


1. Introduction
Van der Woude Syndrome (VWS; NCBI OMIM: 119300) is a dominantly inherited developmental disorder characterized by lower lip, cleft lip and/or cleft palate (Van der Woude, Reference Van der Woude1954; Ali et al., Reference Ali, Singh and Raman2009). It is the most common cleft syndrome, accounting for approximately 2% of all cases and is caused by mutations in the interferon regulatory factor 6 (IRF6) gene, located on 1q32·2 (Ferrero et al., Reference Ferrero, Baldassarre, Panza, Valenzise, Pippucci, Mussa, Pepe, Seri and Silengo2010). Nearly 100 mutations in the coding region of the gene have been reported from several populations in different geographical regions of the globe. So far, IRF6 is the only gene shown to be associated with VWS as a dominant trait with variable penetrance and expressivity (Kayano et al., Reference Kayano, Kure, Suzuki, Kanno, Aoki, Kondo, Schutte, Murray, Yamada and Matsubara2003; Tan et al., Reference Tan, Lim and Lee2013). Ingraham et al. (Reference Ingraham, Kinoshita, Kondo, Yang, Sajan, Trout, Malik, Dunnwald, Goudy, Lovett, Murray and Schutte2006) found that abnormalities of epithelial differentiation that resulted in cleft palate were a consequence of adhesion between the palatal shelves and the tongue in both Irf6-null and homozygous missense mice. Richardson et al. (Reference Richardson, Dixon, Jiang and Dixon2009) showed that Irf6/Jag2 doubly heterozygous mice displayed fully penetrant intraoral epithelial adhesions, resulting in cleft palate. However, direct sequencing of all coding regions and exon–intron boundaries of the IRF6 gene have not shown any mutation in Indian cohorts (Moghe & Mauli, Reference Moghe and Mauli2011). Another Indian study of a VWS pedigree conducted by Moghe et al. (2010) also corroborates the findings that the critical region of 1q32–q41 may not be causal for VWS in multi-ethnic and diverse Indian populations (Moghe et al., Reference Moghe, Kaur, Thomas, Raseswari, Swapna and Rao2010).
A growing body of evidence suggests that structural variations across the genome, including CNVs, are likely to contribute to human disorders (Estivill & Armengo, Reference Estivill and Armengo2007; Conrad et al., Reference Conrad, Pinto, Redon, Feuk, Gokcumen, Zhang, Aerts, Andrews, Barnes, Campbell, Fitzgerald, Hu, Ihm, Kristiansson, Macarthur, Macdonald, Onyiah, Pang, Robson, Stirrups, Valsesia, Walter, Wei, Tyler-Smith, Carter, Lee, Scherer and Hurles2010). CNVs are DNA segments longer than 1 kb with >90% sequence identity that differ in the number of copies between the genomes of different individuals (Freeman et al., Reference Freeman, Perry, Feuk, Redon, McCarroll, Altshuler, Aburatani, Jones, Tyler-Smith, Hurles, Carter, Scherer and Lee2006). They affect more nucleotides per genome than SNPs and contribute significantly to variations in the levels of gene expression and to phenotypes of medical relevance among individuals (Aitman et al., Reference Aitman, Dong, Vyse, Norsworthy, Johnson, Smith, Mangion, Roberton-Lowe, Marshall, Petretto, Hodges, Bhangal, Patel, Sheehan-Rooney, Duda, Cook, Evans, Domin, Flint, Boyle, Pusey and Cook2006). Many reports indicated non-involvement of IRF6 in VWS in the Indian context; therefore, the present study was carried out to examine the involvement of CNVs in a family with members suffering from VWS in South India.
2. Materials and methods
For this study, a family with members suffering from VWS with a four generation family history and a total of seven affected members with or without cleft lip/palate were selected (Fig. 1). Subjects were between the ages of 10–55 years. From the recruited family three affected cases (4S, 5S and 6S) and two healthy controls (2C and 3C) were subjected to a genome-wide scan while the remaining samples could not be genotyped due to non-availability of the samples.

Fig. 1. Illustration of the Van der Woude pedigrees under study. All the seven affected members were found to have lower lip anomalies, among these, two individuals (5S and 6S) also had cleft lip, while only one affected member (4S) was also found to have cleft palate. (a) The pedigree under study consisted of 15 individuals, of which, seven were affected with cleft lip/palate across all the three generations. The proband of this study was a 10 year old boy diagnosed with cleft lip and lower lip anomalies. (b) Shows subject 4S who is an affected female in the second generation of the pedigree with lower lip and cleft palate. (c) Shows subject 5S who is one of the affected daughters of case 4S having lower lip and cleft palate and (d) shows case 6S, who is the affected daughter of 5S with lower lip and cleft lip.
A total of 38 randomly selected non-affected Indian controls that were residing in Karnataka, India, and with an age range of 13–73 years, were genotyped using the Affymetrix Genome-Wide Human SNP Array 6·0 chip. A total of 270 HapMap samples covering CEU (CEPH collection), CHB (Han Chinese in Beijing, China), JPT (Japanese in Tokyo, Japan) and YRI (Yoruba in Ibadan, Nigeria) populations and 31 Tibetan samples were selected as unrelated control subjects. A total of 5 ml EDTA blood was collected from each member of the 12 families from the Indian control group and genomic DNA was extracted using a Promega Wizard® Genomic DNA purification kit. The isolated DNA was quantified by Bio-photometer and gel electrophoresis. This research was approved by the University of Mysore-Institutional Human Ethics Review Committee (IHEC). Written informed consent was obtained from all sample donors and the IHEC approved the sample consent procedure. Written informed consent was obtained from parents/guardians in the cases of participants being minors. They also provided written informed consent for publication of photographs. A total of 38 non-cleft lip/palate control individuals from India, 31 from Tibet, 47 from New World, 269 from HapMap, 64 from Australia, 155 from China and 953 from an Ashkenazi Jewish population, totalling 1557 genomes were selected as controls to screen for CNVs in the IRF6 gene. The data from 270 individuals from the four populations was obtained from the International HapMap Consortium (The International HapMap Consortium, 2003). The HapMap samples come from a total of 270 people: 30 both-parent-and-adult-child trios from YRI subjects, 45 unrelated JPT subjects, 45 unrelated CHB individuals, and 30 both-parent-and-adult-child trios from CEPH. The raw, unprocessed data from the Affymetrix Genome-Wide SNP 6·0 array for the 31 individuals from Tibet, 47 from New World, 269 from HapMap, 64 from Australia, 155 from China and 953 from an Ashkenazi Jewish population, totalling up to 1557 genomes, were obtained from the ArrayExpress Archive at the European Bioinformatics Institute.
(i) Genotyping
Genome-wide genotyping was performed using an Affymetrix Genome-Wide Human SNP Array 6·0 chip and Affymetrix CytoScan® High-Density (HD) Array having 1·8 million and 2·6 million combined SNP and CNV markers, with a median inter-marker distance of 500–600 bases. These chips provide maximum panel power and the highest physical coverage of the genome (Affymetrix, Inc., Data Sheet). Genotyping quality was assessed using Affymetrix Genotyping Console Software. Briefly, all SNPs that were called using the Birdseed v2 algorithm (http://www.broad.mit.edu/mpg/birdsuite/birdseed.html) had a Quality Control (QC) call rate of >97% across members in families. All the members who passed SNP QC procedures were entered into the CNV analysis. The CNV calls were generated using the Canary algorithm. Contrast QC across all samples was >2·5, it was required to be >0·4. A description of the CNV methods used in this study is given below. The copy number analysis method offers two types of segmenting methods, univariate and multivariate. These methods are based on the same algorithm, but use different criteria for determining cut-points denoting CNV boundaries.
Analysing the collated data from both the BirdSuite and Canary algorithms increased the stringency on those meeting the CNV calls with a log10 of odds score greater than or equal to 10 corresponding to a false-discovery rate of ~5%. All SNPs that were called using the Birdseed v2algorithm had a QC call rate of >97% across individuals. All the subjects and members with SNPs that passed SNP QC procedures were entered into the CNV analysis. Filters were set for ID call rates for the overall SNPs to identify IDs with poor quality DNA, if any. The CNV calls were generated using the Canary algorithm. In Affymetrix Genotyping Console Software, contrast QC has to be >0·4 to be included in the CNV analyses. In this study, the observed contrast QC was >2·5 across all samples showing a robust strength. To control for the possibility of spurious or artifact CNVs, we used the EIGENSTRAT approach of Price et al. (Reference Price, Patterson, Plenge, Weinblatt, Shadick and Reich2006). This method derives the principal components of the correlations among gene variants and corrects for those correlations in the testing. We removed one individual from the study group because they were extreme outliers on one or more significant EIGENSTRAT axes and further dropped 54 CNVs in the members selected for the study as they did not meet the required QC measures. CNVs were considered validated when there was a reciprocal overlap of 50% or greater with the reference set. Though the Jaccard statistic is sensitive to the number of CNVs called by each algorithm (ideally each of the two algorithms would detect a similar number of CNV calls), the relative values between the different comparisons of algorithms/platform/site are very informative. All the overlap analyses performed have handled losses and gains separately except when otherwise stated, and were conducted hierarchically. The calls from the algorithms that were called in both were not considered; instead, they were collated so that the relative values between the different comparisons of algorithms/platform/site are still very informative. The genotyping was performed on all subjects using multiple algorithms. The details of genotyping, data analysis, breakpoint validations and other programs used in this study have been described in detail in Veerappa et al., (Reference Veerappa, Padakannaya and Ramachandra2013 a ; Reference Veerappa, Saldanha, Padakannaya and Ramachandra2013 b ; Reference Veerappa, Saldanha, Padakannaya and Ramachandra2013 c ; Reference Veerappa, Vishweswaraiah, Lingaiah, Murthy, Manjegowda, Nayaka and Ramachandra2013 d ).
(ii) Weighted protein interaction network analysis
We used weighted protein network analysis in a first attempt to identify odds ratio gene associated modules and their key constituents. Weighted protein network analysis starts from the level of hundreds of genes, identifies modules of co-expressed proteins and proteins falling under the common pathway and relates these modules to Gene Ontology information. We made use of tools such as GeneMANIA (Warde-Farley et al., Reference Warde-Farley, Donaldson, Comes, Zuberi, Badrawi, Chao, Franz, Grouios, Kazi, Lopes, Maitland, Mostafavi, Montojo, Shao, Wright, Bader and Morris2010), BIOGRID and CYTOSCAPE (Shannon et al., Reference Shannon, Markiel, Ozier, Baliga, Wang, Ramage, Amin, Schwikowski and Ideker2003), which were developed for network studies to assess the functional consequences of the network topology.
(iii) IRF6 Ingenuity Pathway Analysis
Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems; www.ingenuity.com) was used to identify interactions between genes, protein–protein interactions, biological mechanisms, as well as the location and functions of target genes. Genes and chemical search was used to explore the information on protein families, protein signalling and metabolic pathways along with normal cellular activity of proteins. Genes and their protein products are shown based on their location. An edge (line) is used to represent the relationship between two nodes. Each edge in the network was represented by obtaining the information from resource database, Ingenuity Pathways Knowledge Base and also from the cited literature. Ingenuity Pathways Knowledge Base was considered for the analysis. A cascade of protein–protein interactions or protein binding, molecular cleavage, activation, upregulation and downregulation networks were analysed.
3. Results
(i) Genome-wide copy number and phenotype assessment of VWS
The pedigree under study consisted of 15 individuals, of which, seven were affected with cleft lip/palate across all three generations. The proband of this study was a 10 year old boy diagnosed with cleft lip and lower lip anomalies. All seven affected members of the family were recruited after thorough diagnostic testing, and all seven were found to have lower lip anomalies, among these, two individuals (5S and 6S) also had cleft lip, while only one affected member (4S) was also found to have cleft palate (Fig. 1(a)). Three affected and two related unaffected members from the three generation family were screened for CNVs using Affymetrix Cytoscan HD array having 2·6 million CNV and SNP markers. Collated data from the arrays meeting the CNV calls criteria of a log10 of odds score greater than or equal to 10 (corresponding to a false-discovery rate of ~5%) were selected for further investigation. We observed a total of 1198 CNV events across the genomes of both cases and controls, with 74% duplications and 26% deletions. Various CNVs ranging in size from 100 kb to several Mb were observed in multigene families of the alpha-amylase family (AMY1 and AMY2), rRNA genes, immunoglobulins, olfactory receptor genes and in the uridine glucuronosyltransferase genes. However, CNV analysis revealed the presence of rare de novo microduplications and deletions at the 1q32 region (Table 1). This region harbours IRF6 and the CNVs partially disrupted the exon structure (Fig. 2). These duplication and deletion CNVs were found to have a copy number state range of 0–3 in cases, whereas a normal copy number state of 2 was observed in all unaffected individuals.

Fig. 2. Schematic representation of copy number state of Van der Woude cases. An image of the copy number state (5S, 6S and 4S) indicates the quantitative assessments of genotyping used to determine copy number variation in the Van der Woude (VWS) cases. Blue bars indicate duplications and red indicates deletions. Case 4S shows a homozygous deletion copy number variation (CNV) of 3 kb from 209974442 bp to 209977378 bp at the 1q32 region encompassing IRF6. Case 5S shows a contiguous 53 kb heterozygous duplication CNV followed by a 18 kb deletion CNV from 209970522 bp to 209988149 bp in the 1q32 region, but in the homozygous state with the intermediate distance of ~16 kb between the duplication and deletion CNV. The end breakpoint of the duplication CNV and the start point of the deletion CNV partially disrupted IRF6. These two CNVs amounted to ~71 kb in variation. Case 6S shows a deletion CNV of 13 kb from 209974651 bp to 209988149 bp in the 1q32 region in the homozygous state. Though the deletion CNV in both the 5S and 6S cases have varied start points, they both have the same end point, which encompass IRF6.
Table 1. Duplications and deletions in different regions of IRF6 in Van der Woude subjects from a family of South India
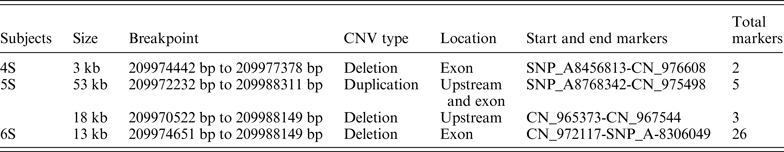
Case 4S is an affected female individual in the second generation of the pedigree (Fig. 1). This individual showed a homozygous deletion CNV of 3 kb in size from 209974442 bp to 209977378 bp at the 1q32 region, which encompasses IRF6. However, Case 5S, one of the affected daughters (Fig. 1) of case 4S, showed a contiguous 53 kb heterozygous duplication CNV from 209972232 bp to 209988311 bp that encompassed upstream and exons of IRF6, this was followed by a 18 kb deletion CNV from 209970522 bp to 209988149 bp in the 1q32 region, but in the homozygous state with the intermediate distance of ~16 kb between the duplication and deletion CNV. The end breakpoint of the duplication CNV and the start point of the deletion CNV partially disrupted IRF6. These two CNVs amounted to ~71 kb in variation. Case 6S, the affected daughter of 5S (Fig. 1), also displayed a deletion CNV of 13 kb from 209974651 bp to 209988149 bp in the 1q32 region in the homozygous state. Though the deletion CNV in both cases 5S and 6S have varied start points, they both have the same end point, which encompasses IRF6.
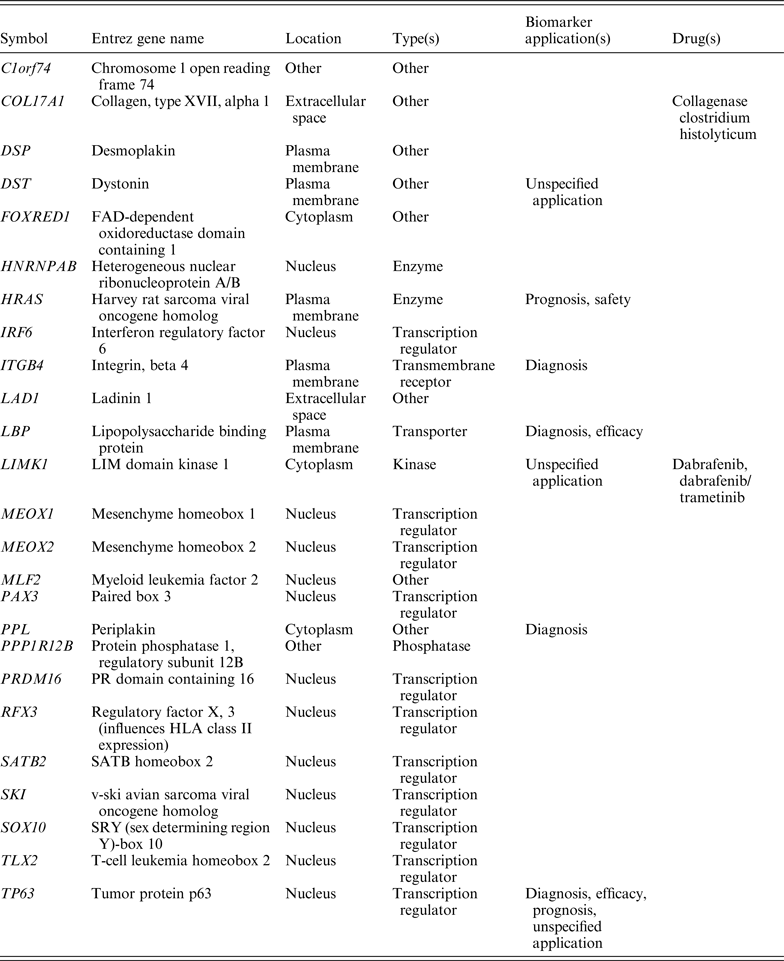
Since the CNVs identified here encompass IRF6 (Fig. 2), we investigated whether IRF6 interacts with any other known facial morphology determining genes using the protein interaction network tool, which uses known functional relationships and interactions between gene products reported in the literature. IRF6 was found to be mapped to a single genetic network (Fig. 3) that included genes interacting with a known candidate genes of facial morphology determining genes such as PAX3, PRDM16, COL17A1, TP63 and HNRNPAB, which are involved in cytokine-mediated signalling pathways, response to interferon-gamma and the interferon-gamma mediated signalling pathways. Protein network and pathway analysis also showed IRF6 interacting with PAX3, PRDM16, COL17A1, TP63 and HNRNPAB proteins directly, or sometimes with intermediate partners essential for response to interferon-gamma and interferon-gamma mediated signalling pathways, which are involved in normal facial morphology. Duplications and deletions in IRF6 identified in this study disrupt the protein production, which in turn fails to interact with PAX3, PRDM16, COL17A1, TP63 and HNRNPAB resulting in the blocked pathway.

Fig. 3. IRF6 protein interaction network establishing the associated gene modules and their key hubs. The figure shows the network of genes involved in cleft lip/palate with hub genes coloured according to their respective Gene Ontology (GO). Network analysis of candidate genes of lip/palate, including the genes identified in this study, has established ~24 genes with protein and genetic interactions that have more than >70 links. Out of these, interactions involving some genes that participate in the facial determination region could be accounted for by the analysis in the present study. These interactions are more specific for interferon signalling as they directly participate in cytokine-mediated signalling pathways, response to interferon-gamma and interferon-gamma mediated signalling pathways. The geometrical establishment of the IRF6 network shows already identified candidate genes interacting with facial morphology genes in the present study. The smaller and larger circles depict network weights that are chosen based on the size of the input gene list. Based on the input gene list, a ‘GO-based weighting, biological process based’ weighting method is chosen. This weighting method assumes the input gene list is related in terms of biological processes (as defined by GO) and assigns weights based on maximizing connectivity between all input genes using the ‘assigned based on query gene’ strategy.
(ii) Protein interaction network of VWS
Network analysis of IRF6 (Fig. 3) has established ~24 genes with protein and genetic interactions, leading to more than 70 links (Table 2). Out of these, interactions involving some genes participating in interferon-gamma mediated signalling pathways could be accounted for by the analysis in the present study. These interactions are more specific for interferon signalling as they directly participate in cytokine-mediated signalling pathways, response to interferon-gamma and interferon-gamma mediated signalling pathways. The geometrical establishment of the IRF6 network also shows already identified candidate genes of facial morphology determinants.
Table 2. Annotation of the genes identified through network and pathway studies based on location, type, biomarker application and drugs
The network analysis of the above genes (Fig. 3) and their interconnecting pathways suggest that they are vital in the following three biological functions: (1) cytokine-mediated signalling pathways, (2) response to interferon-gamma and (3) interferon-gamma mediated signalling pathways. Thus, disruption of the genes involved in these pathways identifies critical regions in normal lip, palate and face morphology formation.
The observed CNV breakpoints in the 1q32 region from this study were validated by genotyping 38 individuals from India, 31 from Tibet, 47 from New World, 269 from HapMap, 64 from Australia, 155 from China and 953 from an Ashkenazi Jewish population, totalling up to 1557 genomes. None of the members among this non-cleft lip/palate control group showed any CNV breakpoints in the 1q32 bearing IRF6 region.
(iii) IPA
IPA revealed interactions between IRF6 and several other transcription factors and DNA binding proteins (Fig. 4). Many of these proteins are major determinants of facial structure and morphology. IRF6 shows interactions with diverse types of functional proteins and these processes include cell migration, signalling, development, and tissue development and morphology (Supplementary Table 1; available online).

Fig. 4. Ingenuity Pathway Analysis of the IRF6 gene and its interactions with target proteins indicating involvement in several biological functions and pathway.
We further examined the proteins interacting with IRF6 to determine functional relevance and disease pathways relevant to VWS. A total of 64 coding genes were included in the IPA. We ranked functional and disease categories based on enrichment for all the genes (Supplementary Table 1). The top 20 ranked categories included functional and disease categories related to cell-to-cell signalling and interaction, cellular movement, cellular growth and proliferation, tissue morphology, and cell death and survival. IPA was also used to search for biological relationships among the IRF6 interacting genes with previously identified facial morphology genes (Fig. 4). Paths between genes represent protein–protein interactions, regulation and phosphorylation activities. IRF6 in the pathway is regulated by several of its partners that if altered result in cleft lip/palate. Supplementary Table 1 indicates the functional annotation of proteins, their significant p-value and the number of genes participating in that particular function.
4. Discussion
So far, globally available reports show that VWS is caused by mutations in the IRF6 gene located on chromosome 1q32·2 (Kondo et al., Reference Kondo, Schutte, Richardson, Bjork, Knight, Watanabe, Howard, de Lima, Daack-Hirsch, Sander, McDonald-McGinn, Zackai, Lammer, Aylsworth, Ardinger, Lidral, Pober, Moreno, Arcos-Burgos, Valencia, Houdayer, Bahuau, Moretti-Ferreira, Richieri-Costa, Dixon and Murray2002; Ferrero et al., Reference Ferrero, Baldassarre, Panza, Valenzise, Pippucci, Mussa, Pepe, Seri and Silengo2010; Leslie & Marazita, Reference Leslie and Marazita2013). Recently, Indian studies on VWS reported that the critical region on 1q32–q41 and the mutations of IRF6 may not be causal for VWS in multi-ethnic and diverse Indian populations (Moghe et al., Reference Moghe, Kaur, Thomas, Raseswari, Swapna and Rao2010; Moghe & Mauli, Reference Moghe and Mauli2011). However, molecular studies on VWS in India are very limited so are not able to confirm or reject these controversial reports. Therefore, a further molecular study involving the identification of CNVs has been carried out by a whole-genome scan to analyse the susceptible region or genes, which may be involved in causing VWS in the South Indian population.
In the present study, CNV analysis in VWS cases revealed the deletion of IRF6 in the critical region 1q32–q41. Even in studies that have shown negative reports of failing to identify mutations in IRF6 in VWS subjects, the role of CNVs cannot be ruled out. Birnbaum et al. (Reference Birnbaum, Reutter, Lauster, Scheer, Schmidt, Saffar, Martini, Hemprich, Henschke, Kramer and Mangold2008) analysed IRF6 in 63 families with what was believed to be isolated cleft lip/palate or cleft palate and identified a deletion/insertion and duplication in two families, respectively, and a history of lip pits in both family members confirmed the diagnosis of VWS. Linkage analyses in five Finnish families linked the 1q32–q41 region with VWS in three of these families (Wong et al., Reference Wong, Koillinen, Rautio, Teh, Ranta, Karsten, Larson, Linder-Aronson, Huggare, Larsson and Kere2001). In a recent study, five independent genetic loci were reported to be associated with different facial phenotypes, suggesting the involvement of five candidate genes: PRDM16, PAX3, TP63, C5orf50 and COL17A1 in the determination of the human face (Liu et al., Reference Liu, van der Lijn, Schurmann, Zhu, Chakravarty, Hysi, Wollstein, Lao, de Bruijne, Ikram, van der Lugt, Rivadeneira, Uitterlinden, Hofman, Niessen, Homuth, de Zubicaray, McMahon, Thompson, Daboul, Puls, Hegenscheid, Bevan, Pausova, Medland, Montgomery, Wright, Wicking, Boehringer, Spector, Paus, Martin, Biffar and Kayser2012). VWS is known to be a Mendelian disorder of variable expressivity with high penetrance. In the present study, we observed CNV breakpoints inherited inconsistently in cases 4S, 5S and 6S. We believe this is due to the unequal crossing-over mechanism since it exhibits contiguous integration of the duplication/deletion CNVs. We put forth that these CNVs have resulted because of the unequal recombination between two 1q32·3 chromosomal regions, resulting in the gain or loss of a part of 1q32·3, which can be seen as heterozygous or homozygous duplication/deletions in the subjects.
(i) Molecular network of IRF6
We propose that the IRF6 gene network model (Fig. 3) controls three major functions: (1) cytokine-mediated signalling pathways, (2) response to interferon-gamma and (3) interferon-gamma mediated signalling pathways. We also identified disease-related networks (modules) and disease-related hub genes and found large protein–protein interaction hubs associated with facial morphology determinants (Fig. 3). We used minimal cut sets (MCSs) in this network study (Klamt, Reference Klamt2006), which helps in studying the structural fragility and to identify knock out strategies in cellular networks. When the MCSs are computed with respect to “physical and genetic” interactions in realistic networks, the experimental block of essential genes will inevitably lead to mutants. Thus, the candidate genes identified in this study are the indicators of major MCSs. The immediate interacting protein partners represents primary level MCSs and the immediate interacting protein partners of the primary level MCSs represent secondary level MCSs. These MCSs are distributed in a fragile interconnected network. This can be demonstrated as follows: IRF6 interacts with TP63 and PAX3 proteins, which are essential for a proper interferon-gamma mediated signalling pathway, whereas PRDM16 and COL17A1 are essential for efficient signalling. Duplication and deletion in IRF6 identified in this study disrupts the protein interaction with TP63, PAX3, PRDM16 and COL17A1 resulting in the blocked pathway. This type of structural fragility in the IRF6 network is given by the size distribution over the total set of MCSs. In such networks, a dysfunction can be caused in higher probability, as a malfunction requires only one or a few network elements to fail. IPA revealed IRF6 involvement in cell migration, signalling, development, and tissue development and morphology. This probably explains the non-migration of certain cells to the lip and palate region and failure to develop facial morphology.
The present study is a maiden report indicating the involvement of CNVs in IRF6 in causing VWS. Genome-wide analysis of CNVs confirms the involvement of the critical 1q32–q41 region and IRF6 in South Indian VWS patients and the CNVs found in the patients were associated with facial morphology. Various studies have shown that direct sequencing of all coding regions and exon–intron boundaries of IRF6 have not shown any mutation in Indian cohorts (Moghe et al., Reference Moghe, Kaur, Thomas, Raseswari, Swapna and Rao2010; Moghe & Mauli, Reference Moghe and Mauli2011) of VWS in multi-ethnic and diverse Indian populations, indicating the presence of a secondary causal gene that can cause VWS. Based on these reports, we hypothesized a role for the PRDM16, PAX3, TP63, C5orf50 and COL17A1 genes (which are known to interact physically with IRF6) in VWS. These genes have not been formally validated but have been found to play a significant role in pathways involving cytokine-mediated signalling, response to interferon-gamma and interferon-gamma mediated signalling pathways. Based on our study of the pathway analysis of IRF6, we identified various forms of interactions with IRF6 proteins that play a key role in VWS manifestation. Hence while considering studies that failed to identify mutations in IRF6, it is necessary to focus on the above-mentioned probable genes. Since CNVs in IRF6/the 1q32·2 region were identified in living cleft lip and cleft palate subjects, and since this gene is not expressed in leukocytes it cannot be analysed from blood; therefore, identifying the changes at expression level was a possible limitation of this study.
This work was supported by the Yenepoya University funding seed grant (YU/Seed Grant/2011-011). The Authors thank participants and their family for participating in this study.
5. Declaration of interest
None
6. Authors' contributions
DSM and AMV performed experiments and analysis. DSM, AMV, MP and NBR conceived of the study, designed the experiments, interpreted the results and prepared the manuscript. DSM, MP, AMV and NBR read and approved the final manuscript.
7. Supplementary material
The online supplementary material can be found available at http://journals.cambridge.org/GRH






