}\quad \text{as}\ z\rightarrow+\infty, \end{equation}
\begin{equation} \rho_{i}\rightarrow\rho_{\infty,i}\quad \text{as}\ z\rightarrow+\infty, \end{equation}
where  $\rho _{\infty,i}$ corresponds to a stable or metastable state. Finally, (6.9) used in the previous subsection for 1-D drops and bubbles applies to the present case as well.
$\rho _{\infty,i}$ corresponds to a stable or metastable state. Finally, (6.9) used in the previous subsection for 1-D drops and bubbles applies to the present case as well.
The boundary-value problem (6.9), (6.15) and (6.16) was solved numerically for a van der Waals pure fluid with
 \begin{equation} T=0.2,\quad \rho_{0,1}=0.3. \end{equation}
\begin{equation} T=0.2,\quad \rho_{0,1}=0.3. \end{equation}
Its typical solutions are illustrated in figure 6 ( $\rho _{0,i}$ varies,
$\rho _{0,i}$ varies,  $\rho _{\infty,1}$ is fixed) and in figure 7 (
$\rho _{\infty,1}$ is fixed) and in figure 7 ( $\rho _{0,i}$ is fixed,
$\rho _{0,i}$ is fixed,  $\rho _{\infty,1}$ varies). The following features can be observed.
$\rho _{\infty,1}$ varies). The following features can be observed.
(i) For a given
$\rho _{\infty,1}$, there exists a certain pool of $\rho _{0,i}$ that can be ‘connected’ to this $\rho _{\infty,1}$. If, for example, $\rho _{\infty,1}$ equals the saturated vapour density (the case illustrated in figure 6a), then $\rho _{0,1}$ must be smaller than the saturated liquid density (and vice versa: if $\rho _{\infty,1}=\rho _{1}^{(l)}$, then $\rho _{0,i}>\rho _{1}^{(v)}$; see figure 6b). When $\rho _{\infty,1}$ approaches the pool's boundary, a clearly visible liquid/vapour interface emerges in the solution and gradually moves away from the substrate.(ii) It follows from above that, for some pairs
$( \rho _{0,1} , \rho _{\infty,1})$, no solution exists. As illustrated in figure 7, some such pairs involve unstable values of $\rho _{\infty,1}$ (and, thus, are unimportant), but there are also ones with a metastable $\rho _{\infty,1}$.
Figure 6. Examples of solid/fluid interfaces for a pure van der Waals fluid with parameters (6.17a,b), for a given
$\rho _{\infty,1}$: (a) $\rho _{\infty,1}=\rho _{1}^{(v)}$, $\rho _{0,1}$ varies from $0$ to $\rho _{1}^{(l)}$; (b) $\rho _{\infty,1} =\rho _{1}^{(l)}$, $\rho _{0,1}$ varies from $1$ to $\rho _{1}^{(v)}$. The arrows show the direction towards the boundary of the pool of allowable values of $\rho _{0,1}$.Figure 7. Examples of solid/fluid interfaces for a pure van der Waals fluid with parameters (6.17a,b). The fluid at infinity is: (1) undersaturated vapour, (2) oversaturated vapour with two possible solutions (the unstable one is shown as a thin line), (3) oversaturated vapour with a single solution, (4) undersaturated liquid with a single solution, etc. Observe that there are no solutions between curves (3) and (4).
To clarify what happens in such cases, the evolution was simulated numerically (using the model and method used in the previous two subsections). It turned out that, if the boundary-value problem (6.9), (6.15) and (6.16) does not have a solution, the substrate triggers off a spontaneous phase change. This result can be readily interpreted physically: if a sufficiently hydrophilic substrate (with a sufficiently small
$\rho _{0,1}$) touches oversaturated vapour, it triggers off spontaneous condensation, whereas a sufficiently hydrophobic substrate touching undersaturated liquid triggers off evaporation. In either case, a shock wave of phase change propagates away from the substrate, and the solid/fluid interface cannot be stationary.(iii) For some pairs
$( \rho _{0,1}, \rho _{\infty,1})$, there are two different solutions: a monotonic and non-monotonic one (see figure 7). It is unclear which of the two occurs in reality.It turned out that only monotonic solutions of the boundary-value problem (6.9), (6.15) and (6.16) maximise the entropy, whereas non-monotonic solutions do not (the former claim is proved in the general case, but the latter, only for
$N=1$; see Appendix C.3). That is, non-monotonic solutions cannot be ruled out with certainty for $N\geq 2$ – yet the mere fact that the general stability proof that works for monotonic solutions cannot be extended to non-monotonic ones seems to resolve the dilemma in favour of the former.(iv) Another physically important conclusion follows from the fact that the boundary-value problem (6.9), (6.15) and (6.16) has no more than one stable solution, and, thus, does not admit solutions describing a liquid layer of a finite thickness on a substrate, with saturated vapour above it. All such layers, regardless of their thickness, evaporate, and the Kelvin effect cannot be responsible for this effect (because the liquid/vapour interface is flat). The evaporation in this case can only be caused by long-range interaction between the interface and the substrate.
(v) It is interesting to compare the interfacial profiles shown in figures 6 and 7 to those computed by Evans, Stewart & Wilding (Reference Evans, Stewart and Wilding2017) via the density functional theory and Monte Carlo method for a Lennard-Jones fluid bounded by a single wall or contained between two parallel walls. The single-wall profiles of Evans et al. (Reference Evans, Stewart and Wilding2017) (see their figure 3) are qualitatively similar to those computed in this paper, but their two-wall profiles are riddled with short-scale oscillations (see figures 6, 14 and 20 of Evans et al. Reference Evans, Stewart and Wilding2017). It is not clear whether the oscillations are caused by ‘interference’ of the walls: on the one hand, the distance between the walls exceeds the spatial scale of wall–molecule interaction by a factor of
$30$ (hence, the ‘interference’ should be weak), but, on the other hand, no oscillations occur when this distance is infinite.Whatever the nature of the oscillations is, one should not expect the DIM to describe them due to the omission of the higher-order derivatives of
$\rho _{i}$ when obtaining expression (2.43) for the van der Waals force.





























7. Surface tension and contact angle
Consider a fluid bounded below by a horizontal substrate, and an oblique liquid/vapour interface intersecting the substrate; see figure 8. Note that this figure depicts a hydrophilic substrate, such that the contact angle  $\theta$ is smaller than
$\theta$ is smaller than  ${\rm \pi} /2$.
${\rm \pi} /2$.



If in equilibrium, the setting outlined is described by the two-dimensional reduction of (3.12). Given expression (3.13) for  $\eta _{i}$, one obtains
$\eta _{i}$, one obtains
 \begin{equation} G_{i}-\sum_{j}K_{ij}\left( \frac{\partial^{2}\rho_{j}}{\partial x^{2}} +\frac{\partial^{2}\rho_{j}}{\partial z^{2}}\right) +G_{\infty,i}=0.\end{equation}
\begin{equation} G_{i}-\sum_{j}K_{ij}\left( \frac{\partial^{2}\rho_{j}}{\partial x^{2}} +\frac{\partial^{2}\rho_{j}}{\partial z^{2}}\right) +G_{\infty,i}=0.\end{equation}Impose also the boundary condition
 \begin{equation} \rho_{i}\rightarrow\rho_{i}^{(v)}\quad \text{as} z\rightarrow+\infty, \end{equation}
\begin{equation} \rho_{i}\rightarrow\rho_{i}^{(v)}\quad \text{as} z\rightarrow+\infty, \end{equation}
which implies that the constant in (7.1) is  $G_{\infty,i}=G_{i}(\rho _{1}^{(v)}\ldots \rho _{N}^{(v)},T)$. At the substrate, the standard boundary condition
$G_{\infty,i}=G_{i}(\rho _{1}^{(v)}\ldots \rho _{N}^{(v)},T)$. At the substrate, the standard boundary condition
 \begin{equation} \rho_{i}=\rho_{0,i}\quad \text{at}\ z=0 \end{equation}
\begin{equation} \rho_{i}=\rho_{0,i}\quad \text{at}\ z=0 \end{equation}is assumed.
To close the boundary-value problem (7.1)–(7.3), one should set boundary conditions as  $x\rightarrow \pm \infty$. The setting depicted in figure 8 corresponds to
$x\rightarrow \pm \infty$. The setting depicted in figure 8 corresponds to
 $$\begin{gather} \rho_{i} \rightarrow\rho_{i}^{(s/v)}(z)\quad \text{as}\ x\rightarrow-\infty, \end{gather}$$
$$\begin{gather} \rho_{i} \rightarrow\rho_{i}^{(s/v)}(z)\quad \text{as}\ x\rightarrow-\infty, \end{gather}$$ $$\begin{gather}\rho_{i} \rightarrow\rho_{i}^{(s/l)}(z)+\rho_{i}^{(l/v)}(\xi )\quad \text{as}\ x\rightarrow+\infty. \end{gather}$$
$$\begin{gather}\rho_{i} \rightarrow\rho_{i}^{(s/l)}(z)+\rho_{i}^{(l/v)}(\xi )\quad \text{as}\ x\rightarrow+\infty. \end{gather}$$
Here, the function  $\rho _{i}^{(s/v)}(z)$ describes a solid/vapour interface (i.e. satisfies the boundary-value problem (6.9), (6.15) and (6.16) with
$\rho _{i}^{(s/v)}(z)$ describes a solid/vapour interface (i.e. satisfies the boundary-value problem (6.9), (6.15) and (6.16) with  $\rho _{\infty,i}=\rho _{i}^{(v)}$); the function
$\rho _{\infty,i}=\rho _{i}^{(v)}$); the function  $\rho _{i}^{(s/l)}(z)$ describes a solid/liquid interface; and
$\rho _{i}^{(s/l)}(z)$ describes a solid/liquid interface; and  $\rho _{i}^{(l/v)} (\xi )$ (where
$\rho _{i}^{(l/v)} (\xi )$ (where  $\xi =z\cos \theta -x\sin \theta$) describes a liquid/vapour interface tilted at an angle
$\xi =z\cos \theta -x\sin \theta$) describes a liquid/vapour interface tilted at an angle  $\theta$ (it satisfies the boundary-value problem (6.1)–(6.3) with
$\theta$ (it satisfies the boundary-value problem (6.1)–(6.3) with  $z$ changed to
$z$ changed to  $\xi$).
$\xi$).
In the framework of the DIM, the contact angle  $\theta$ is not arbitrary, but is fully determined by the fluid's thermodynamic properties, the Korteweg matrix
$\theta$ is not arbitrary, but is fully determined by the fluid's thermodynamic properties, the Korteweg matrix  $K_{ij}$ and the near-wall density
$K_{ij}$ and the near-wall density  $\rho _{0,i}$. To derive an expression for
$\rho _{0,i}$. To derive an expression for  $\theta$ (similar to that derived by Pismen & Pomeau (Reference Pismen and Pomeau2000) for pure fluids), consider
$\theta$ (similar to that derived by Pismen & Pomeau (Reference Pismen and Pomeau2000) for pure fluids), consider
 \begin{equation} \sum_{i}\int_{0}^{\infty} (7.1) \times\frac{\partial \rho_{i}}{\partial x}\,\mathrm{d}z. \end{equation}
\begin{equation} \sum_{i}\int_{0}^{\infty} (7.1) \times\frac{\partial \rho_{i}}{\partial x}\,\mathrm{d}z. \end{equation}After straightforward algebra involving integration by parts and the use of boundary conditions (7.2) and (7.3) and identity (2.7), one obtains
 \begin{align} \frac{\partial}{\partial x}\int_{0}^{\infty}\left[ \sum_{j}\rho_{j}( G_{j}-G_{j,\infty}) -p+p_{\infty}+\frac{1}{2}\sum_{ij}K_{ij}\left( \frac{\partial\rho_{i}}{\partial z}\frac{\partial\rho_{j}}{\partial z} -\frac{\partial\rho_{i}}{\partial x}\frac{\partial\rho_{j}}{\partial x}\right) \right] \,\mathrm{d}z=0, \end{align}
\begin{align} \frac{\partial}{\partial x}\int_{0}^{\infty}\left[ \sum_{j}\rho_{j}( G_{j}-G_{j,\infty}) -p+p_{\infty}+\frac{1}{2}\sum_{ij}K_{ij}\left( \frac{\partial\rho_{i}}{\partial z}\frac{\partial\rho_{j}}{\partial z} -\frac{\partial\rho_{i}}{\partial x}\frac{\partial\rho_{j}}{\partial x}\right) \right] \,\mathrm{d}z=0, \end{align}
where  $p_{\infty }=p(\rho _{1}^{(v)}\ldots \rho _{N}^{(v)},T)$. Finally, integrating the above expression from
$p_{\infty }=p(\rho _{1}^{(v)}\ldots \rho _{N}^{(v)},T)$. Finally, integrating the above expression from  $x=-\infty$ to
$x=-\infty$ to  $x=+\infty$ and recalling boundary conditions (7.4) and (7.5), one obtains
$x=+\infty$ and recalling boundary conditions (7.4) and (7.5), one obtains
 \begin{align} &\int_{0}^{\infty}\left\{ \left[ \sum_{i}\rho_{i}( G_{i}-G_{\infty ,i}) -p+p_{\infty}\right] _{\rho_{i}=\rho_{i}^{(s/v)}}+\frac{1}{2} \sum_{ij}K_{ij}\frac{\mathrm{d}\rho_{i}^{(s/v)}}{\mathrm{d}z}\frac {\mathrm{d}\rho_{j}^{(s/v)}}{\mathrm{d}z}\right\}\, \mathrm{d}z\nonumber\\ &\quad =\int_{0}^{\infty}\left\{ \left[ \sum_{i}\rho_{i}( G_{i}-G_{\infty ,i}) -p+p_{\infty}\right] _{\rho_{i}=\rho_{i}^{(s/l)}}+\frac{1}{2} \sum_{ij}K_{ij}\frac{\mathrm{d}\rho_{i}^{(s/l)}}{\mathrm{d}z}\frac {\mathrm{d}\rho_{j}^{(s/l)}}{\mathrm{d}z}\right\}\, \mathrm{d}z\nonumber\\ &\qquad+\int_{0}^{\infty}\left\{ \left[ \sum_{i}\rho_{i}( G_{i}-G_{\infty ,i}) -p+p_{\infty}\right] _{\rho_{i}=\rho_{i}^{(l/v)}} \vphantom{\left[ \sum_{i}\right] _{\rho_{i}=\rho_{i}^{(l/v)}}\frac{\mathrm{d}\rho _{i}^{(l/v)}}{\mathrm{d}\xi}}\right.\nonumber\\ &\qquad +\left. \frac{1}{2}( \cos^{2}\theta-\sin^{2}\theta) \sum _{ij}K_{ij}\frac{\mathrm{d}\rho_{i}^{(l/v)}}{\mathrm{d}\xi} \frac{\mathrm{d}\rho_{i}^{(l/v)}}{\mathrm{d}\xi} \vphantom{\left[ \sum_{i}\right] _{\rho_{i}=\rho_{i}^{(l/v)}}\frac{\mathrm{d}\rho _{i}^{(l/v)}}{\mathrm{d}\xi}}\right\} \frac{\mathrm{d}\xi}{\cos\theta}. \end{align}
\begin{align} &\int_{0}^{\infty}\left\{ \left[ \sum_{i}\rho_{i}( G_{i}-G_{\infty ,i}) -p+p_{\infty}\right] _{\rho_{i}=\rho_{i}^{(s/v)}}+\frac{1}{2} \sum_{ij}K_{ij}\frac{\mathrm{d}\rho_{i}^{(s/v)}}{\mathrm{d}z}\frac {\mathrm{d}\rho_{j}^{(s/v)}}{\mathrm{d}z}\right\}\, \mathrm{d}z\nonumber\\ &\quad =\int_{0}^{\infty}\left\{ \left[ \sum_{i}\rho_{i}( G_{i}-G_{\infty ,i}) -p+p_{\infty}\right] _{\rho_{i}=\rho_{i}^{(s/l)}}+\frac{1}{2} \sum_{ij}K_{ij}\frac{\mathrm{d}\rho_{i}^{(s/l)}}{\mathrm{d}z}\frac {\mathrm{d}\rho_{j}^{(s/l)}}{\mathrm{d}z}\right\}\, \mathrm{d}z\nonumber\\ &\qquad+\int_{0}^{\infty}\left\{ \left[ \sum_{i}\rho_{i}( G_{i}-G_{\infty ,i}) -p+p_{\infty}\right] _{\rho_{i}=\rho_{i}^{(l/v)}} \vphantom{\left[ \sum_{i}\right] _{\rho_{i}=\rho_{i}^{(l/v)}}\frac{\mathrm{d}\rho _{i}^{(l/v)}}{\mathrm{d}\xi}}\right.\nonumber\\ &\qquad +\left. \frac{1}{2}( \cos^{2}\theta-\sin^{2}\theta) \sum _{ij}K_{ij}\frac{\mathrm{d}\rho_{i}^{(l/v)}}{\mathrm{d}\xi} \frac{\mathrm{d}\rho_{i}^{(l/v)}}{\mathrm{d}\xi} \vphantom{\left[ \sum_{i}\right] _{\rho_{i}=\rho_{i}^{(l/v)}}\frac{\mathrm{d}\rho _{i}^{(l/v)}}{\mathrm{d}\xi}}\right\} \frac{\mathrm{d}\xi}{\cos\theta}. \end{align}
This equality can be simplified using identity (6.6) (with  $\rho _{i}$ changed to
$\rho _{i}$ changed to $\rho _{i}^{(l/v)}$), and similar identities for
$\rho _{i}^{(l/v)}$), and similar identities for  $\rho _{i}^{(s/v)}$ and
$\rho _{i}^{(s/v)}$ and  $\rho _{i}^{(s/l)}$. After straightforward algebra involving re-denoting
$\rho _{i}^{(s/l)}$. After straightforward algebra involving re-denoting  $\xi \rightarrow z$, one obtains the DIM version of Young's formula,
$\xi \rightarrow z$, one obtains the DIM version of Young's formula,
 \begin{equation} \cos\theta=\frac{\sigma^{(s/v)}-\sigma^{(s/l)}}{\sigma^{(l/v)}},\end{equation}
\begin{equation} \cos\theta=\frac{\sigma^{(s/v)}-\sigma^{(s/l)}}{\sigma^{(l/v)}},\end{equation}where
 \begin{align} \left.\begin{array}{c} \displaystyle \sigma^{(s/v)}=\sum_{ij}K_{ij} {\int_{0}^{\infty}} \dfrac{\mathrm{d}\rho_{i}^{(s/v)}}{\mathrm{d}z}\dfrac{\mathrm{d}\rho _{j}^{(s/v)}}{\mathrm{d}z}\,\mathrm{d}z,\quad \sigma^{(s/l)}=\sum_{ij}K_{ij} {\displaystyle\int_{0}^{\infty}} \dfrac{\mathrm{d}\rho_{i}^{(s/l)}}{\mathrm{d}z}\dfrac{\mathrm{d}\rho _{j}^{(s/l)}}{\mathrm{d}z}\,\mathrm{d}z,\\ \displaystyle \sigma^{(l/v)}=\sum_{ij}K_{ij} {\int_{-\infty}^{\infty}} \dfrac{\mathrm{d}\rho_{i}^{(l/v)}}{\mathrm{d}z}\dfrac{\mathrm{d}\rho _{j}^{(l/v)}}{\mathrm{d}z}\,\mathrm{d}z , \end{array}\right\} \end{align}
\begin{align} \left.\begin{array}{c} \displaystyle \sigma^{(s/v)}=\sum_{ij}K_{ij} {\int_{0}^{\infty}} \dfrac{\mathrm{d}\rho_{i}^{(s/v)}}{\mathrm{d}z}\dfrac{\mathrm{d}\rho _{j}^{(s/v)}}{\mathrm{d}z}\,\mathrm{d}z,\quad \sigma^{(s/l)}=\sum_{ij}K_{ij} {\displaystyle\int_{0}^{\infty}} \dfrac{\mathrm{d}\rho_{i}^{(s/l)}}{\mathrm{d}z}\dfrac{\mathrm{d}\rho _{j}^{(s/l)}}{\mathrm{d}z}\,\mathrm{d}z,\\ \displaystyle \sigma^{(l/v)}=\sum_{ij}K_{ij} {\int_{-\infty}^{\infty}} \dfrac{\mathrm{d}\rho_{i}^{(l/v)}}{\mathrm{d}z}\dfrac{\mathrm{d}\rho _{j}^{(l/v)}}{\mathrm{d}z}\,\mathrm{d}z , \end{array}\right\} \end{align}
are the surface tension coefficients of the solid/vapour, solid/liquid and liquid/vapour interfaces, respectively. Treating hydrophobic substrates ( $\theta >\frac {1}{2}{\rm \pi}$) in a similar fashion, one can show that (7.9) applies to that case as well.
$\theta >\frac {1}{2}{\rm \pi}$) in a similar fashion, one can show that (7.9) applies to that case as well.
To calculate the surface tension – say,  $\sigma ^{(s/l)}$ – one first needs to solve the boundary-value problem (6.1)–(6.4) that determines the function
$\sigma ^{(s/l)}$ – one first needs to solve the boundary-value problem (6.1)–(6.4) that determines the function  $\rho _{i}=\rho _{i}^{(l/v)}(z)$. For a pure fluid, however, the expression for
$\rho _{i}=\rho _{i}^{(l/v)}(z)$. For a pure fluid, however, the expression for  $\sigma ^{(s/l)}$ can be rewritten as a closed-form integral. To do so, let
$\sigma ^{(s/l)}$ can be rewritten as a closed-form integral. To do so, let  $N=1$ in (6.6), which yields
$N=1$ in (6.6), which yields
 \begin{equation} \frac{\mathrm{d}\rho_{1}}{\mathrm{d}z}={-}\sqrt{\frac{2}{K_{11}}\{ \rho_{1}[ G_{1}(\rho_{1},T)-G_{1}(\rho_{1}^{(v)},T)] -p(\rho _{1},T)+p(\rho_{1}^{(v)},T)\}}. \end{equation}
\begin{equation} \frac{\mathrm{d}\rho_{1}}{\mathrm{d}z}={-}\sqrt{\frac{2}{K_{11}}\{ \rho_{1}[ G_{1}(\rho_{1},T)-G_{1}(\rho_{1}^{(v)},T)] -p(\rho _{1},T)+p(\rho_{1}^{(v)},T)\}}. \end{equation}
Using this equation to change the variable of integration in the expression for  $\sigma ^{(l/v)}$ in (7.10), one obtains
$\sigma ^{(l/v)}$ in (7.10), one obtains
 \begin{equation} \sigma^{(l/v)}=\sqrt{2K_{11}} {\displaystyle\int_{\rho_{1}^{(v)}}^{\rho_{1}^{(l)}}} \sqrt{\rho_{1}[ G_{1}(\rho_{1},T)-G_{1}(\rho_{1}^{(v)},T)] -p(\rho_{1},T)+p(\rho_{1}^{(v)},T)}\,\mathrm{d}\rho. \end{equation}
\begin{equation} \sigma^{(l/v)}=\sqrt{2K_{11}} {\displaystyle\int_{\rho_{1}^{(v)}}^{\rho_{1}^{(l)}}} \sqrt{\rho_{1}[ G_{1}(\rho_{1},T)-G_{1}(\rho_{1}^{(v)},T)] -p(\rho_{1},T)+p(\rho_{1}^{(v)},T)}\,\mathrm{d}\rho. \end{equation}
Thus, to calculate  $\sigma ^{(l/v)}$, one should first use the given
$\sigma ^{(l/v)}$, one should first use the given  $p(\rho _{1},T)$ and
$p(\rho _{1},T)$ and  $G_{1}(\rho _{1},T)$ to find
$G_{1}(\rho _{1},T)$ to find  $\rho _{1}^{(l)}$ and
$\rho _{1}^{(l)}$ and  $\rho _{1}^{(v)}$ (via the Maxwell construction) and then evaluate integral (7.12).
$\rho _{1}^{(v)}$ (via the Maxwell construction) and then evaluate integral (7.12).
8. Parameterising the diffuse-interface model for water/air interfaces
To use the DIM in applications, one needs the following external parameters: the fluid's thermodynamic properties, the dependence of the viscosity and transport coefficients on  $( \rho _{1}\ldots \rho _{N},T)$, and the Korteweg matrix. In this section an approach to specifying these parameters is described and applied to water/air interfaces.
$( \rho _{1}\ldots \rho _{N},T)$, and the Korteweg matrix. In this section an approach to specifying these parameters is described and applied to water/air interfaces.
For reasons described in § 2.2, the fluid's thermodynamic properties will be approximated by the EV model. It involves an undetermined matrix  $a_{ij}$ and an undetermined function
$a_{ij}$ and an undetermined function  $\varTheta (\rho _{1}\ldots \rho _{N})$, which should be fixed as the best fits of the empiric characteristics of the fluid under consideration.
$\varTheta (\rho _{1}\ldots \rho _{N})$, which should be fixed as the best fits of the empiric characteristics of the fluid under consideration.
In §§ 8.1 and 8.2 we will explain how the fitting should be carried out for a pure fluid (in application to water, nitrogen and oxygen). An approach to determining  $K_{11}$ for a pure fluid will be outlined in § 8.3. Section 8.4 describes how
$K_{11}$ for a pure fluid will be outlined in § 8.3. Section 8.4 describes how  $a_{ij}$,
$a_{ij}$,  $\varTheta$ and
$\varTheta$ and  $K_{ij}$ can be determined for a water/air mixture, and its viscosity and transport coefficients are dealt with in § 8.5.
$K_{ij}$ can be determined for a water/air mixture, and its viscosity and transport coefficients are dealt with in § 8.5.
All these tasks will be carried out in terms of the original (dimensional) variables.
8.1. The van der Waals parameter of pure water, nitrogen and oxygen
For a pure fluid, the EV expression (2.14) for the internal energy yields (the subscript  ${1}$ is omitted)
${1}$ is omitted)
 \begin{equation} e=cT-a\rho, \end{equation}
\begin{equation} e=cT-a\rho, \end{equation}
which suggests that the van der Waals parameter  $a$ can be determined as the linear fit of the empiric dependence of
$a$ can be determined as the linear fit of the empiric dependence of  $cT-e$ on
$cT-e$ on  $\rho$. The heat capacity
$\rho$. The heat capacity  $c$ in this expression should be the same as that in the EV kinetic theory, i.e.
$c$ in this expression should be the same as that in the EV kinetic theory, i.e.  $3R$ for water and
$3R$ for water and  $5R/2$ for nitrogen and oxygen. For simplicity, the fitting was carried out using only the data on the critical isobar
$5R/2$ for nitrogen and oxygen. For simplicity, the fitting was carried out using only the data on the critical isobar  $p=p_{cr}$ (as done by Benilov & Benilov Reference Benilov and Benilov2019), but the resulting straight line fits the isobars
$p=p_{cr}$ (as done by Benilov & Benilov Reference Benilov and Benilov2019), but the resulting straight line fits the isobars  $p=p_{cr}/2$ and
$p=p_{cr}/2$ and  $p=2p_{cr}$ reasonably well too (see the top panels of figure 9). The parameters of the critical points for the fluids under consideration, as well as the other parameters needed here and hereinafter, can be found in tables 1 and 2.
$p=2p_{cr}$ reasonably well too (see the top panels of figure 9). The parameters of the critical points for the fluids under consideration, as well as the other parameters needed here and hereinafter, can be found in tables 1 and 2.

Figure 9. Thermodynamic properties of  $\mathrm {H}_{2}\mathrm {O}$ (left-hand panels) and
$\mathrm {H}_{2}\mathrm {O}$ (left-hand panels) and  $\mathrm {N}_{2}$ (right-hand panels). The non-connected symbols show the empiric data from Lindstrom & Mallard (Reference Lindstrom and Mallard1997) presented in isobaric form, for three values of the pressure
$\mathrm {N}_{2}$ (right-hand panels). The non-connected symbols show the empiric data from Lindstrom & Mallard (Reference Lindstrom and Mallard1997) presented in isobaric form, for three values of the pressure  $p$ relative to its critical value,
$p$ relative to its critical value,  $p_{cr}$ (see the legend). The gap between
$p_{cr}$ (see the legend). The gap between  $\rho ^{(v)}$ and
$\rho ^{(v)}$ and  $\rho ^{(l)}$ in the empiric data for
$\rho ^{(l)}$ in the empiric data for  $p=p_{cr}/2$ reflects the impossibility (difficulty) of measurements in an unstable (metastable) fluid. The dotted lines show the parameters calculated via the EV fluid model.
$p=p_{cr}/2$ reflects the impossibility (difficulty) of measurements in an unstable (metastable) fluid. The dotted lines show the parameters calculated via the EV fluid model.
Table 1. The parameters of the triple and critical points (subscripts  $_{tr}$ and
$_{tr}$ and  $_{cr}$, respectively) of
$_{cr}$, respectively) of  $\mathrm {H}_{2}\mathrm {O}$,
$\mathrm {H}_{2}\mathrm {O}$,  $\mathrm {N}_{2}$ and
$\mathrm {N}_{2}$ and  $\mathrm {O}_{2}$ (Lindstrom & Mallard Reference Lindstrom and Mallard1997).
$\mathrm {O}_{2}$ (Lindstrom & Mallard Reference Lindstrom and Mallard1997).

Table 2. The parameters of  $\mathrm {H}_{2}\mathrm {O}$,
$\mathrm {H}_{2}\mathrm {O}$,  $\mathrm {N}_{2}$,
$\mathrm {N}_{2}$,  $\mathrm {O}_{2}$ and air:
$\mathrm {O}_{2}$ and air:  $m$ is the molecular mass,
$m$ is the molecular mass,  $R$ is the specific gas constant,
$R$ is the specific gas constant,  $a$ is the van der Waals parameter,
$a$ is the van der Waals parameter,  $K$ is the Korteweg constant. The parameters of air are calculated as the 79-21 weighted averages of the corresponding parameters of nitrogen and oxygen, respectively.
$K$ is the Korteweg constant. The parameters of air are calculated as the 79-21 weighted averages of the corresponding parameters of nitrogen and oxygen, respectively.

The resulting values of the van der Waals parameter  $a$ for
$a$ for  $\mathrm {H} _{2}\mathrm {O}$,
$\mathrm {H} _{2}\mathrm {O}$,  $\mathrm {N}_{2}$ and
$\mathrm {N}_{2}$ and  $\mathrm {O}_{2}$ are presented in table 2. Interestingly,
$\mathrm {O}_{2}$ are presented in table 2. Interestingly,  $a$ of water exceeds significantly those of nitrogen and oxygen. This is likely to be caused by the difference in the molecular structure of these fluids: the water molecule has a non-zero dipolar moment, whereas nitrogen and oxygen molecules are symmetric – hence, do not. Since the van der Waals force is of an electric nature, one can assume that dipolar molecules interact stronger than neutral ones.
$a$ of water exceeds significantly those of nitrogen and oxygen. This is likely to be caused by the difference in the molecular structure of these fluids: the water molecule has a non-zero dipolar moment, whereas nitrogen and oxygen molecules are symmetric – hence, do not. Since the van der Waals force is of an electric nature, one can assume that dipolar molecules interact stronger than neutral ones.
8.2. The equation of state of pure $\mathrm {H}_{2}\mathrm {O}$, $\mathrm {N}_{2}$, and $\mathrm {O}_{2}$



Pure fluids will be described by the EV model (2.14) and (2.15) with
 \begin{equation} \varTheta=R\left[{-}q^{(0)}\ln\left( 1-0.99\frac{\rho}{\rho_{tp}}\right) +\sum_{n=1}^{4}q^{(n)}\left( \frac{\rho}{\rho_{tp}}\right)^{n}\right] , \end{equation}
\begin{equation} \varTheta=R\left[{-}q^{(0)}\ln\left( 1-0.99\frac{\rho}{\rho_{tp}}\right) +\sum_{n=1}^{4}q^{(n)}\left( \frac{\rho}{\rho_{tp}}\right)^{n}\right] , \end{equation}
where  $q^{(0)}\ldots q^{(4)}$ are undetermined coefficients and
$q^{(0)}\ldots q^{(4)}$ are undetermined coefficients and  $\rho _{tp}$ is the fluid's density at the triple point (
$\rho _{tp}$ is the fluid's density at the triple point ( $\rho _{tp}$ is just a convenient density scale; the fact that, at the triple point, the liquid and vapour are in equilibrium with the solid phase is irrelevant, as solids are not described by the DIM). Note that the first term in expression (8.2) sets the maximum density at
$\rho _{tp}$ is just a convenient density scale; the fact that, at the triple point, the liquid and vapour are in equilibrium with the solid phase is irrelevant, as solids are not described by the DIM). Note that the first term in expression (8.2) sets the maximum density at  $\rho _{tp}/0.99$.
$\rho _{tp}/0.99$.
The coefficients  $q^{(n)}$ were determined for
$q^{(n)}$ were determined for  $\mathrm {H}_{2}\mathrm {O}$,
$\mathrm {H}_{2}\mathrm {O}$,  $\mathrm {N}_{2}$ and
$\mathrm {N}_{2}$ and  $\mathrm {O}_{2}$ by ensuring that the expressions for
$\mathrm {O}_{2}$ by ensuring that the expressions for  $p(\rho,T)$ and
$p(\rho,T)$ and  $G(\rho,T)$ corresponding to (8.2) yield the ‘correct’ – i.e. empiric – values for the critical density, temperature and pressure, as well as the liquid and vapour densities at the triple-point temperature
$G(\rho,T)$ corresponding to (8.2) yield the ‘correct’ – i.e. empiric – values for the critical density, temperature and pressure, as well as the liquid and vapour densities at the triple-point temperature  $T=T_{tp}$ (five equations for the five unknown coefficients). All the necessary empiric data can be found in tables 1 and 2, and the computed values of
$T=T_{tp}$ (five equations for the five unknown coefficients). All the necessary empiric data can be found in tables 1 and 2, and the computed values of  $q^{(n)}$ are listed in table 3. Such an approach to calibrating the EV fluid model is a slight modification of that of Benilov (Reference Benilov2020b), which is, in turn, a modification of the approach of Benilov & Benilov (Reference Benilov and Benilov2018).
$q^{(n)}$ are listed in table 3. Such an approach to calibrating the EV fluid model is a slight modification of that of Benilov (Reference Benilov2020b), which is, in turn, a modification of the approach of Benilov & Benilov (Reference Benilov and Benilov2018).
Table 3. The coefficients of the equation of state (8.2) for  $\mathrm {H}_{2}\mathrm {O}$,
$\mathrm {H}_{2}\mathrm {O}$,  $\mathrm {N}_{2}$ and
$\mathrm {N}_{2}$ and  $\mathrm {O}_{2}$.
$\mathrm {O}_{2}$.

To illustrate how well the EV model describes real fluids, the isobaric (with  $p$ fixed) dependence of the temperature on the density is plotted for
$p$ fixed) dependence of the temperature on the density is plotted for  $\mathrm {H}_{2}\mathrm {O}$ and
$\mathrm {H}_{2}\mathrm {O}$ and  $\mathrm {N}_{2}$ in the lower panels of figure 9. The results for
$\mathrm {N}_{2}$ in the lower panels of figure 9. The results for  $\mathrm {O}_{2}$ (not presented) are similar to those for
$\mathrm {O}_{2}$ (not presented) are similar to those for  $\mathrm {N}_{2}$.
$\mathrm {N}_{2}$.
Note that the DIM has been coupled with realistic equations of state before (e.g. Caupin Reference Caupin2005; Magaletti, Gallo & Casciola Reference Magaletti, Gallo and Casciola2021); for pure water, this was typically done using the IAPWS-95 equation (Wagner & Pruß Reference Wagner and Pruß2002). The EV model used here is undoubtedly less accurate than the IAPWS-95 model, but it allows one to describe consistently all of the fluids involved ( $\mathrm {H} _{2}\mathrm {O}$,
$\mathrm {H} _{2}\mathrm {O}$,  $\mathrm {N}_{2}$ and
$\mathrm {N}_{2}$ and  $\mathrm {O}_{2}$).
$\mathrm {O}_{2}$).
8.3. The Korteweg parameter of pure $\mathrm {H}_{2}\mathrm {O}$, $\mathrm {N}_{2}$, and $\mathrm {O}_{2}$



Benilov (Reference Benilov2020b) proposed to deduce  $K$ from the requirement that the DIM predicts the correct value of the surface tension
$K$ from the requirement that the DIM predicts the correct value of the surface tension  $\sigma ^{(l/v)}$ of the liquid/vapour interface at the triple point. The same was done in the present work:
$\sigma ^{(l/v)}$ of the liquid/vapour interface at the triple point. The same was done in the present work:  $\sigma ^{(l/v)}$ was calculated via the DIM formula (7.12) with
$\sigma ^{(l/v)}$ was calculated via the DIM formula (7.12) with  $G$ and
$G$ and  $p$ of the EV fluid, and the value of
$p$ of the EV fluid, and the value of  $K$ was chosen for which (7.12) yields the same result as the empiric formula of Somayajulu (Reference Somayajulu1988). The resulting
$K$ was chosen for which (7.12) yields the same result as the empiric formula of Somayajulu (Reference Somayajulu1988). The resulting  $K$ for
$K$ for  $\mathrm {H}_{2}\mathrm {O}$,
$\mathrm {H}_{2}\mathrm {O}$,  $\mathrm {N}_{2}$ and
$\mathrm {N}_{2}$ and  $\mathrm {O}_{2}$ can be found in table 2.
$\mathrm {O}_{2}$ can be found in table 2.
It should be emphasised that the values of  $K$ computed via the above approach depend on the chosen fluid model. This explains the difference between the result for
$K$ computed via the above approach depend on the chosen fluid model. This explains the difference between the result for  $K$ in this paper and the one computed by Benilov (Reference Benilov2020b): the former is based on the EV fluid model and the latter on the van der Waals model. The resulting 30 % difference in
$K$ in this paper and the one computed by Benilov (Reference Benilov2020b): the former is based on the EV fluid model and the latter on the van der Waals model. The resulting 30 % difference in  $\sigma ^{(l/v)}$ reflects the fact that the latter model is much less accurate.
$\sigma ^{(l/v)}$ reflects the fact that the latter model is much less accurate.
As a test of the whole approach based on the DIM and EV models, the theoretical values for the saturation characteristics and  $\sigma ^{(l/v)}$ have been compared with their empiric counterparts for the whole temperature range where liquid/vapour interfaces exist, i.e. between the triple and critical points. The results are shown in figure 10: evidently, the theoretical predictions are reasonably accurate.
$\sigma ^{(l/v)}$ have been compared with their empiric counterparts for the whole temperature range where liquid/vapour interfaces exist, i.e. between the triple and critical points. The results are shown in figure 10: evidently, the theoretical predictions are reasonably accurate.

Figure 10. Comparison of the results obtained via the DIM for an EV fluid (solid line) with the corresponding empiric data (dashed line): (a) the densities of saturated vapour and liquid, (b) saturated pressure, (c) surface tension of liquid/vapour interface. The empiric data in panels (a) and (b) are from Lindstrom & Mallard (Reference Lindstrom and Mallard1997), and those in panel (c) are from Somayajulu (Reference Somayajulu1988).
8.4. Parameters of water/air interaction
Generally, the parameters of a fluid should be deduced from measurements of its equation of state, surface tension, etc., but these are rarely known for multicomponent fluids. The water/air mixture at normal conditions is an exception in this respect: the density of air is small in this case, and its equation of state is close to that of an ideal gas. In addition, air will be treated as a pure fluid with parameters equal to the weighted averages of those of nitrogen and oxygen (see table 2).
Thus, let the function  $\varTheta$ (the non-ideal part of the entropy of an EV fluid) be independent of the air density
$\varTheta$ (the non-ideal part of the entropy of an EV fluid) be independent of the air density  $\rho _{2}$, so that expressions (2.17) and (2.18) with
$\rho _{2}$, so that expressions (2.17) and (2.18) with  $N=2$ yield
$N=2$ yield
 \begin{equation} p=T\left[ R_{1}\rho_{1}+R_{2}\rho_{2}+( \rho_{1}+\rho_{2}) \frac{\mathrm{d}\varTheta(\rho_{1})}{\mathrm{d}\rho_{1}}\right] -a_{11}\rho _{1}^{2}-2a_{12}\rho_{1}\rho_{2}, \end{equation}
\begin{equation} p=T\left[ R_{1}\rho_{1}+R_{2}\rho_{2}+( \rho_{1}+\rho_{2}) \frac{\mathrm{d}\varTheta(\rho_{1})}{\mathrm{d}\rho_{1}}\right] -a_{11}\rho _{1}^{2}-2a_{12}\rho_{1}\rho_{2}, \end{equation} \begin{align} G_{1}&=T\left[ R_{1}\ln\rho_{1}+\varTheta(\rho_{1})+( \rho_{1}+\rho _{2})\frac{\mathrm{d}\varTheta(\rho_{1})}{\mathrm{d}\rho_{1}}\right]\nonumber\\ &\quad -2(a_{11}\rho_{1}+a_{12}\rho_{2}) +T[ R_{1}+c_{1}( 1-\ln T) ] , \end{align}
\begin{align} G_{1}&=T\left[ R_{1}\ln\rho_{1}+\varTheta(\rho_{1})+( \rho_{1}+\rho _{2})\frac{\mathrm{d}\varTheta(\rho_{1})}{\mathrm{d}\rho_{1}}\right]\nonumber\\ &\quad -2(a_{11}\rho_{1}+a_{12}\rho_{2}) +T[ R_{1}+c_{1}( 1-\ln T) ] , \end{align} \begin{equation} G_{2}=T[ R_{2}\ln\rho_{2}+\varTheta(\rho_{1})] -2a_{12}\rho _{1}+T[ R_{2}+c_{2}( 1-\ln T) ] . \end{equation}
\begin{equation} G_{2}=T[ R_{2}\ln\rho_{2}+\varTheta(\rho_{1})] -2a_{12}\rho _{1}+T[ R_{2}+c_{2}( 1-\ln T) ] . \end{equation}
Here, the function  $\varTheta (\rho _{1})$ is the one given by (8.2), with
$\varTheta (\rho _{1})$ is the one given by (8.2), with  $\rho$ changed to
$\rho$ changed to  $\rho _{1}$, and with its coefficients corresponding to water.
$\rho _{1}$, and with its coefficients corresponding to water.
Before using expressions (8.3)–(8.5), one needs to fix the non-diagonal term  $a_{12}$ of the matrix
$a_{12}$ of the matrix  $a_{ij}$, responsible for the interaction of water and air molecules. It can be deduced from
$a_{ij}$, responsible for the interaction of water and air molecules. It can be deduced from  $\rho _{2} ^{(l)}$ (the amount of air dissolved in water): one should choose such
$\rho _{2} ^{(l)}$ (the amount of air dissolved in water): one should choose such  $a_{12}$ that the Maxwell construction based on (8.3)–(8.5) predicts the correct value of
$a_{12}$ that the Maxwell construction based on (8.3)–(8.5) predicts the correct value of  $\rho _{2}^{(l)}$. Since
$\rho _{2}^{(l)}$. Since  $a_{12}$ is supposed to not depend on
$a_{12}$ is supposed to not depend on  $T$, such a calculation should be done for a single temperature and the atmospheric pressure. For
$T$, such a calculation should be done for a single temperature and the atmospheric pressure. For  $T=25\,^{\circ }\mathrm {C}$ and
$T=25\,^{\circ }\mathrm {C}$ and  $p=1\ \mathrm {atm}$, for example,
$p=1\ \mathrm {atm}$, for example,  $\rho _{2}^{(l)}=0.0227\ \mathrm {kg\ m}^{-3}$ (The Engineering Toolbox 2004). It can then be deduced that the Maxwell construction based on (8.3)–(8.5) yields the correct value of
$\rho _{2}^{(l)}=0.0227\ \mathrm {kg\ m}^{-3}$ (The Engineering Toolbox 2004). It can then be deduced that the Maxwell construction based on (8.3)–(8.5) yields the correct value of  $\rho _{2}^{(l)}$ if
$\rho _{2}^{(l)}$ if
 \begin{equation} a_{12}=208\ \mathrm{m}^{5}\ \mathrm{s}^{{-}2}\ \mathrm{kg}^{{-}1}, \end{equation}
\begin{equation} a_{12}=208\ \mathrm{m}^{5}\ \mathrm{s}^{{-}2}\ \mathrm{kg}^{{-}1}, \end{equation}
i.e. approximately equal to  $a_{22}$ and ten times smaller than
$a_{22}$ and ten times smaller than  $a_{11}$.
$a_{11}$.
The accuracy of expressions (8.3)–(8.5) can be illustrated by the corresponding value of the boiling point ( $T\approx 109\,^{\circ }\mathrm {C}$, as opposed to the exact value of
$T\approx 109\,^{\circ }\mathrm {C}$, as opposed to the exact value of  $T=100\,^{\circ }\mathrm {C}$). Furthermore, at ‘room temperature’ (say,
$T=100\,^{\circ }\mathrm {C}$). Furthermore, at ‘room temperature’ (say,  $T=25\,^{\circ }\mathrm {C}$), the error should be even smaller, because the room temperature is close to the triple point of water (
$T=25\,^{\circ }\mathrm {C}$), the error should be even smaller, because the room temperature is close to the triple point of water ( $T\approx 0\,^{\circ }\mathrm {C}$) where (8.3)–(8.5) were calibrated.
$T\approx 0\,^{\circ }\mathrm {C}$) where (8.3)–(8.5) were calibrated.
It still remains to determine the non-diagonal term  $K_{12}$ of the Korteweg matrix
$K_{12}$ of the Korteweg matrix  $K_{ij}$.
$K_{ij}$.
Since  $K_{ij}$ is supposed to be positive definite,
$K_{ij}$ is supposed to be positive definite,  $K_{12}$ should satisfy
$K_{12}$ should satisfy
 \begin{equation} -1<\frac{K_{12}}{( K_{11}K_{22})^{1/2}}<1. \end{equation}
\begin{equation} -1<\frac{K_{12}}{( K_{11}K_{22})^{1/2}}<1. \end{equation}
One would think that  $K_{12}$ could be determined by comparing the surface tension of liquid-water/air interface to that of liquid-water/vapour-water interface. It turns out, however, that the difference between these is so small that the existing experimental techniques cannot detect it (see Vargaftik, Volkov & Voljak (Reference Vargaftik, Volkov and Voljak1983) and references therein). This can be due to smallness of
$K_{12}$ could be determined by comparing the surface tension of liquid-water/air interface to that of liquid-water/vapour-water interface. It turns out, however, that the difference between these is so small that the existing experimental techniques cannot detect it (see Vargaftik, Volkov & Voljak (Reference Vargaftik, Volkov and Voljak1983) and references therein). This can be due to smallness of  $K_{12}$, but more likely, because the density of air is small. Either way, the determination of
$K_{12}$, but more likely, because the density of air is small. Either way, the determination of  $K_{12}$ via measurements of surface tension is impossible.
$K_{12}$ via measurements of surface tension is impossible.
To at least confirm that air cannot affect significantly the surface tension of water, the boundary-value problem (6.1)–(6.4) was solved for the parameters of water and air at  $T=25\,^{\circ }\mathrm {C}$, and
$T=25\,^{\circ }\mathrm {C}$, and  $K_{12}$ varying through range (8.7). It turned out that the resulting variation of the surface tension is only
$K_{12}$ varying through range (8.7). It turned out that the resulting variation of the surface tension is only  $7\,\%$.
$7\,\%$.
Even though the dependence of the surface tension (which is a global characteristic) on  $K_{12}$ is weak, this parameter does influence the local structure of the interface.
$K_{12}$ is weak, this parameter does influence the local structure of the interface.
Figure 11 depicts the density profiles  $\rho _{1}(z)$ and
$\rho _{1}(z)$ and  $\rho _{2}(z)$ for the water/air interface at room temperature (i.e. the above-mentioned solution of the boundary-value problem (6.1)–(6.4)). One can see that, as
$\rho _{2}(z)$ for the water/air interface at room temperature (i.e. the above-mentioned solution of the boundary-value problem (6.1)–(6.4)). One can see that, as  $K_{12}$ approaches the right endpoint of range (8.7), the profile of the water density is getting steeper, and a layer of high air density is developing inside the interface. For curve 5 of figure 11(b), which is half-way through range (8.7), the maximum of
$K_{12}$ approaches the right endpoint of range (8.7), the profile of the water density is getting steeper, and a layer of high air density is developing inside the interface. For curve 5 of figure 11(b), which is half-way through range (8.7), the maximum of  $\rho _{2}(z)$ exceeds the atmospheric air density by a factor of approximately
$\rho _{2}(z)$ exceeds the atmospheric air density by a factor of approximately  $20$. Since the amount of air drawn into the interface grows with
$20$. Since the amount of air drawn into the interface grows with  $K_{12}$, one concludes that the air is ‘pulled in’ by the van der Waals attraction exerted by the bulk of the liquid.
$K_{12}$, one concludes that the air is ‘pulled in’ by the van der Waals attraction exerted by the bulk of the liquid.

Figure 11. The spatial structure of water/air interfaces for  $T=25\,^{\circ }\mathrm {C}$ and
$T=25\,^{\circ }\mathrm {C}$ and  $K_{12}=( K_{11}K_{22})^{1/2}\times n/10$, where
$K_{12}=( K_{11}K_{22})^{1/2}\times n/10$, where  $n$ is the curve number: (a) density of water; (b) density of air. The horizontal scales of panels (a) and (b) differ by a factor of 5; the latter and the horizontal scale of the shaded inset in panel (b) differ by a factor of 1500. The inset shows that the concentration of the gas dissolved in liquid is non-zero.
$n$ is the curve number: (a) density of water; (b) density of air. The horizontal scales of panels (a) and (b) differ by a factor of 5; the latter and the horizontal scale of the shaded inset in panel (b) differ by a factor of 1500. The inset shows that the concentration of the gas dissolved in liquid is non-zero.
This suggests a possibility of deducing  $K_{12}$ from empiric data on evaporation of drops (which depends on both global and local characteristics of the interface Benilov Reference Benilov2022b). Such an approach, however, requires an extension of the evaporation model of Benilov (Reference Benilov2022b) to multicomponent fluids, which is currently in progress.
$K_{12}$ from empiric data on evaporation of drops (which depends on both global and local characteristics of the interface Benilov Reference Benilov2022b). Such an approach, however, requires an extension of the evaporation model of Benilov (Reference Benilov2022b) to multicomponent fluids, which is currently in progress.
Observe that the interfaces depicted in figure 11 are such that  $\rho _{2}(z)$ is not monotonic – hence, the sufficient stability criterion from § 6.1 and Appendix C.1 does not apply. This does not, however, mean that this interface is unstable. It is, in fact, difficult to imagine that a microscopic non-monotonicity of the density of air dissolved in water could destabilise the whole interface, but this issue is still of interest theoretically and, thus, deserves further investigation.
$\rho _{2}(z)$ is not monotonic – hence, the sufficient stability criterion from § 6.1 and Appendix C.1 does not apply. This does not, however, mean that this interface is unstable. It is, in fact, difficult to imagine that a microscopic non-monotonicity of the density of air dissolved in water could destabilise the whole interface, but this issue is still of interest theoretically and, thus, deserves further investigation.
8.5. The viscosity and thermal conductivity of water/air mixture
There is a large body of work on the viscosity of multicomponent fluids, with even the simplest theoretical models, e.g. that of Enskog–Chapman for dilute gases (e.g. Ferziger & Kaper Reference Ferziger and Kaper1972, yielding a fairly complicated dependence of  $\mu _{s}$ and
$\mu _{s}$ and  $\mu _{b}$ on
$\mu _{b}$ on  $\rho _{i}$. Phenomenological results, on the other hand, tend to include many ad hoc parameters specific to the fluid under consideration (e.g. Davidson (Reference Davidson1993) and the references therein). Overall, the simplest option seems to be the expression for the shear viscosity proposed on a phenomenological basis by Hind, McLaughlin & Ubbelohde (Reference Hind, McLaughlin and Ubbelohde1960) and justified, under certain approximations, via statistical mechanics by Bearman & Jones (Reference Bearman and Jones1960),
$\rho _{i}$. Phenomenological results, on the other hand, tend to include many ad hoc parameters specific to the fluid under consideration (e.g. Davidson (Reference Davidson1993) and the references therein). Overall, the simplest option seems to be the expression for the shear viscosity proposed on a phenomenological basis by Hind, McLaughlin & Ubbelohde (Reference Hind, McLaughlin and Ubbelohde1960) and justified, under certain approximations, via statistical mechanics by Bearman & Jones (Reference Bearman and Jones1960),
 \begin{equation} \mu_{s}=\mu_{s,1}f_{1}^{2}+( \mu_{s,1}+\mu_{s,2}) f_{1}f_{2} +\mu_{s,2}f_{2}^{2}, \end{equation}
\begin{equation} \mu_{s}=\mu_{s,1}f_{1}^{2}+( \mu_{s,1}+\mu_{s,2}) f_{1}f_{2} +\mu_{s,2}f_{2}^{2}, \end{equation}
where  $\mu _{s,i}$ is the shear viscosity of the
$\mu _{s,i}$ is the shear viscosity of the  $i$th species and
$i$th species and
 \begin{equation} f_{i}=\frac{\rho_{i}/m_{i}}{\rho_{1}/m_{1}+\rho_{2}/m_{2}} \end{equation}
\begin{equation} f_{i}=\frac{\rho_{i}/m_{i}}{\rho_{1}/m_{1}+\rho_{2}/m_{2}} \end{equation}
is its mole fraction. Expression (8.8) does not include any fluid-specific parameters, but is capable of predicting the shear viscosity with a reasonable accuracy. If, for example, air is treated as a mixture of  $\mathrm {N}_{2}$ and
$\mathrm {N}_{2}$ and  $\mathrm {O}_{2}$, the error of (8.8) for the range
$\mathrm {O}_{2}$, the error of (8.8) for the range  $T=0-100\,^{\circ }\mathrm {C}$ is less than 1 %. A similar ‘mixture rule’ can be assumed for the bulk viscosity and thermal conductivity,
$T=0-100\,^{\circ }\mathrm {C}$ is less than 1 %. A similar ‘mixture rule’ can be assumed for the bulk viscosity and thermal conductivity,
 $$\begin{gather} \mu_{b}=\mu_{b,1}f_{1}^{2}+( \mu_{b,1}+\mu_{b,2}) f_{1}f_{2} +\mu_{b,2}f_{2}^{2}, \end{gather}$$
$$\begin{gather} \mu_{b}=\mu_{b,1}f_{1}^{2}+( \mu_{b,1}+\mu_{b,2}) f_{1}f_{2} +\mu_{b,2}f_{2}^{2}, \end{gather}$$ $$\begin{gather}\kappa=\kappa_{1}f_{1}^{2}+( \kappa_{1}+\kappa_{2}) f_{1} f_{2}+\kappa_{2}f_{2}^{2}. \end{gather}$$
$$\begin{gather}\kappa=\kappa_{1}f_{1}^{2}+( \kappa_{1}+\kappa_{2}) f_{1} f_{2}+\kappa_{2}f_{2}^{2}. \end{gather}$$Generally, various mechanical properties of a multicomponent fluid are often described by the same mixture rule, in which case it is referred to as ‘generalized’.
It still remains to fix the dependence of  $\mu _{s,i}$,
$\mu _{s,i}$,  $\mu _{b,i}$ and
$\mu _{b,i}$ and  $\kappa _{i}$ on
$\kappa _{i}$ on  $( \rho _{1},\rho _{2},T)$. In application to air, which can be treated as a dilute gas, one can assume these parameter depend only on
$( \rho _{1},\rho _{2},T)$. In application to air, which can be treated as a dilute gas, one can assume these parameter depend only on  $T$ (as predicted by the kinetic theory of dilute gases), i.e.
$T$ (as predicted by the kinetic theory of dilute gases), i.e.
 \begin{equation} \mu_{s,2}(\rho_{2},T)=\mu_{s,2}(0,T),\quad \mu_{b,2}(\rho_{2},T)=\mu _{b,2}(0,T),\quad \kappa_{2}(\rho_{2},T)=\kappa_{2}(0,T), \end{equation}
\begin{equation} \mu_{s,2}(\rho_{2},T)=\mu_{s,2}(0,T),\quad \mu_{b,2}(\rho_{2},T)=\mu _{b,2}(0,T),\quad \kappa_{2}(\rho_{2},T)=\kappa_{2}(0,T), \end{equation}
where  $\mu _{s,2}(0,T)$,
$\mu _{s,2}(0,T)$,  $\mu _{b,2}(0,T)$ and
$\mu _{b,2}(0,T)$ and  $\kappa _{2}(0,T)$ are the small-density limiting values of the corresponding parameters.
$\kappa _{2}(0,T)$ are the small-density limiting values of the corresponding parameters.
For water, whose liquid phase cannot be treated as a dilute gas, such an approximation is inapplicable. Aiming again for simplicity, one can approximate both viscosities by a parabola passing through two reference points, the zero-density limit and the saturated liquid state,
 $$\begin{gather} \mu_{s,1}(\rho_{1},T)=\mu_{s,1}(0,T)+[ \mu_{s,1}(\rho_{1}^{(l)} ,T)-\mu_{s,1}(0,T)] \left[ \frac{\rho_{1}}{\rho_{1}^{(l)}(T)}\right] ^{2}, \end{gather}$$
$$\begin{gather} \mu_{s,1}(\rho_{1},T)=\mu_{s,1}(0,T)+[ \mu_{s,1}(\rho_{1}^{(l)} ,T)-\mu_{s,1}(0,T)] \left[ \frac{\rho_{1}}{\rho_{1}^{(l)}(T)}\right] ^{2}, \end{gather}$$ $$\begin{gather}\mu_{b,1}(\rho_{1},T)=\mu_{b,1}(0,T)+[ \mu_{b,1}(\rho_{1}^{(l)} ,T)-\mu_{b,1}(0,T)] \left[ \frac{\rho_{1}}{\rho_{1}^{(l)}(T)}\right] ^{2}. \end{gather}$$
$$\begin{gather}\mu_{b,1}(\rho_{1},T)=\mu_{b,1}(0,T)+[ \mu_{b,1}(\rho_{1}^{(l)} ,T)-\mu_{b,1}(0,T)] \left[ \frac{\rho_{1}}{\rho_{1}^{(l)}(T)}\right] ^{2}. \end{gather}$$A similar approximation, but by a linear function, will be adopted for the thermal conductivity,
 \begin{equation} \kappa_{1}(\rho_{1},T)=\kappa_{1}(0,T)+[ \kappa_{1}(\rho_{1} ^{(l)},T)-\kappa_{1}(0,T)] \frac{\rho_{1}}{\rho_{1}^{(l)}(T)}.\end{equation}
\begin{equation} \kappa_{1}(\rho_{1},T)=\kappa_{1}(0,T)+[ \kappa_{1}(\rho_{1} ^{(l)},T)-\kappa_{1}(0,T)] \frac{\rho_{1}}{\rho_{1}^{(l)}(T)}.\end{equation}
The accuracy of the above approximations for  $\mu _{s,1}$ and
$\mu _{s,1}$ and  $\kappa _{1}$ are illustrated in figure 12.
$\kappa _{1}$ are illustrated in figure 12.

Figure 12. The parameters of pure water at  $T=327\,^{\circ }\mathrm {C}$: (a) shear viscosity, (b) thermal conductivity. The empiric data (Lindstrom & Mallard Reference Lindstrom and Mallard1997) are shown as a solid line and approximations (8.13) and (8.15) as a dashed line. The dots mark the reference points. The gap in the empiric data reflects the impossibility (difficulty) of measurements in an unstable (metastable) fluid.
$T=327\,^{\circ }\mathrm {C}$: (a) shear viscosity, (b) thermal conductivity. The empiric data (Lindstrom & Mallard Reference Lindstrom and Mallard1997) are shown as a solid line and approximations (8.13) and (8.15) as a dashed line. The dots mark the reference points. The gap in the empiric data reflects the impossibility (difficulty) of measurements in an unstable (metastable) fluid.
To use (8.8)–(8.15), one needs to know how the viscosities and thermal conductivities of water and air depend on  $T$. For
$T$. For  $\mu _{s}$ and
$\mu _{s}$ and  $\kappa$, such data are widely available (e.g. Lindstrom & Mallard Reference Lindstrom and Mallard1997; White Reference White2005). Measurements of
$\kappa$, such data are widely available (e.g. Lindstrom & Mallard Reference Lindstrom and Mallard1997; White Reference White2005). Measurements of  $\mu _{b}$, on the other hand, are scarce, but can still be found in Holmes, Parker & Povey (Reference Holmes, Parker and Povey2011), Cramer (Reference Cramer2012) and Shang et al. (Reference Shang, Wu, Wang, Yang, Ye, Hu, Tao and He2019) for liquid water, vapour water and air, respectively.
$\mu _{b}$, on the other hand, are scarce, but can still be found in Holmes, Parker & Povey (Reference Holmes, Parker and Povey2011), Cramer (Reference Cramer2012) and Shang et al. (Reference Shang, Wu, Wang, Yang, Ye, Hu, Tao and He2019) for liquid water, vapour water and air, respectively.
The empiric formulae proposed in these papers will not be discussed in detail. The characteristic values they yield for normal conditions are listed in table 4.
Table 4. The empiric viscosity and thermal conductivity of liquid water, water vapour and air at  $25\,^{\circ }\mathrm {C}$, and the corresponding references.
$25\,^{\circ }\mathrm {C}$, and the corresponding references.

8.6. The transport coefficients
Recalling restrictions (2.32a,b), one can deduce that the extended transport matrix for a two-component fluid is
 \begin{equation} D_{ij}^{(ext)}= \begin{bmatrix} D_{11} & -D_{11} & \zeta_{1}\\ -D_{11} & D_{11} & -\zeta_{1}\\ \zeta_{1} & -\zeta_{1} & \kappa \end{bmatrix}. \end{equation}
\begin{equation} D_{ij}^{(ext)}= \begin{bmatrix} D_{11} & -D_{11} & \zeta_{1}\\ -D_{11} & D_{11} & -\zeta_{1}\\ \zeta_{1} & -\zeta_{1} & \kappa \end{bmatrix}. \end{equation}
Evidently, it involves only three independent coefficients, one of which (the thermal conductivity  $\kappa$) has already been discussed in § 8.5. The other two,
$\kappa$) has already been discussed in § 8.5. The other two,  $D_{11}$ and
$D_{11}$ and  $\zeta _{1}$, are discussed below.
$\zeta _{1}$, are discussed below.
It can be safely assumed that diffusion is of importance only where the water density is comparable to that of air. In the region where the former is high, the latter is miniscule, and so is its influence on the global dynamics. This effectively means that the diffusivities can be represented using the Enskog–Chapman method; even though it does not apply to liquid water, the error due to using it anyway is negligible.
According to the leading-order Enskog–Chapman formula (e.g. Ferziger & Kaper Reference Ferziger and Kaper1972),
 \begin{equation} D_{11}=\frac{\rho_{1}\rho_{2}( \rho_{1}m_{2}+\rho_{2}m_{1}) }{( \rho_{1}+\rho_{2})^{2}k_{B}T}\mathfrak{D}(T), \end{equation}
\begin{equation} D_{11}=\frac{\rho_{1}\rho_{2}( \rho_{1}m_{2}+\rho_{2}m_{1}) }{( \rho_{1}+\rho_{2})^{2}k_{B}T}\mathfrak{D}(T), \end{equation}
where  $\mathfrak {D}(T)$ does not depend on
$\mathfrak {D}(T)$ does not depend on  $\rho _{1}$ and
$\rho _{1}$ and  $\rho _{2}$. Its dependence on
$\rho _{2}$. Its dependence on  $T$ can be found in The Engineering Toolbox (2018), from which the following characteristic value can be deduced,
$T$ can be found in The Engineering Toolbox (2018), from which the following characteristic value can be deduced,
 \begin{equation} \mathfrak{D}(25\,^{{\circ}}\mathrm{C})=2.49\times10^{{-}5}\ \mathrm{m}^{2} \ \mathrm{s}^{{-}1}. \end{equation}
\begin{equation} \mathfrak{D}(25\,^{{\circ}}\mathrm{C})=2.49\times10^{{-}5}\ \mathrm{m}^{2} \ \mathrm{s}^{{-}1}. \end{equation}The Enskog–Chapman expression for the thermodiffusivity is fairly bulky; as a result, one often uses the simpler formula of de Groot & Mazur (Reference de Groot and Mazur1962),
 \begin{equation} \zeta_{1}=( \rho_{1}+\rho_{2}) \frac{( \rho_{1} /m_{1}) ( \rho_{2}/m_{2}) }{( \rho_{1}/m_{1} +\rho_{2}/m_{2})^{2}}\mathfrak{U}(T). \end{equation}
\begin{equation} \zeta_{1}=( \rho_{1}+\rho_{2}) \frac{( \rho_{1} /m_{1}) ( \rho_{2}/m_{2}) }{( \rho_{1}/m_{1} +\rho_{2}/m_{2})^{2}}\mathfrak{U}(T). \end{equation}
Unfortunately, no data on  $\mathfrak {U}(T)$ for water/air mixture are available in the literature; the author of this paper was able to find only an estimate for a single temperature value (Lidon, Perrot & Stroock Reference Lidon, Perrot and Stroock2021),
$\mathfrak {U}(T)$ for water/air mixture are available in the literature; the author of this paper was able to find only an estimate for a single temperature value (Lidon, Perrot & Stroock Reference Lidon, Perrot and Stroock2021),
 \begin{equation} \mathfrak{U}(21\,^{{\circ}}\mathrm{C})={-}4.98\times10^{{-}6}\ \mathrm{m}^{2} \mathrm{s}^{{-}1}. \end{equation}
\begin{equation} \mathfrak{U}(21\,^{{\circ}}\mathrm{C})={-}4.98\times10^{{-}6}\ \mathrm{m}^{2} \mathrm{s}^{{-}1}. \end{equation}
It is shown later, however, that thermodiffusion in water/air interfaces is negligible, so the lack of data on  $\mathfrak {U}(T)$ is unimportant.
$\mathfrak {U}(T)$ is unimportant.
In the next subsection characteristic values of the coefficients  $D_{11}$ and
$D_{11}$ and  $\zeta _{1}$ will be needed. These will be estimated for the particular case
$\zeta _{1}$ will be needed. These will be estimated for the particular case  $\rho _{1}=\rho _{2}=1.17\ \mathrm {kg\ m}^{-3}$, i.e. when the water density matches that of air at normal conditions. Substituting these values and the molecular masses of water and air from table 2 into (8.17)–(8.20), one obtains
$\rho _{1}=\rho _{2}=1.17\ \mathrm {kg\ m}^{-3}$, i.e. when the water density matches that of air at normal conditions. Substituting these values and the molecular masses of water and air from table 2 into (8.17)–(8.20), one obtains
 \begin{equation} D_{11}\approx1.37\times10^{{-}10}\ \mathrm{m}^{3}\ \mathrm{s\ kg},\quad \zeta _{1}\approx{-}2.76\times10^{{-}6}\ \mathrm{m}^{{-}1}\ \mathrm{s}^{{-}1}\ \mathrm{kg}. \end{equation}
\begin{equation} D_{11}\approx1.37\times10^{{-}10}\ \mathrm{m}^{3}\ \mathrm{s\ kg},\quad \zeta _{1}\approx{-}2.76\times10^{{-}6}\ \mathrm{m}^{{-}1}\ \mathrm{s}^{{-}1}\ \mathrm{kg}. \end{equation}
Even though these values apply to different temperatures ( $T=25\,^{\circ }\mathrm {C}$ and
$T=25\,^{\circ }\mathrm {C}$ and  $T=21\,^{\circ }\mathrm {C}$), they will be used simultaneously in the same qualitative estimate (under the implied assumption that the four-degree difference does not matter).
$T=21\,^{\circ }\mathrm {C}$), they will be used simultaneously in the same qualitative estimate (under the implied assumption that the four-degree difference does not matter).
8.7. The non-dimensional parameters
To understand which effects are important for water/air interfaces under normal conditions, one should estimate the non-dimensional parameters (4.13a–e). They will be calculated using the characteristics of water from tables 2–4 and estimates (8.21a,b). The characteristic pressure scale will be assumed to be  $P=a\varrho ^{2}$, where
$P=a\varrho ^{2}$, where  $\varrho =997\ \mathrm {kg\ m}^{-3}$ is the density of liquid water at
$\varrho =997\ \mathrm {kg\ m}^{-3}$ is the density of liquid water at  $25\,^{\circ }\mathrm {C}$ and
$25\,^{\circ }\mathrm {C}$ and  $a$ equals the van der Waals parameter of water from table 2. The following expression for the viscosity scale will be used:
$a$ equals the van der Waals parameter of water from table 2. The following expression for the viscosity scale will be used:
 \begin{equation} \mu=\tfrac{4}{3}\mu_{s}+\mu_{b} \end{equation}
\begin{equation} \mu=\tfrac{4}{3}\mu_{s}+\mu_{b} \end{equation}
that arises naturally in problems involving liquid films (Benilov Reference Benilov2020c, Reference Benilov2022a) and drops (Benilov Reference Benilov2022b). In the estimates for this paper, the viscosities  $\mu _{s}$ and
$\mu _{s}$ and  $\mu _{b}$ are those for water at
$\mu _{b}$ are those for water at  $25\,^{\circ }\mathrm {C}$.
$25\,^{\circ }\mathrm {C}$.
The following values of parameters (4.13a–e) have been obtained:
 \begin{equation} \alpha\approx0.00139,\quad \tau\approx0.0653,\quad \beta\approx0.0593,\quad \nu\approx0.0321,\quad \delta\approx0.0176. \end{equation}
\begin{equation} \alpha\approx0.00139,\quad \tau\approx0.0653,\quad \beta\approx0.0593,\quad \nu\approx0.0321,\quad \delta\approx0.0176. \end{equation}
The smallness of the microscopic Reynolds number  $\alpha$ suggests that inertia plays little role in the dynamics of water/air interfaces (hence, one can use the Stokes approximation). The smallness of the non-dimensional temperature
$\alpha$ suggests that inertia plays little role in the dynamics of water/air interfaces (hence, one can use the Stokes approximation). The smallness of the non-dimensional temperature  $\tau$ does not have physical implications, but enables one to use asymptotic tools when calculating, say, the profile of the equilibrium interface (Benilov Reference Benilov2020c). The smallness of the Nusselt number
$\tau$ does not have physical implications, but enables one to use asymptotic tools when calculating, say, the profile of the equilibrium interface (Benilov Reference Benilov2020c). The smallness of the Nusselt number  $\nu$ implies that the Soret and Dufour effects are negligible, and so the lack of empiric data on them for water and air is unimportant. The smallness of
$\nu$ implies that the Soret and Dufour effects are negligible, and so the lack of empiric data on them for water and air is unimportant. The smallness of  $\beta$, in turn, implies that the flow is almost isothermal, whereas the small value of
$\beta$, in turn, implies that the flow is almost isothermal, whereas the small value of  $\delta$ suggests that diffusion dominates advection.
$\delta$ suggests that diffusion dominates advection.
Similar estimates have also been carried out for the parameters of air at  $p=1\ \mathrm {atm}$ and
$p=1\ \mathrm {atm}$ and  $T=25\,^{\circ }\mathrm {C}$. It turned out that
$T=25\,^{\circ }\mathrm {C}$. It turned out that  $\alpha$,
$\alpha$,  $\beta$,
$\beta$,  $\nu$ and
$\nu$ and  $\delta$ are even smaller than those for water, but the non-dimensional temperature
$\delta$ are even smaller than those for water, but the non-dimensional temperature  $\tau$ for air is order one. The latter is not surprising, as the room temperature is much higher than the freezing temperatures of nitrogen and oxygen, but is close to that for water.
$\tau$ for air is order one. The latter is not surprising, as the room temperature is much higher than the freezing temperatures of nitrogen and oxygen, but is close to that for water.
All these observations should be helpful when using the DIM to examine asymptotically the problems of evaporation of drops and moving contact lines.
One should keep in mind, however, that parameters (4.13a–e) are sensitive to the choice of the viscosity scale  $\mu$. Benilov (Reference Benilov2020a), for example, chose
$\mu$. Benilov (Reference Benilov2020a), for example, chose  $\mu$ equal to the half-sum of the shear viscosities of liquid water and vapour water; as a result,
$\mu$ equal to the half-sum of the shear viscosities of liquid water and vapour water; as a result,  $\alpha$ and
$\alpha$ and  $\beta$ were noticeably larger than those calculated above. More generally, one should choose the viscosity scale, as well as the other characteristic scales, to essentially reflect the physics of the setting at hand.
$\beta$ were noticeably larger than those calculated above. More generally, one should choose the viscosity scale, as well as the other characteristic scales, to essentially reflect the physics of the setting at hand.
Note also that estimates (8.23a–e) apply to water under normal conditions, but in industrial applications the governing non-dimensional parameters can be very different. In steam turbines, for example, the temperature can be as high as  $540\,^{\circ }\mathrm {C}$, and the pressure can exceed
$540\,^{\circ }\mathrm {C}$, and the pressure can exceed  $230\ \mathrm {atm}$ (Vasserman & Shutenko Reference Vasserman and Shutenko2017).
$230\ \mathrm {atm}$ (Vasserman & Shutenko Reference Vasserman and Shutenko2017).
9. Concluding remarks
The results obtained in this paper can be briefly summarised as follows.
(a) The entropy principle and conservation laws of the multicomponent DIM have been used to examine the stability of liquid/vapour interfaces. Several physically important results are reported as follows.
(i) Flat liquid/vapour interfaces in an unbounded fluid are stable if the density profiles of all species are monotonic.
(ii) There can exist up to two different solutions describing a solid/vapour interface, one monotonic and one non-monotonic. The former is stable and the latter is likely to be unstable (definitely unstable for pure fluids). Similar conclusions apply to solid/liquid interfaces.
(iii) For certain values of the near-substrate density (which is an external parameter in DIM, linked to the contact angle of the substrate), no steady solution exists describing a solid/vapour interface. Physically, such substrates are too hydrophilic and, thus, trigger off spontaneous condensation of adjacent vapour. Similarly, some substrates are too hydrophobic and trigger off spontaneous evaporation of adjacent liquid.
(iv) A liquid layer between a flat substrate and a semi-space filled with vapour can exist as a steady state only if the vapour is oversaturated. All such layers are unstable, however, depending on the perturbation, they either fully evaporate or grow indefinitely due to condensation of vapour on its upper boundary.
If the vapour above the layer is saturated or undersaturated, the liquid evaporates and no steady solution exists.
Similar conclusions apply to 1-D vapour layers between a flat substrate and a semi-space filled with liquid.
(v) Similar conclusions to those in the previous point apply to a liquid layer with vapour both above and below it, and a vapour layer with liquid below and above it. Such solutions can be viewed as 1-D drops and bubbles, respectively.
(b) The multicomponent DIM has been fully parameterised for water/air interfaces under normal conditions. It is shown that the Soret and Dufour effects are weak in this case, which agrees with the results of Jiang, Studer & Podvin (Reference Jiang, Studer and Podvin2020). It is also argued that the interfacial flow in this case is isothermal.
These are of importance when studying evaporation of water drops or the dynamics of their contact lines.
Declaration of interests
The author reports no conflict of interests.
Appendix A. The Gibbs relation
We introduce the volumetric densities of energy  $\mathcal {U}(\rho _{1}\ldots \rho _{N},T)$, entropy
$\mathcal {U}(\rho _{1}\ldots \rho _{N},T)$, entropy  $\mathcal {S}(\rho _{1}\ldots \rho _{N},T)$ and partial chemical potentials
$\mathcal {S}(\rho _{1}\ldots \rho _{N},T)$ and partial chemical potentials  $\mathcal {G}_{i}(\rho _{1}\ldots \rho _{N},T)$, related to the corresponding specific quantities by
$\mathcal {G}_{i}(\rho _{1}\ldots \rho _{N},T)$, related to the corresponding specific quantities by
 \begin{equation} \mathcal{U}=e\rho,\quad \mathcal{S}=s\rho,\quad \mathcal{G}_{i}=G_{i}. \end{equation}
\begin{equation} \mathcal{U}=e\rho,\quad \mathcal{S}=s\rho,\quad \mathcal{G}_{i}=G_{i}. \end{equation}
Now, definition (2.3) of  $G_{i}$ can be rewritten in the form
$G_{i}$ can be rewritten in the form
 \begin{equation} \mathcal{G}_{i}=\frac{\partial\mathcal{U}}{\partial\rho_{i}}-T\frac {\partial\mathcal{S}}{\partial\rho_{i}}. \end{equation}
\begin{equation} \mathcal{G}_{i}=\frac{\partial\mathcal{U}}{\partial\rho_{i}}-T\frac {\partial\mathcal{S}}{\partial\rho_{i}}. \end{equation}The standard volumetric version of the Gibbs relation (e.g. (2.4) of Giovangigli & Matuszewski Reference Giovangigli and Matuszewski2013) is
 \begin{equation} \mathrm{d}\mathcal{U}=T\,\mathrm{d}\mathcal{S}+\sum_{i}\mathcal{G} _{i}\,\mathrm{d}\rho_{i}. \end{equation}
\begin{equation} \mathrm{d}\mathcal{U}=T\,\mathrm{d}\mathcal{S}+\sum_{i}\mathcal{G} _{i}\,\mathrm{d}\rho_{i}. \end{equation}
Substituting (A1a–c) and (A2) into this equality and rewriting it in terms of  $\mathrm {d}T$ and
$\mathrm {d}T$ and  $\mathrm {d}\rho _{i}$ (instead of
$\mathrm {d}\rho _{i}$ (instead of  $\mathrm {d} \mathcal {S}$ and
$\mathrm {d} \mathcal {S}$ and  $\mathrm {d}\rho _{i}$), one obtains (2.4) as required.
$\mathrm {d}\rho _{i}$), one obtains (2.4) as required.
Appendix B. The general boundary condition at a solid wall
Consider the following generalization of the boundary condition (2.47):
 \begin{equation} \sum_{j}C_{ij}( \boldsymbol{n}\boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{j}) +\rho _{i}=\rho_{0,i}\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D}. \end{equation}
\begin{equation} \sum_{j}C_{ij}( \boldsymbol{n}\boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{j}) +\rho _{i}=\rho_{0,i}\ \text{at}\ \boldsymbol{r}\in\partial\mathcal{D}. \end{equation}
Here 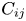 $C_{ij}$ is a symmetric matrix, depending generally on
$C_{ij}$ is a symmetric matrix, depending generally on  $\rho _{1} \ldots \rho _{N}$. Equation (B1) is a multicomponent extension of a boundary condition often used for pure fluids (e.g. Madruga & Thiele Reference Madruga and Thiele2009; Gallo, Magaletti & Casciola Reference Gallo, Magaletti and Casciola2021).
$\rho _{1} \ldots \rho _{N}$. Equation (B1) is a multicomponent extension of a boundary condition often used for pure fluids (e.g. Madruga & Thiele Reference Madruga and Thiele2009; Gallo, Magaletti & Casciola Reference Gallo, Magaletti and Casciola2021).
To understand how the energy conservation law is affected by switching to a different boundary condition, observe that the governing equations (2.19)–(2.24) and the other boundary conditions, (2.44)–(2.46), imply that
 \begin{align} &\frac{\mathrm{d}}{\mathrm{d}t}\int_{\mathcal{D}}\left[ \rho e+\frac{1}{2} \rho\vert \boldsymbol{v}\vert^{2}+\frac{1}{2}\sum_{ij}K_{ij}( \boldsymbol{\nabla} \rho_{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \rho_{j}) \right] \,\mathrm{d}^{3}\boldsymbol{r}\nonumber\\ &\quad +\int_{\partial\mathcal{D}}\sum_{i}(\boldsymbol{n}\boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{i}) \sum_{j}K_{ij}\frac{\partial\rho_{j}}{\partial t} \,\mathrm{d}A=0, \end{align}
\begin{align} &\frac{\mathrm{d}}{\mathrm{d}t}\int_{\mathcal{D}}\left[ \rho e+\frac{1}{2} \rho\vert \boldsymbol{v}\vert^{2}+\frac{1}{2}\sum_{ij}K_{ij}( \boldsymbol{\nabla} \rho_{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \rho_{j}) \right] \,\mathrm{d}^{3}\boldsymbol{r}\nonumber\\ &\quad +\int_{\partial\mathcal{D}}\sum_{i}(\boldsymbol{n}\boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{i}) \sum_{j}K_{ij}\frac{\partial\rho_{j}}{\partial t} \,\mathrm{d}A=0, \end{align}
where  $\mathrm {d}A$ is the elemental aria on
$\mathrm {d}A$ is the elemental aria on  $\partial \mathcal {D}$. The ‘old’ boundary condition (2.47) implies that the second integral in this equality vanishes, making the integral in the first term be a conserved quantity (the energy) in this case.
$\partial \mathcal {D}$. The ‘old’ boundary condition (2.47) implies that the second integral in this equality vanishes, making the integral in the first term be a conserved quantity (the energy) in this case.
Next, assume that the ‘new’ boundary condition (B1) is imposed. Differentiate it with respect to  $t$, change the indices – first
$t$, change the indices – first  $j\rightarrow k$, then
$j\rightarrow k$, then  $i\rightarrow j$ – and use the resulting equality to rearrange (B2) in the form
$i\rightarrow j$ – and use the resulting equality to rearrange (B2) in the form
 \begin{align} &\frac{\mathrm{d}}{\mathrm{d}t}\int_{\mathcal{D}}\left[ \rho e+\frac{1}{2} \rho\vert \boldsymbol{v}\vert^{2}+\frac{1}{2}\sum_{ij}K_{ij}( \boldsymbol{\nabla} \rho_{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \rho_{j}) \right] \mathrm{d}^{3}\boldsymbol{r}\nonumber\\ &\quad -\int_{\partial\mathcal{D}}\sum_{ij}K_{ij}^{\prime}( \boldsymbol{n} \boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{i}) \frac{\partial( \boldsymbol{n} \boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{j})}{\partial t}\,\mathrm{d}A=0, \end{align}
\begin{align} &\frac{\mathrm{d}}{\mathrm{d}t}\int_{\mathcal{D}}\left[ \rho e+\frac{1}{2} \rho\vert \boldsymbol{v}\vert^{2}+\frac{1}{2}\sum_{ij}K_{ij}( \boldsymbol{\nabla} \rho_{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \rho_{j}) \right] \mathrm{d}^{3}\boldsymbol{r}\nonumber\\ &\quad -\int_{\partial\mathcal{D}}\sum_{ij}K_{ij}^{\prime}( \boldsymbol{n} \boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{i}) \frac{\partial( \boldsymbol{n} \boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{j})}{\partial t}\,\mathrm{d}A=0, \end{align}where
 \begin{equation} K_{ij}^{\prime}=\sum_{k}K_{ik}C_{kj}. \end{equation}
\begin{equation} K_{ij}^{\prime}=\sum_{k}K_{ik}C_{kj}. \end{equation}Equation (B3) implies that the following quantity is conserved:
 \begin{align} E&=\int_{\mathcal{D}}\left[ \rho e+\frac{1}{2}\rho\vert \boldsymbol{v} \vert^{2}+\frac{1}{2}\sum_{ij}K_{ij}( \boldsymbol{\nabla} \rho _{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \rho_{j}) \right]\, \mathrm{d}^{3}\boldsymbol{r}\nonumber\\ &\quad -\frac{1}{2}\int_{\partial\mathcal{D}}\sum_{ij}K_{ij}^{\prime}( \boldsymbol{n}\boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{i}) ( \boldsymbol{n} \boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{j}) \,\mathrm{d}A. \end{align}
\begin{align} E&=\int_{\mathcal{D}}\left[ \rho e+\frac{1}{2}\rho\vert \boldsymbol{v} \vert^{2}+\frac{1}{2}\sum_{ij}K_{ij}( \boldsymbol{\nabla} \rho _{i}) \boldsymbol{{\cdot}}( \boldsymbol{\nabla} \rho_{j}) \right]\, \mathrm{d}^{3}\boldsymbol{r}\nonumber\\ &\quad -\frac{1}{2}\int_{\partial\mathcal{D}}\sum_{ij}K_{ij}^{\prime}( \boldsymbol{n}\boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{i}) ( \boldsymbol{n} \boldsymbol{{\cdot}}\boldsymbol{\nabla} \rho_{j}) \,\mathrm{d}A. \end{align}The second term in this expression is the surface energy corresponding to the new boundary condition (B1).
By comparison to the Dirichlet boundary condition (2.47), the mixed condition (B1) includes additional  $N( N-1) /2$ adjustable parameters. Even though the extra parameters may enable the DIM to be more accurate quantitatively, one should think that a physics-based model does not need many adjustable parameters to capture the qualitative nature of the phenomenon it is applied to.
$N( N-1) /2$ adjustable parameters. Even though the extra parameters may enable the DIM to be more accurate quantitatively, one should think that a physics-based model does not need many adjustable parameters to capture the qualitative nature of the phenomenon it is applied to.
Appendix C. Stability of 1-D steady states
In this appendix, the stability of three families of 1-D solutions are examined:
(i) liquid/vapour interfaces,
(ii) 1-D drops and bubbles,
(iii) solid/fluid interfaces.
C.1. Liquid/vapour interfaces
As mentioned before, (3.12) describes all steady states in the problem at hand, and expression (3.15) describes the corresponding second variation of the entropy. To adapt the latter for flat liquid/vapour interfaces, one needs to replace the three-dimensional integral over the domain  $\mathcal {D}$ with a 1-D integral over the domain
$\mathcal {D}$ with a 1-D integral over the domain  $-\infty < z<\infty$,
$-\infty < z<\infty$,
 \begin{equation} \delta^{2}H=\frac{1}{T}\int_{-\infty}^{\infty}\sum_{ij}\left[ -\frac{\partial G_{i}}{\partial\rho_{j}}( \delta\rho_{i}) ( \delta\rho _{j}) -K_{ij}\frac{\mathrm{d}( \delta\rho_{i})}{\mathrm{d}z}\frac{\mathrm{d}( \delta\rho_{j})}{\mathrm{d} z}\right] \,\mathrm{d}z. \end{equation}
\begin{equation} \delta^{2}H=\frac{1}{T}\int_{-\infty}^{\infty}\sum_{ij}\left[ -\frac{\partial G_{i}}{\partial\rho_{j}}( \delta\rho_{i}) ( \delta\rho _{j}) -K_{ij}\frac{\mathrm{d}( \delta\rho_{i})}{\mathrm{d}z}\frac{\mathrm{d}( \delta\rho_{j})}{\mathrm{d} z}\right] \,\mathrm{d}z. \end{equation}This expression can be rewritten in the form
 \begin{equation} \delta^{2}H=\frac{1}{T}\int_{-\infty}^{\infty}\sum_{ij}\delta\rho _{i}{\hat{O}}_{ij}\delta\rho_{j}\,\mathrm{d}z, \end{equation}
\begin{equation} \delta^{2}H=\frac{1}{T}\int_{-\infty}^{\infty}\sum_{ij}\delta\rho _{i}{\hat{O}}_{ij}\delta\rho_{j}\,\mathrm{d}z, \end{equation}where
 \begin{equation} {\hat{O}}_{ij}={-}\frac{\partial G_{i}}{\partial\rho_{j}}+K_{ij} \frac{\mathrm{d}^{2}}{\mathrm{d}z^{2}} \end{equation}
\begin{equation} {\hat{O}}_{ij}={-}\frac{\partial G_{i}}{\partial\rho_{j}}+K_{ij} \frac{\mathrm{d}^{2}}{\mathrm{d}z^{2}} \end{equation}
is a second-order differential operator. Since the matrices  $\partial G_{i}/\partial \rho _{j}$ and
$\partial G_{i}/\partial \rho _{j}$ and  $K_{ij}$ are symmetric (see definition (2.3) of
$K_{ij}$ are symmetric (see definition (2.3) of  $G_{i}$ and § 2.5, respectively),
$G_{i}$ and § 2.5, respectively),  ${\hat {O}} _{ij}$ is self-adjoint – hence, its spectrum is real. Note also that
${\hat {O}} _{ij}$ is self-adjoint – hence, its spectrum is real. Note also that  ${\hat {O}}_{ij}$ depends on the steady state
${\hat {O}}_{ij}$ depends on the steady state  $\rho _{i}(z)$ via the coefficient
$\rho _{i}(z)$ via the coefficient  $\partial G_{i}/\partial \rho _{j}$.
$\partial G_{i}/\partial \rho _{j}$.
It follows from (C2) that the functional  $H$ has a maximum at
$H$ has a maximum at  $\rho _{i}(z)$ if and only if the corresponding operator
$\rho _{i}(z)$ if and only if the corresponding operator  ${\hat {O}} _{ij}$ is negative definite or, equivalently, its spectrum (the set of all discrete and continuous eigenvalues) is negative.
${\hat {O}} _{ij}$ is negative definite or, equivalently, its spectrum (the set of all discrete and continuous eigenvalues) is negative.
Theorem C.1 If the solution  $\rho _{i}(z)$ of the boundary-value problem (6.1)–(6.3) is monotonic for all
$\rho _{i}(z)$ of the boundary-value problem (6.1)–(6.3) is monotonic for all  $i$, the spectrum of the corresponding operator
$i$, the spectrum of the corresponding operator  ${\hat {O}}_{ij}$ is negative.
${\hat {O}}_{ij}$ is negative.
Proof. Let  $\varLambda$ be an eigenvalue of the discrete spectrum (if any) and
$\varLambda$ be an eigenvalue of the discrete spectrum (if any) and  $\psi _{i}$, the corresponding eigenfunction,
$\psi _{i}$, the corresponding eigenfunction,
 $$\begin{gather} \sum_{j}\left( -\frac{\partial G_{i}}{\partial\rho_{j}}\psi_{j}+K_{ij} \frac{\mathrm{d}^{2}\psi_{j}}{\mathrm{d}z^{2}}\right) =\varLambda\psi_{i}, \end{gather}$$
$$\begin{gather} \sum_{j}\left( -\frac{\partial G_{i}}{\partial\rho_{j}}\psi_{j}+K_{ij} \frac{\mathrm{d}^{2}\psi_{j}}{\mathrm{d}z^{2}}\right) =\varLambda\psi_{i}, \end{gather}$$ $$\begin{gather}\psi_{i}\rightarrow0\ \text{as}\ z\rightarrow\pm\infty. \end{gather}$$
$$\begin{gather}\psi_{i}\rightarrow0\ \text{as}\ z\rightarrow\pm\infty. \end{gather}$$We introduce
 \begin{equation} \phi_{i}=\left( \frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\right)^{{-}1}\psi_{i}, \end{equation}
\begin{equation} \phi_{i}=\left( \frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\right)^{{-}1}\psi_{i}, \end{equation}
and observe that, since  $\rho _{i}(z)$ is monotonic,
$\rho _{i}(z)$ is monotonic,  $\mathrm {d}\rho _{i}/\mathrm {d}z$ never vanishes and
$\mathrm {d}\rho _{i}/\mathrm {d}z$ never vanishes and  $\phi _{i}(z)$ is non-singular.
$\phi _{i}(z)$ is non-singular.
Rewriting the boundary-value problem (C4) and (C5) in terms of  $\phi _{i}$ and keeping in mind that
$\phi _{i}$ and keeping in mind that  $\rho _{i}(z)$ satisfies (6.3), one obtains
$\rho _{i}(z)$ satisfies (6.3), one obtains
 $$\begin{gather} \sum_{j}K_{ij}\left( 2\frac{\mathrm{d}^{2}\rho_{j}}{\mathrm{d}z^{2}} \frac{\mathrm{d}\phi_{j}}{\mathrm{d}z}+\frac{\mathrm{d} \rho_{j}}{\mathrm{d} z}\frac{\mathrm{d}^{2} \phi_{j}}{\mathrm{d}z^{2}}\right) =\varLambda \frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\phi_{i}, \end{gather}$$
$$\begin{gather} \sum_{j}K_{ij}\left( 2\frac{\mathrm{d}^{2}\rho_{j}}{\mathrm{d}z^{2}} \frac{\mathrm{d}\phi_{j}}{\mathrm{d}z}+\frac{\mathrm{d} \rho_{j}}{\mathrm{d} z}\frac{\mathrm{d}^{2} \phi_{j}}{\mathrm{d}z^{2}}\right) =\varLambda \frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\phi_{i}, \end{gather}$$ $$\begin{gather}\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\phi_{i}\rightarrow0 \text{as}\ z\rightarrow\pm\infty. \end{gather}$$
$$\begin{gather}\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\phi_{i}\rightarrow0 \text{as}\ z\rightarrow\pm\infty. \end{gather}$$
Let  $( K_{ij})^{-1}$ be the inverse matrix to
$( K_{ij})^{-1}$ be the inverse matrix to  $K_{ij}$ (the latter is positive definite – hence, invertible) and rewrite (C7) in the form
$K_{ij}$ (the latter is positive definite – hence, invertible) and rewrite (C7) in the form
 \begin{equation} \frac{\mathrm{d}}{\mathrm{d}z}\left[ \left( \frac{\mathrm{d}\rho_{j} }{\mathrm{d}z}\right)^{2}\frac{\mathrm{d}\phi_{j}}{\mathrm{d}z}\right] =\varLambda\sum_{i}\left( K_{ij}\right)^{{-}1}\frac{\mathrm{d}\rho_{j} }{\mathrm{d}z}\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\phi_{i}. \end{equation}
\begin{equation} \frac{\mathrm{d}}{\mathrm{d}z}\left[ \left( \frac{\mathrm{d}\rho_{j} }{\mathrm{d}z}\right)^{2}\frac{\mathrm{d}\phi_{j}}{\mathrm{d}z}\right] =\varLambda\sum_{i}\left( K_{ij}\right)^{{-}1}\frac{\mathrm{d}\rho_{j} }{\mathrm{d}z}\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\phi_{i}. \end{equation}
Multiplying this equation by  $\phi _{j}$, summing it with respect to
$\phi _{j}$, summing it with respect to  $j$, integrating from
$j$, integrating from  $z=0$ to
$z=0$ to  $z=\infty$, integrating the left-hand side by parts and recalling boundary conditions (C8), one obtains
$z=\infty$, integrating the left-hand side by parts and recalling boundary conditions (C8), one obtains
 \begin{equation} -\int_{-\infty}^{\infty}\sum_{j}\left( \frac{\mathrm{d}\rho_{j}}{\mathrm{d} z}\right)^{2}\left( \frac{\mathrm{d}\phi_{j}}{\mathrm{d}z}\right) ^{2}\,\mathrm{d}z=\varLambda\int_{-\infty}^{\infty}\sum_{ij}( K_{ij})^{{-}1}\left( \phi_{i}\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\right) \left( \phi_{j}\frac{\mathrm{d}\rho_{j}}{\mathrm{d}z}\right) \mathrm{d}z. \end{equation}
\begin{equation} -\int_{-\infty}^{\infty}\sum_{j}\left( \frac{\mathrm{d}\rho_{j}}{\mathrm{d} z}\right)^{2}\left( \frac{\mathrm{d}\phi_{j}}{\mathrm{d}z}\right) ^{2}\,\mathrm{d}z=\varLambda\int_{-\infty}^{\infty}\sum_{ij}( K_{ij})^{{-}1}\left( \phi_{i}\frac{\mathrm{d}\rho_{i}}{\mathrm{d}z}\right) \left( \phi_{j}\frac{\mathrm{d}\rho_{j}}{\mathrm{d}z}\right) \mathrm{d}z. \end{equation}
It can be shown that the integrands of both integrals in (C10) decay exponentially as  $z\rightarrow +\infty$ – hence, the integrals converge. Keeping also in mind that
$z\rightarrow +\infty$ – hence, the integrals converge. Keeping also in mind that  $K_{ij}$ is positive definite (hence,
$K_{ij}$ is positive definite (hence,  $( K_{ij})^{-1}$ is such), one concludes that equality (C10) implies that
$( K_{ij})^{-1}$ is such), one concludes that equality (C10) implies that  $\varLambda <0$, as required.
$\varLambda <0$, as required.
Next, let  $\varLambda$ be an eigenvalue of the continuous spectrum and
$\varLambda$ be an eigenvalue of the continuous spectrum and  $\psi (z)$ the corresponding eigenfunction. They satisfy (C4) and the boundary conditions
$\psi (z)$ the corresponding eigenfunction. They satisfy (C4) and the boundary conditions
 \begin{equation} \psi_{i}\sim A_{i\pm}\,{\rm e}^{{\rm i}k_{{\pm}}z}\quad \text{as} z\rightarrow\pm\infty, \end{equation}
\begin{equation} \psi_{i}\sim A_{i\pm}\,{\rm e}^{{\rm i}k_{{\pm}}z}\quad \text{as} z\rightarrow\pm\infty, \end{equation}
where  $A_{i\pm }$ and
$A_{i\pm }$ and  $k_{\pm }$ are undetermined constants (the latter is real). Substituting asymptotics (C11) into (C4), one obtains
$k_{\pm }$ are undetermined constants (the latter is real). Substituting asymptotics (C11) into (C4), one obtains
 $$\begin{gather} \sum_{j}\left[ -\left( \frac{\partial G_{i}}{\partial\rho_{j}}\right) _{\rho_{1}=\rho_{1}^{(l)}\ldots \rho_{N}=\rho_{N}^{(l)}}-k_{-}^{2}K_{ij}\right] A_{j-}=\varLambda A_{-}, \end{gather}$$
$$\begin{gather} \sum_{j}\left[ -\left( \frac{\partial G_{i}}{\partial\rho_{j}}\right) _{\rho_{1}=\rho_{1}^{(l)}\ldots \rho_{N}=\rho_{N}^{(l)}}-k_{-}^{2}K_{ij}\right] A_{j-}=\varLambda A_{-}, \end{gather}$$ $$\begin{gather}\sum_{j}\left[ -\left( \frac{\partial G_{i}}{\partial\rho_{j}}\right) _{\rho_{1}=\rho_{1}^{(v)}\ldots \rho_{N}=\rho_{N}^{(v)}}-k_{+}^{2}K_{ij}\right] A_{j+}=\varLambda A_{+}. \end{gather}$$
$$\begin{gather}\sum_{j}\left[ -\left( \frac{\partial G_{i}}{\partial\rho_{j}}\right) _{\rho_{1}=\rho_{1}^{(v)}\ldots \rho_{N}=\rho_{N}^{(v)}}-k_{+}^{2}K_{ij}\right] A_{j+}=\varLambda A_{+}. \end{gather}$$
Evidently,  $\varLambda$ is a common eigenvalue of the matrices
$\varLambda$ is a common eigenvalue of the matrices
 \begin{equation} -G_{ij-}^{\prime}-k_{-}^{2}K_{ij}\quad \text{and}\quad -G_{ij+}^{\prime} -k_{+}^{2}K_{ij}, \end{equation}
\begin{equation} -G_{ij-}^{\prime}-k_{-}^{2}K_{ij}\quad \text{and}\quad -G_{ij+}^{\prime} -k_{+}^{2}K_{ij}, \end{equation}where
 \begin{equation} G_{ij-}^{\prime}=\left( \frac{\partial G_{i}}{\partial\rho_{j}}\right) _{\rho_{1}=\rho_{1}^{(l)}\ldots \rho_{N}=\rho_{N}^{(l)}},\quad G_{ij+}^{\prime }=\left( \frac{\partial G_{i}}{\partial\rho_{j}}\right) _{\rho_{1}=\rho _{1}^{(v)}\ldots \rho_{N}=\rho_{N}^{(v)}}. \end{equation}
\begin{equation} G_{ij-}^{\prime}=\left( \frac{\partial G_{i}}{\partial\rho_{j}}\right) _{\rho_{1}=\rho_{1}^{(l)}\ldots \rho_{N}=\rho_{N}^{(l)}},\quad G_{ij+}^{\prime }=\left( \frac{\partial G_{i}}{\partial\rho_{j}}\right) _{\rho_{1}=\rho _{1}^{(v)}\ldots \rho_{N}=\rho_{N}^{(v)}}. \end{equation}
Recall that, at infinity, the liquid and vapour are supposed to be stable, which implies that  $G_{ij\pm }^{\prime }$ are both positive definite. The Korteweg matrix
$G_{ij\pm }^{\prime }$ are both positive definite. The Korteweg matrix  $K_{ij}$ is also positive definite – hence, all eigenvalues of
$K_{ij}$ is also positive definite – hence, all eigenvalues of  $-G_{ij\pm }^{\prime }-k_{\pm }^{2}K_{ij}$ are negative, as required.
$-G_{ij\pm }^{\prime }-k_{\pm }^{2}K_{ij}$ are negative, as required.
C.2. One-dimensional drops and bubbles
The entropy maximization problem in this case can again be reduced to the analysis of the operator  ${\hat {O}}_{ij}$. Furthermore, the second part of the proof of Theorem C.1 can be applied to 1-D drops and bubbles without modifications, and so the continuous spectrum of
${\hat {O}}_{ij}$. Furthermore, the second part of the proof of Theorem C.1 can be applied to 1-D drops and bubbles without modifications, and so the continuous spectrum of  ${\hat {O}} _{ij}$ is, again, negative.
${\hat {O}} _{ij}$ is, again, negative.
The discrete-spectrum part of Theorem C.1, however, cannot be generalized for non-monotonic  $\rho _{i}(z)$. Indeed, if
$\rho _{i}(z)$. Indeed, if  $\mathrm {d}\rho _{i}/\mathrm {d}z$ vanishes somewhere, the function
$\mathrm {d}\rho _{i}/\mathrm {d}z$ vanishes somewhere, the function  $\phi _{i}(z)$ defined by (C6) is singular, and the integral on the left-hand side of (C10) diverges. Thus, for 1-D drops and bubbles,
$\phi _{i}(z)$ defined by (C6) is singular, and the integral on the left-hand side of (C10) diverges. Thus, for 1-D drops and bubbles,  ${\hat {O}}_{ij}$ may have positive discrete eigenvalues, but proving that it definitely does is not easy. In what follows, such a proof is presented only for
${\hat {O}}_{ij}$ may have positive discrete eigenvalues, but proving that it definitely does is not easy. In what follows, such a proof is presented only for  $N=1$.
$N=1$.
Theorem C.2 The operator  ${\hat {O}}_{11}$ with
${\hat {O}}_{11}$ with  $\rho _{1}(z)$ satisfying the boundary-value problem (6.8) and (6.9) with
$\rho _{1}(z)$ satisfying the boundary-value problem (6.8) and (6.9) with  $N=1$, has at least one positive discrete eigenvalue (corresponding to an even eigenfunction).
$N=1$, has at least one positive discrete eigenvalue (corresponding to an even eigenfunction).
Proof. For  $N=1$, the boundary-value problem (6.8) and (6.9) becomes
$N=1$, the boundary-value problem (6.8) and (6.9) becomes
 $$\begin{gather} K_{11}\frac{\mathrm{d}^{2}\rho_{1}}{\mathrm{d}z^{2}}=G_{1}-G_{\infty,1}, \end{gather}$$
$$\begin{gather} K_{11}\frac{\mathrm{d}^{2}\rho_{1}}{\mathrm{d}z^{2}}=G_{1}-G_{\infty,1}, \end{gather}$$ $$\begin{gather}\rho_{1}\rightarrow\rho_{\infty,1}\quad \text{as} z\rightarrow\pm\infty. \end{gather}$$
$$\begin{gather}\rho_{1}\rightarrow\rho_{\infty,1}\quad \text{as} z\rightarrow\pm\infty. \end{gather}$$It can be readily shown that
 \begin{equation} \rho_{1}(z)\sim\rho_{\infty,1}+A\operatorname{e}^{\sqrt{G^{\prime}}z} \quad \text{as}\ z\rightarrow-\infty, \end{equation}
\begin{equation} \rho_{1}(z)\sim\rho_{\infty,1}+A\operatorname{e}^{\sqrt{G^{\prime}}z} \quad \text{as}\ z\rightarrow-\infty, \end{equation}
where  $G^{\prime }=( \partial G_{11}/\partial \rho _{1}) _{\rho _{1}=\rho _{\infty,1}}$ and
$G^{\prime }=( \partial G_{11}/\partial \rho _{1}) _{\rho _{1}=\rho _{\infty,1}}$ and  $A$ is a real constant (which can be expressed via a certain integral, but its exact value is unimportant). Note that
$A$ is a real constant (which can be expressed via a certain integral, but its exact value is unimportant). Note that  $A$ is positive for drops (i.e. solutions of the kind illustrated in figure 4a) and negative for bubbles (solutions illustrated in figure 4b).
$A$ is positive for drops (i.e. solutions of the kind illustrated in figure 4a) and negative for bubbles (solutions illustrated in figure 4b).
Next, the eigenvalue problem (C4) and (C5) with  $N=1$ has the form
$N=1$ has the form
 $$\begin{gather} -\frac{\partial G_{1}}{\partial\rho_{1}}\psi_{1}+K_{11} \frac{\mathrm{d}^{2}\psi_{1}}{\mathrm{d}z^{2}}=\varLambda\psi_{1}, \end{gather}$$
$$\begin{gather} -\frac{\partial G_{1}}{\partial\rho_{1}}\psi_{1}+K_{11} \frac{\mathrm{d}^{2}\psi_{1}}{\mathrm{d}z^{2}}=\varLambda\psi_{1}, \end{gather}$$ $$\begin{gather}\psi_{1}\rightarrow0\ \text{as}\ z\rightarrow-\infty, \end{gather}$$
$$\begin{gather}\psi_{1}\rightarrow0\ \text{as}\ z\rightarrow-\infty, \end{gather}$$ $$\begin{gather}\psi_{1}\rightarrow0\ \text{as}\ z\rightarrow+\infty. \end{gather}$$
$$\begin{gather}\psi_{1}\rightarrow0\ \text{as}\ z\rightarrow+\infty. \end{gather}$$
Note that, for 1-D drops and bubbles,  $\rho _{1}(z)$ is even – hence, so is
$\rho _{1}(z)$ is even – hence, so is  $\partial G_{1}/\partial \rho _{1}$ that appears in (C19). Since all other coefficients in this (linear second-order) equation are constants, one concludes that
$\partial G_{1}/\partial \rho _{1}$ that appears in (C19). Since all other coefficients in this (linear second-order) equation are constants, one concludes that  $\psi _{1}$ is either even or odd. Assuming the latter, one can reduce the interval where (C19) is to be solved to
$\psi _{1}$ is either even or odd. Assuming the latter, one can reduce the interval where (C19) is to be solved to  $( 0,\infty )$ and replace boundary condition (C21) with
$( 0,\infty )$ and replace boundary condition (C21) with
 \begin{equation} \frac{\mathrm{d}\psi_{1}}{\mathrm{d}z}=0\quad \text{at}\ z=0. \end{equation}
\begin{equation} \frac{\mathrm{d}\psi_{1}}{\mathrm{d}z}=0\quad \text{at}\ z=0. \end{equation}
Next, define an auxiliary function  $\chi (z)$ that satisfies (C19) and boundary condition (C20), but not necessarily condition (C22). To still have two boundary conditions, we require that
$\chi (z)$ that satisfies (C19) and boundary condition (C20), but not necessarily condition (C22). To still have two boundary conditions, we require that
 \begin{equation} \chi(z)\sim A\sqrt{G^{\prime}+\varLambda}\,{\rm e}^{\sqrt{G^{\prime }+\varLambda}z}\ \text{as}\ z\rightarrow-\infty, \end{equation}
\begin{equation} \chi(z)\sim A\sqrt{G^{\prime}+\varLambda}\,{\rm e}^{\sqrt{G^{\prime }+\varLambda}z}\ \text{as}\ z\rightarrow-\infty, \end{equation}
where  $A$ is the constant from asymptotics (C16). Note that
$A$ is the constant from asymptotics (C16). Note that  $\chi (z)$ exists for any value
$\chi (z)$ exists for any value  $\varLambda$ (depends on it as a parameter). In particular,
$\varLambda$ (depends on it as a parameter). In particular,
 \begin{equation} \chi(z)=\frac{\mathrm{d}\rho_{1}}{\mathrm{d}z}\quad \text{for}\, \varLambda=0, \end{equation}
\begin{equation} \chi(z)=\frac{\mathrm{d}\rho_{1}}{\mathrm{d}z}\quad \text{for}\, \varLambda=0, \end{equation}
which can be verified by comparing (C19) with the derivative of (C16) and asymptotics (C23) with the derivative of asymptotics (C18). For a large  $\varLambda$, on the other hand, it can be deduced from (C19) and boundary condition (C23) that
$\varLambda$, on the other hand, it can be deduced from (C19) and boundary condition (C23) that
 \begin{equation} \chi(z)\sim A\sqrt{\varLambda}\, {\rm e}^{\sqrt{\varLambda}z}\quad \text{as}\ \varLambda\rightarrow+\infty. \end{equation}
\begin{equation} \chi(z)\sim A\sqrt{\varLambda}\, {\rm e}^{\sqrt{\varLambda}z}\quad \text{as}\ \varLambda\rightarrow+\infty. \end{equation}
This asymptotics applies to all finite  $z$.
$z$.
Now, consider a drop solution (e.g. one of those illustrated in figure 4a). It is evident from the figure (and common sense) that  $( \mathrm {d}^{2}\rho _{1}/\mathrm {d}z^{2}) _{z=0}<0$ and
$( \mathrm {d}^{2}\rho _{1}/\mathrm {d}z^{2}) _{z=0}<0$ and  $A>0$ – hence, (C24) and (C25) imply that
$A>0$ – hence, (C24) and (C25) imply that
 $$\begin{gather} \left( \frac{\mathrm{d}\chi}{\mathrm{d}z}\right) _{z=0}<0\quad \text{for} \varLambda=0, \end{gather}$$
$$\begin{gather} \left( \frac{\mathrm{d}\chi}{\mathrm{d}z}\right) _{z=0}<0\quad \text{for} \varLambda=0, \end{gather}$$ $$\begin{gather}\left( \frac{\mathrm{d}\chi}{\mathrm{d}z}\right) _{z=0}>0\quad \text{as}\ \varLambda\rightarrow+\infty. \end{gather}$$
$$\begin{gather}\left( \frac{\mathrm{d}\chi}{\mathrm{d}z}\right) _{z=0}>0\quad \text{as}\ \varLambda\rightarrow+\infty. \end{gather}$$
These two inequalities indicate that there exists a  $\varLambda \in ( 0,+\infty )$ such that
$\varLambda \in ( 0,+\infty )$ such that  $( \mathrm {d}\chi /\mathrm {d}z) _{z=0}=0$ – hence, the corresponding function
$( \mathrm {d}\chi /\mathrm {d}z) _{z=0}=0$ – hence, the corresponding function  $\chi (z)$ satisfies boundary condition (C22) (as well as condition (C20) and (C19)). The corresponding (positive)
$\chi (z)$ satisfies boundary condition (C22) (as well as condition (C20) and (C19)). The corresponding (positive)  $\varLambda$ is, obviously, an eigenvalue, as required.
$\varLambda$ is, obviously, an eigenvalue, as required.
The case of bubble solutions is similar to that of drops (examined above) and yields the same conclusion.
C.3. Solid/fluid interfaces
In this case,  $\rho _{i}(z)$ satisfies (6.9) and boundary conditions (6.15) and (6.16).
$\rho _{i}(z)$ satisfies (6.9) and boundary conditions (6.15) and (6.16).
Theorem C.3 The spectrum of the operator  ${\hat {O}}_{ij}$ corresponding to a monotonic
${\hat {O}}_{ij}$ corresponding to a monotonic  $\rho _{i}(z)$ is real and negative.
$\rho _{i}(z)$ is real and negative.
The proof of this theorem is similar to that of Theorem C.1. The only difference is in the boundary conditions: since the density is constrained to a fixed value at the substrate, its variation is zero, and the eigenfunctions of the operator  ${\hat {O}}_{ij}$ should satisfy
${\hat {O}}_{ij}$ should satisfy
 \begin{equation} \psi_{i}=0\quad \text{at}\ z=0. \end{equation}
\begin{equation} \psi_{i}=0\quad \text{at}\ z=0. \end{equation}This boundary condition should hold for both discrete and continuous spectra.
Theorem C.4 If  $N=1$, the operator
$N=1$, the operator  ${\hat {O}}_{11}$ with non-monotonic
${\hat {O}}_{11}$ with non-monotonic  $\rho _{1}(z)$ has at least one positive discrete eigenvalue.
$\rho _{1}(z)$ has at least one positive discrete eigenvalue.
The proof of this theorem is similar to that of Theorem C.2.
Appendix D. One-dimensional pure-fluid reduction of (4.18), (4.19) and (4.9)
To adapt asymptotic (4.18) and (4.19) for a pure fluid, recall that the transport coefficients  $D_{11}$ and
$D_{11}$ and  $\zeta _{1}$ are zero in this case (because this is the only possibility to satisfy restrictions (2.32a,b) for
$\zeta _{1}$ are zero in this case (because this is the only possibility to satisfy restrictions (2.32a,b) for  $N=1$). Thus, one obtains
$N=1$). Thus, one obtains
 $$\begin{gather} \frac{\partial\rho_{1}}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho _{1}\boldsymbol{v}) =0, \end{gather}$$
$$\begin{gather} \frac{\partial\rho_{1}}{\partial t}+\boldsymbol{\nabla} \boldsymbol{{\cdot}}( \rho _{1}\boldsymbol{v}) =0, \end{gather}$$ $$\begin{gather}\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi}+\rho_{1}\boldsymbol{\nabla} ( K_{11}\nabla^{2}\rho_{1}-G_{1}) =0. \end{gather}$$
$$\begin{gather}\boldsymbol{\nabla} \boldsymbol{{\cdot}}\boldsymbol{\varPi}+\rho_{1}\boldsymbol{\nabla} ( K_{11}\nabla^{2}\rho_{1}-G_{1}) =0. \end{gather}$$
Let the flow be one dimensional, so that the unknowns depend only on  $z$ and
$z$ and  $t$, and the velocity has only one component
$t$, and the velocity has only one component  $\boldsymbol {v}=[ 0,0,w]$. Then, (D1) and (D2) and expression (4.9) for the viscous stress yield
$\boldsymbol {v}=[ 0,0,w]$. Then, (D1) and (D2) and expression (4.9) for the viscous stress yield
 $$\begin{gather} \frac{\partial\rho_{1}}{\partial t}+\frac{\partial( \rho_{1}w) }{\partial z}=0, \end{gather}$$
$$\begin{gather} \frac{\partial\rho_{1}}{\partial t}+\frac{\partial( \rho_{1}w) }{\partial z}=0, \end{gather}$$ $$\begin{gather}\frac{\partial}{\partial z}\left[ \left( \frac{4}{3}\mu_{s}+\mu_{b}\right) \frac{\partial w}{\partial z}\right] +\rho_{1}\frac{\partial}{\partial z}\left( K_{11}\frac{\partial^{2}\rho_{1}}{\partial z^{2}}-G_{1}\right) =0. \end{gather}$$
$$\begin{gather}\frac{\partial}{\partial z}\left[ \left( \frac{4}{3}\mu_{s}+\mu_{b}\right) \frac{\partial w}{\partial z}\right] +\rho_{1}\frac{\partial}{\partial z}\left( K_{11}\frac{\partial^{2}\rho_{1}}{\partial z^{2}}-G_{1}\right) =0. \end{gather}$$
Let the substrate be located at  $z=0$, so that boundary conditions (2.44) and (2.47) yield
$z=0$, so that boundary conditions (2.44) and (2.47) yield
 \begin{equation} w=0,\quad \rho_{1}\rightarrow\rho_{0,1}\quad \text{at}\ z=0. \end{equation}
\begin{equation} w=0,\quad \rho_{1}\rightarrow\rho_{0,1}\quad \text{at}\ z=0. \end{equation}At infinity, the density tends to a fixed value and there should be no stress – hence,
 \begin{equation} \rho_{1}\rightarrow\rho_{\infty,1},\quad \frac{\partial w}{\partial z}\rightarrow0\quad \text{as}\ z\rightarrow+\infty. \end{equation}
\begin{equation} \rho_{1}\rightarrow\rho_{\infty,1},\quad \frac{\partial w}{\partial z}\rightarrow0\quad \text{as}\ z\rightarrow+\infty. \end{equation}
Using identity (2.6), one can replace in (D4),  $\rho _{1}\partial G_{1}/\partial z\rightarrow \partial p/\partial z$ and then integrate this equation with respect to
$\rho _{1}\partial G_{1}/\partial z\rightarrow \partial p/\partial z$ and then integrate this equation with respect to  $z$. Fixing the constant of integration via boundary conditions (D6a,b) and (D7), one obtains
$z$. Fixing the constant of integration via boundary conditions (D6a,b) and (D7), one obtains
 \begin{equation} \left( \frac{4}{3}\mu_{s}+\mu_{b}\right) \frac{\partial w}{\partial z}+K_{11}\left[ \rho_{1}\frac{\partial^{2}\rho_{1}}{\partial z^{2}}-\frac {1}{2}\left( \frac{\partial\rho_{1}}{\partial z}\right)^{2}\right] =0. \end{equation}
\begin{equation} \left( \frac{4}{3}\mu_{s}+\mu_{b}\right) \frac{\partial w}{\partial z}+K_{11}\left[ \rho_{1}\frac{\partial^{2}\rho_{1}}{\partial z^{2}}-\frac {1}{2}\left( \frac{\partial\rho_{1}}{\partial z}\right)^{2}\right] =0. \end{equation}
Given a suitable initial condition, (D3) and (D7), with boundary conditions (D5a,b) fully determine  $\rho _{1}(z,t)$ and
$\rho _{1}(z,t)$ and  $w(z,t)$.
$w(z,t)$.
For numerical simulations, (D3) and (D7) were discretised in  $z$ using central differences, and the resulting set of ordinary differential equations were solved using the MATLAB function ODE23tb (which can handle stiff problems). This approach is usually referred to as the ‘method of lines’ (Schiesser Reference Schiesser1978).
$z$ using central differences, and the resulting set of ordinary differential equations were solved using the MATLAB function ODE23tb (which can handle stiff problems). This approach is usually referred to as the ‘method of lines’ (Schiesser Reference Schiesser1978).







































































 Open access
Open access