


Globalisation and human encroachment into native wildlife habitats will probably continue to cause an increase in emerging zoonotic viral diseases. In recent years, members of the Paramyxoviridae viral family have caused some of the deadliest emerging zoonoses. The Paramyxoviridae family comprises important old and new human and animal viral pathogens, and Nipah (NiV) and Hendra (HeV) viruses make up the new Henipavirus genus within this family (Refs 1, Reference Luby2, Reference Luby3). HeV was first identified in 1994 in Australia, and NiV was discovered in 1998 in Malaysia and Singapore; both caused epidemics that concerned national and international authorities because of the high mortality and morbidity rates in affected animals and humans (Refs Reference Chua4, Reference Halpin5). For most paramyxoviruses, the host range is narrow and cross-species transmission events are rare; hence, the recent emergence of the henipaviruses with high virulence and a broad host range is alarming.
Other paramyxoviruses, with lower mortality rates or fewer incidents in humans, have also emerged in recent years, including Menangle virus, Tioman virus, avian paramyxovirus 1 and human metapneumovirus (HMPV). Nonetheless, the incidence of HMPV in human populations approaches 100%, and causes 5–20% of young children to be hospitalised with respiratory tract infections (reviewed in Ref. Reference Virtue, Marsh and Wang6). In addition, although other emerging paramyxoviruses such as the Beilong or J viruses have not been reported to cross species from their putative rodent reservoirs, the ability of Beilong virus to cross-contaminate human cell cultures from rodent cell cultures in the same laboratory raises the spectre of zoonotic spread to humans (Refs Reference Jun, Karabatsos and Johnson7, Reference Li8, Reference Mesina9).
Therefore, understanding the mechanistic underpinnings of viral entry, replication and assembly of these emerging paramyxoviruses is of critical importance. This review focuses primarily on henipaviruses because most recent molecular and mechanistic studies that inform potential antiviral strategies have been directed against this most lethal group of paramyxoviruses. We do not cover vaccine approaches, because they have been recently reviewed elsewhere (Refs Reference Bossart and Broder10,Reference Vigant and Lee11, Reference Williamson and Torres-Velez12).
The Paramyxoviridae family
The Paramyxoviridae family has been divided into two subfamilies: Paramyxovirinae and Pneumovirinae (Fig. 1). The Paramyxovirinae subfamily comprises five genera: Respirovirus, Rubulavirus, Avulavirus, Morbillivirus and Henipavirus. This subfamily includes the important measles, mumps, Newcastle disease and parainfluenza viruses, as well as HeV and NiV, although some of the emerging Paramyxovirinae members (e.g. Menangle, Tioman, Beilong and J) do not formally cluster into these five main genera. Some viruses within this subfamily have caused important human diseases for millennia. For example, reports of symptoms such as those caused by the measles virus date back to the seventh century. Although the measles virus has now been eradicated from most developed countries through vaccination, it still produces a significant number of deaths globally, with 197 000 deaths reported in 2007 (Ref. 13).

Figure 1. Phylogenetic tree of the Paramyxoviridae family, built using a fusion-protein sequence comparison. The tree was generated from a COBALT (NCBI) multiple fusion-protein sequence alignment, by the fast minimum evolution method, and visualised using the Fig Tree program (http://www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi?CMD=Get&cobaltRID=M93UBRKP212&SEQ_NUMBER=14&UNIQ_OBJ_NAME=A_CobaltResults_1PjIvj_2LC4_3DdVPpg5IK_GTJe2_NDktU&link_loc=FromRes, http://www.ncbi.nlm.nih.gov/blast/treeview/treeView.cgi). Representative members of each genus of the Paramyxovirinae and Pneumovirinae subfamilies are shown (genera are shown in blue type). Abbreviations: APIV-1, avian parainfluenza virus 1; CDV, canine distemper virus; HeV, Hendra virus; HMPV, human metapneumovirus; HPIV-3, human parainfluenza virus 3; HRSV, human respiratory syncytial virus; MeV, measles virus; NDV, Newcastle disease virus; NiV, Nipah virus; PIV-5, parainfluenza virus 5.
The second subfamily, the Pneumovirinae, consists of two genera: Pneumovirus and Metapneumovirus (Fig. 1). This subfamily also includes important old and new human and animal pathogens, such as the human and bovine respiratory syncytial viruses (RSVs) that specifically affect bovine, caprine and ovine species, and the human and avian metapneumoviruses, among others. Human RSV (HRSV) is an important pathogen within this subfamily, causing 64 million infections and 160 000 deaths, primarily infant, per year (Ref. 14).
The emerging Henipavirus genus
HeV and NiV have been classified in a new genus because their genomic lengths and protein homologies are sufficiently different from extant genera of paramyxoviruses (Ref. Reference Chua4). Their particularly broad tropism and extreme virulence compared with other paramyxoviruses also sets them apart. The henipaviruses naturally infect flying foxes (bats of the genus Pteropus), and transmit to humans either by an intermediate host, usually horses for HeV and swine for NiV, or directly from bat to human or from human to human, as reported for post-2004 epidemics for NiV in Bangladesh (Refs 1, 15, 16, Reference Gurley17).
HeV has reportedly caused the death of dozens of horses and three humans in Australia, through several outbreaks since 1994 (Refs Reference Halpin5, 18, Reference Field19, Reference Field20, Reference Young21). By contrast, NiV has caused the death of almost 200 humans and high numbers of animals, with 1.1 million pigs culled in the first 1998 Malaysian epidemic alone (Ref. Reference Chua4). Since then, flying foxes seropositive for NiV have been detected in Cambodia, Thailand, India, and as far west as Madagascar and Ghana in West Africa (Refs Reference Drexler22, Reference Iehle23). NiV causes respiratory and neurological symptoms that often lead to encephalitis and mortality rates from 40% to 92% in humans (Refs Reference Luby2, Reference Lam24, Reference Tan and Wong25). Additionally, NiV can spread efficiently and cause morbidity in economically important livestock (Ref. Reference Lam24).
As a result of their high virulence and the absence of therapeutics or vaccines to control them, henipaviruses are classified as Biosafety Level 4 pathogens, and NiV is classified as a Category C Priority Pathogen by the US NIAID Biodefense Research Agenda for its bio- and agro-terrorism potential (Ref. Reference Lam24). These characteristics of the henipaviruses underscore the need for research and treatment development against these perilous pathogens.
Molecular advancements in emerging paramyxovirus biology: implications for drug development
The development of antiviral therapeutic agents for other viral infections has been facilitated by elucidation of the molecular mechanisms underlying various steps of their viral life cycles. As an example, insights into the life cycle of human immunodeficiency virus 1 (HIV-1) have led to approved antiretroviral drugs that target distinct steps: co-receptor antagonists and fusion inhibitors target viral entry, nucleoside and non-nucleoside inhibitors target viral reverse transcriptase, integrase inhibitors target integration, and protease inhibitors target viral maturation (reviewed in Refs Reference Menendez-Arias26, Reference Stellbrink27). Among the emerging paramyxoviruses, the henipaviruses have been studied most extensively because of their relatively high morbidity rates. Recent discoveries have shed light on the molecular mechanisms underpinning several steps of their life cycle, including host receptor usage, membrane fusion and viral entry, viral replication, interferon (IFN) responses, assembly and budding, and each step represents a potential target for the development of antiviral drugs (Fig. 2). These research advances and antiviral therapeutic strategies are discussed here, with most focus on the viral entry and assembly steps, carried out by the fusion, attachment and matrix viral proteins. The molecular mechanisms and antiviral approaches that target the functions of other nonstructural paramyxovirus proteins, particularly the gene products P, V, C and W, have been previously reviewed in detail elsewhere (Refs Reference Vigant and Lee11, Reference Eaton28, Reference Fuentes29, Reference Rodriguez and Horvath30).
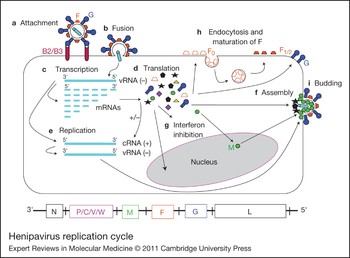
Figure 2. Henipavirus replication cycle. After attachment to the ephrinB2/B3 receptor (a) and fusion (b), the virus enters the cell. The negative RNA genome [vRNA(−)] is a template for transcription of viral mRNAs following a 3′ to 5′ attenuation gradient from N to L (c). N and L are depicted on the henipavirus genomic RNA, represented in its 3′ to 5′ orientation, at the bottom of the figure. mRNAs are translated into proteins (d), while the vRNA(−) is also a template for cRNA(+), which in turn is a template for vRNA(−) genomes during replication (e). New vRNA(−) genomes will be incorporated into new virions during viral assembly (f). Following translation (d), various viral proteins function in interferon (IFN) signalling pathways (g), and the precursor fusion protein (F0) will be endocytosed and matured (F1/2) (h). Assembly (f) and budding (i) are orchestrated primarily by the M (matrix) protein, and N, P, C, M, F (fusion) and G (attachment) proteins are incorporated into virions.
In general, after virus binding to the host cell receptor, paramyxoviruses require the cooperation of their separate attachment and fusion transmembrane glycoproteins (reviewed in Refs Reference Dutch31, Reference Lamb and Jardetzky32, Reference Lamb, Paterson and Jardetzky33, Reference Smith34). However, how the attachment glycoprotein activates the fusion protein, or how the fusion protein senses that it is the right time and place for carrying out its host–virus membrane fusion function, is still a matter of intense investigation. The regulation of the molecular choreography that leads to productive membrane fusion provides a particularly fertile area for the development of therapeutics that can thwart this process.
Molecular mechanisms and antiviral strategies targeting the attachment glycoprotein
The paramyxovirus attachment proteins are type II transmembrane proteins on the surface of virions that mediate attachment of the virus to the cell-surface receptor. This interaction between the viral attachment protein and the host receptor has an important role in determining cell tropism. There are several conserved features among all known paramyxovirus attachment proteins (G, H or HN). They contain a head domain linked to the viral membrane by a stalk domain, and a cytoplasmic tail that is intraviral, or intracellular when the proteins are expressed at the cell surface (Fig. 3). The globular head of HeV-G and NiV-G (HNV-G) has a six-bladed β-propeller structure common to the head domains of multiple paramyxovirus attachment proteins (Refs Reference Bowden37, Reference Xu38). The oligomeric structure of HNV-G (dimers of dimers) (Ref. Reference Bishop39) is also thought to resemble that of the attachment glycoproteins of other Paramyxovirinae (Refs Reference Lamb, Paterson and Jardetzky33, Reference Bowden40), and it is likely that a finely balanced stoichiometry is required for optimal fusion because endogenous lectins such as galectin-1 (see below) that cause inappropriate oligomerisation of henipavirus envelope proteins can be detrimental to the fusion process (Ref. Reference Levroney41).

Figure 3. Henipavirus membrane fusion and viral entry. The attachment and membrane fusion steps necessary for viral entry [steps (a) and (b) from Fig. 2] are depicted here in greater detail in three major stages. (a–c) The fusion protein F is depicted in its pre-fusion, pre-hairpin intermediate and post-fusion forms. (a) EphrinB2 or ephrinB3 binding to NiV-G initiates a conformational cascade in F. (b) After F is triggered, it forms a pre-hairpin intermediate, in which the fusion peptide (FP) is harpooned into the host cell membrane. The pre-hairpin intermediate can be captured by peptides that mimic the NiV HR1 (orange-ended cylinder) or HR2 (green-ended cylinder) regions and bind the F HR2 or HR1 regions, respectively. (c) The HR1 and HR2 regions in the pre-hairpin intermediate coalesce to form the six-helix bundle conformation, bringing the viral and cell membranes together and facilitating viral–host membrane fusion and viral entry. (d) Ribbon structure of the monomer of NiV-G (blue) head domain (pdb code 2VSM) and its interaction with its ephrinB2 receptor (red), drawn using PYMOL (http://www.pymol.org) and modelled by aligning the G–B2 monomer with each monomer of the HPIV-3 haemagglutinin–neuraminidase dimer (pdb code 1V2I) similarly to Ref. Reference Aguilar35. The second monomer is shown in grey. According to this model, the flexible region in the NiV-G ectodomain (green and orange) might interact with the same region in another monomer and might be involved in receptor-induced G-mediated NiV-F triggering (Ref. Reference Aguilar35). (e) Representation of the structure of the NiV-F protein modelled using the HPIV-3-F crystal structure (pdb code 1ztm) by the Phyre threading program, as performed in Ref. Reference Aguilar36. (f) Representation of the trimer of NiV-F monomers from (e), also modelled using the HPIV-3-F crystal structure as in Ref. Reference Aguilar36. Abbreviations: HR1, heptad repeat 1; HR2, heptad repeat 2; HPIV-3, human parainfluenza virus 3; NiV-G, Nipah virus attachment protein.
Emerging paramyxovirus receptors
The host receptors for Menangle virus, Tioman virus, HMPV, and Beilong or J viruses, which are considered as emerging paramyxoviruses with lower morbidities in humans, are unknown (reviewed in Ref. Reference Virtue, Marsh and Wang6). By contrast, the receptors for the henipaviruses were discovered in 2005 and 2006 to be ephrinB2 and ephrinB3, respectively (Refs Reference Bonaparte42, Reference Negrete43, Reference Negrete44). These transmembrane proteins are receptor tyrosine kinases that interact with their endogenous receptors on opposing cells and have critical roles in cell–cell signalling, particularly during angiogenic and neuronal development (Ref. Reference Pasquale45). The distribution of ephrinB2 and ephrinB3 is consistent with the respiratory and neurological symptoms of henipavirus infections, because ephrinB2 and ephrinB3 are highly expressed in endothelial cells that line the microvasculature and in neurons (Refs Reference Bonaparte42, Reference Negrete43, Reference Negrete44). In the central nervous system, ephrinB3 but not ephrinB2 is expressed in the brain stem, and ephrinB3-mediated entry might account for the brain stem dysfunction that is the ultimate cause of death from NiV encephalitis (Refs Reference Negrete44, Reference Goh46). The identification of NiV and HeV receptors greatly facilitates the rational development of strategies and therapeutics that block virus–receptor binding.
Mechanisms of fusion triggering by the attachment protein
With very few exceptions, the attachment protein of paramyxoviruses is essential for viral entry (Fig. 3). Even for HRSV, whose attachment protein is not required for membrane fusion, fusion is enhanced in the presence of the attachment glycoprotein. Interestingly, HMPV membrane fusion, and sometimes replication, is not enhanced by the presence of the attachment protein (reviewed in Refs Reference Dutch31, Reference Lamb, Paterson and Jardetzky33). Thus, the specific role of the attachment protein in promoting viral entry is a subject of intense study (reviewed in Refs Reference Lamb and Jardetzky32, Reference Lamb, Paterson and Jardetzky33, Reference White47).
Several studies in various paramyxoviruses implicate the attachment glycoprotein stalk domain in interaction with and triggering of the fusion glycoprotein, which is the ultimate protein that mediates membrane fusion (Refs Reference Aguilar35, Reference Deng48, Reference Lee49, Reference Melanson and Iorio50, Reference Paal51, Reference Tanabayashi and Compans52, Reference Tsurudome53). Biochemical and biophysical studies suggest that a receptor-induced conformational change in NiV-G, which involves crucial residues at the base of the NiV-G head domain and the presence of an intact stalk domain, is important for allosteric triggering of the fusion protein (Ref. Reference Aguilar35). Although no dramatic differences were found between the apo- and receptor-bound structures of NiV-G (Refs Reference Xu38, Reference Bowden54), the stalk domain was not apparent in any of these structures. Perhaps the presence of the stalk allows for proper disassembly of higher-ordered oligomers on receptor binding, which might lead to the exposure of neo-epitopes that functionally trigger the fusion protein. Although the specifics of how HNV-G triggers its own fusion protein are beyond the scope of this review, it is likely that this triggering process is finely tuned (Ref. Reference Aguilar35) and therefore vulnerable to disruption. A better understanding of this triggering process might lead to therapeutics that target conserved features, which might limit the development of resistance. For example, anti-HNV-G antibodies that recognise conserved neo-epitopes exposed after receptor binding might be good candidates for passive immunisation strategies (Ref. Reference Aguilar35).
Antiviral strategies that target the attachment protein
There have been several monoclonal antibodies (mAbs) produced against NiV-G and HeV-G, with a range of in vitro neutralisation activities (IC50 of ~40–600 ng/ml) (Refs Reference Aguilar35, Reference Bossart55, Reference Zhu56, Reference Zhu57). One of these human mAbs (m102.4), which engages the receptor-binding site in NiV- or HeV-G, appears to be protective in a lethal challenge ferret model when administered intravenously 10 h post-infection but not 24 h pre-infection (Ref. Reference Bossart55). This difference could be due to the relatively low serum stability of m102.4 when administered intravenously, but nevertheless bodes well for the development of m102.4 as a post-exposure therapeutic in resource-sufficient settings. In comparison, palivizumab (Synagis®, MedImmune Inc.), a mAb therapeutic approved by the US Food and Drug Administration (FDA) that targets the fusion protein of HRSV, has an in vitro IC50 of 363.7 ng/ml (Ref. Reference Wu58) and monthly administration (for HRSV prophylaxis) by intramuscular injections can maintain serum concentrations of 100-fold (>40 µg/ml) above its in vitro IC50 in most patients (Ref. Reference Fenton, Scott and Plosker59). It would be interesting to see whether intramuscular injection will increase the effective half-life of m102.4 in vivo.
Soluble ephrinB2 or ephrinB3, or soluble HNV-G proteins, have also been shown to block virus entry and cell–cell fusion (Refs Reference Bonaparte42, Reference Negrete43, Reference Negrete44, Reference Bossart60), although the likely interference with ephrinB function and the antigenicity of HNV-G itself limits the practical utility of these molecules as antivirals. However, the structure of the ephrinB2- or ephrinB3-bound HNV-G complex shows a large protein–protein interface and also a lock-and-key binding pocket that might be targeted by small-molecule therapeutics (Refs Reference Bowden37, Reference Xu38). For example, Trp125 and Phe120 in the G–H loop of ephrinB2 interact differently with ephrinB4 than with HNV-G, suggesting a ‘druggable’ pocket to disrupt B2/B3–G interactions specifically (Ref. Reference Lee, Ataman and Jin61). A likely caveat to this approach is that a small molecule designed to fit the B2/B3–G binding pocket specifically might still not be able to overcome the strong avidity of oligomeric B2/B3–G interactions. For example, ephrinB2 binds to NiV-G with a subnanomolar affinity (K d ~0.06 nm) (Ref. Reference Negrete44), suggesting that a drug would have to bind at picomolar concentrations or have a very slow off-rate to compete with B2–G interactions.
Molecular mechanisms and antiviral strategies targeting the fusion glycoprotein
The fusion (F) glycoproteins are synthesised as type I transmembrane trimeric precursors that are activated by protease cleavage into a metastable pre-fusion conformation, poised for enabling membrane fusion (Fig. 3). Cleavage generates a new hydrophobic N-terminus, the fusion peptide, which is buried in the metastable pre-fusion F conformation. On attachment-protein–receptor binding, the fusion cascade is triggered and the fusion peptide is harpooned into the target cell membrane in the pre-hairpin intermediate conformation (Fig. 3b). Two helical regions present in the pre-hairpin intermediate, HR1 and HR2, have high affinities for each other and coalesce to form the six-helix bundle (6HB), which brings the viral and target cell membranes together in close apposition, allowing virus–target-cell membrane fusion and viral entry.
Maturation of the fusion protein
Important differences in viral entry and membrane fusion mechanisms carried out by the F protein have been highlighted for the emerging paramyxoviruses (Refs Reference Eaton28, Reference Dutch31, Reference Smith34). First, although many paramyxoviral F proteins are cleaved (once or twice) by furin-like cellular proteases during transport through the trans-Golgi network (Refs Reference Begona Ruiz-Arguello62, Reference Garten63, Reference Gonzalez-Reyes64, Reference Ortmann65, Reference Watanabe66), HMPV and Sendai virus F proteins are cleaved by tissue-specific extracellular proteases such as mini-plasmin or tryptase Clara (Refs Reference Murakami67, Reference van den Hoogen68), and cell-surface henipavirus F is cleaved by cathepsin L on endocytosis (Refs Reference Diederich69, Reference Diederich, Thiel and Maisner70, Reference Pager71, Reference Pager and Dutch72). Specific inhibition of these proteases by antiviral compounds could be envisioned. For example, the lack of an acutely lethal phenotype in cathepsin-L-knockout mice suggests that short-term inhibition of cathepsin L in the context of a highly pathogenic virus infection might be a clinically viable option. Recently, a small-molecule oxocarbazate-specific inhibitor of cathepsin L was reported to be effective against Ebola and severe acute respiratory syndrome (SARS) viruses at subnanomolar concentrations in vitro (Ref. Reference Shah73). Although Ebola and SARS viruses directly require cathepsin L cleavage during viral entry, this compound could also prove useful in treating henipavirus infections by preventing the generation of mature F protein. However, past in vitro versus in vivo discrepancies between drugs that indirectly inhibit cathepsin L cleavage have been observed. Chloroquine, normally used to treat malaria, has been shown to inhibit pseudotyped NiV entry, presumably by inhibiting endosomal acidification and indirectly cathepsin L activity (Ref. Reference Porotto74). However, chloroquine treatment did not prevent NiV infection or disease in ferrets (Ref. Reference Pallister75), and combined chloroquine and ribavirin treatments did not prevent death in a hamster model of NiV and HeV infection (Ref. Reference Freiberg76). These in vitro versus in vivo discrepancies suggest that we need to improve our understanding of the role of endocytosis and cathepsin L cleavage in henipavirus infection.
N-glycans in henipavirus fusion protein, and galectin-1
Another characteristic of emerging paramyxoviral F proteins is their atypical use of N-glycans. For most paramyxovirus F proteins, specific N-glycans are necessary for proper protein folding and N-glycan removal is deleterious to the fusion process (Refs Reference Bagai and Lamb77, Reference von Messling and Cattaneo78). Surprisingly, removal of specific individual or multiple N-glycans from NiV- and HeV-F resulted in marked hyperfusogenicity manifested in fusion and viral entry assays (Refs Reference Aguilar36, Reference Carter79). However, N-glycan removal also increased the sensitivity of NiV-F to antibody neutralisation; it thus seems that N-glycans in henipavirus F are kept (at least partially) to serve as a shield against antibody neutralisation (Ref. Reference Aguilar36).
NiV-F N-glycans were also found to mediate binding to galectin-1, an innate immune lectin with many functions that binds to specific galactose-containing carbohydrates on the surface of mammalian cells or pathogens (reviewed in Ref. Reference Camby80). Galectin-1 inhibits NiV-mediated cell–cell fusion and syncytia formation, a hallmark of NiV pathogenicity (Ref. Reference Levroney41). Interestingly, the individual N-glycan in NiV-F (F3) whose removal resulted in the highest level of hyperfusogenicity also gave rise to the most optimal N-glycan moiety that mediates galectin-1 binding to NiV-F. Endogenous levels of galectin-1 in endothelial cells were sufficient to inhibit NiV-envelope-mediated syncytia, and galectin-1 binding to the F3 N-glycan in NiV-F inhibited maturation, mobility and triggering of the F protein (Ref. Reference Garner81). Although it is unlikely that galectin-1 can be developed as an antiviral therapeutic because of its pleiotropic effects, these reports shed light on the innate immune defences based on recognition of pathogen-associated molecular patterns. Furthermore, 14 single-nucleotide polymorphisms have been identified in the genomic locus of galectin-1 (Ref. Reference Iida82), which raises the intriguing possibility that genetic variability at this locus might contribute to the range in pathophysiology seen in henipavirus infections.
Blocking the membrane fusion cascade
Blocking viral entry by trapping one of the fusion protein intermediates during the membrane fusion cascade is a therapeutic approach that has been pursued and used for class I fusion protein enveloped viruses. For example, enfuvirtide, sifuvirtide and their analogues are peptides that mimic the C-terminal heptad-repeat region (HR2) of class I fusion proteins, and are approved for HIV-1 treatment (reviewed in Refs Reference He83, Reference Makinson and Reynes84, Reference Poveda, Briz and Soriano85). Because paramyxoviral F proteins undergo equivalent class I fusion protein conformational changes, including pre-hairpin intermediate formation (Refs Reference Eaton28, Reference Dutch31, Reference Lamb, Paterson and Jardetzky33, Reference Smith34, Reference Xu86), the paramyxovirus HR2 (also known as HRC) peptide has been used to trap the pre-hairpin intermediate (Refs Reference Aguilar35, Reference Aguilar36, Reference Aguilar87–Reference Russell, Jardetzky and Lamb93) (Fig. 3b). Although a peptide mimicking the N-terminal HR1 also inhibits fusion, it is generally a less efficient inhibitor (Ref. Reference Bossart89), even when artificially trimerised to mimic the trimeric HR1 core (Ref. Reference Aguilar35).
HR2 peptides
For the henipaviruses, the HR2 peptide has been shown to inhibit cell–cell membrane fusion and viral entry in a pseudotyped viral system at nanomolar concentrations (Refs Reference Aguilar36, Reference Aguilar88, Reference Bossart89, Reference Porotto91). Surprisingly, higher levels of inhibition of HeV fusion were observed when using a human parainfluenza virus 3 (HPIV-3)-F- versus a HeV-F-derived HR2 peptide, although the mechanism for this phenomenon is unclear (Ref. Reference Porotto92). Additionally, a second generation of capped and PEGylated HR2 peptides resulted in increased solubility in water, stability, synthesis yields and possibilities for their use as antiviral agents in vivo (Ref. Reference Bossart89). Another strategy for increasing HR2 peptide inhibition efficacy has been the addition of cholesterol to the peptide C-terminus. This approach probably brings the peptide into close proximity to the membrane site of action where fusion occurs, reducing the IC50 of HPIV-3-derived peptides on pseudotyped HeV and NiV infections from 10–100 nm to near 1 nm (Ref. Reference Porotto94). However, the IC50 values for inhibition of live HeV and NiV viruses in vitro were close to 100 nm, and relatively large amounts of HR2–cholesterol peptides (2 mg/kg) were needed to achieve ≤60% survival of hamsters infected with NiV, when used simultaneously to or before NiV infection. It is likely that large HR2 peptide amounts are needed in order to efficiently ‘coat’ the surfaces of target cells in the host (Ref. Reference Porotto95).
Anti-F mAbs
Another approach to inhibiting membrane fusion is the blocking of the fusion protein conformational changes required for the fusion cascade by the use of mAbs. Two anti-NiV-F antibodies have been reported to neutralise NiV and HeV in vitro (1.6–20 ng) and in a hamster model (180–520 µg/animal) (Ref. Reference Guillaume96). Although the binding epitopes of these antibodies have not been characterised, their cross-reactivity suggests they might target a conserved region in HNV-F, which might limit the generation of escape variants. Moreover, antibodies that bind conformational epitopes critical for membrane fusion are highly desirable, because mutations that annul both mAb binding and the need of conformational changes would be relatively rare. Conformational mAbs against the henipaviruses that preferably bind hyper- or hypo-fusogenic mutants have been reported, but their neutralisation activities or their binding epitopes have not been shown (Ref. Reference Aguilar88).
Small-molecule inhibitors
Quinolone derivatives designed based on structure similarities among paramyxovirus F proteins in their HR1/HR2-binding motifs were tested for inhibition of NiV- and measles-virus-induced cell fusion. Two of 18 compounds tested were moderately active as inhibitors of NiV-induced cell–cell fusion and NiV-infection-induced syncytia at an EC50 of 1–3 µm. These compounds also showed some cytotoxicity in Vero cells [CC50 of 10 to >20 µm using the MTT (cytotoxity) test], resulting in a selectivity index (SI; CC50/IC50) of ~13 for the compound with the lowest toxicity (Ref. Reference Niedermeier97). This SI is relatively poor for a lead compound but might be improved by further structure–activity relation analysis. Mutants that cause resistance to HR2 peptide binding have been detected for HIV (Refs Reference He83, Reference Makinson and Reynes84, Reference Poveda, Briz and Soriano85), and similar mutants might occur after the use of these small-molecule inhibitors that target HR1–HR2 interactions.
Molecular mechanisms and antiviral strategies targeting the matrix protein
Paramyxoviral matrix (M) proteins are structural proteins that directly underlie the viral envelope, and are important for the assembly and budding of viral particles (Refs Reference Harrison, Sakaguchi and Schmitt98, Reference Takimoto and Portner99). Infectious paramyxoviral particles form after all the structural viral components have assembled at selected sites on the cell membrane, and M proteins are known to organise the assembly process. The position of M proteins underneath the cellular plasma membrane allows them to interact with ribonucleoproteins [RNA genomes bound to nucleocapsid (N or NP) proteins] as well as viral glycoproteins through their cytoplasmic tails (Refs Reference Harrison, Sakaguchi and Schmitt98, Reference Takimoto and Portner99). Recently, the atomic structure of the paramyxovirus HRSV M protein was solved and shown to contain two β-sheet-rich domains, joined by a short unstructured linker (Ref. Reference Money100). This structure is similar to that of the filovirus Ebola M (Ref. Reference Dessen101). The joined domains share an extensive positively charged surface, which probably binds to the negatively charged membrane phospholipid head groups (Ref. Reference Money100). For many paramyxoviruses, transient expression of M proteins alone, without the expression of other viral proteins, is sufficient to form and release viral-like particles (VLPs); this is the case for HPIV-1 (Ref. Reference Coronel102), Sendai virus (Ref. Reference Sugahara103), Newcastle disease virus (Ref. Reference Pantua104), measles virus (Refs Reference Pohl105, Reference Runkler106) and NiV (Refs Reference Ciancanelli and Basler107, Reference Patch108). However, in some cases, M-dependent VLP production is enhanced in the presence of other viral proteins, such as the glycoproteins, the nucleocapsid protein or the C protein (reviewed in Ref. Reference Harrison, Sakaguchi and Schmitt98).
Antivirals against M
Because the M protein is crucial in paramyxoviral assembly and budding, antiviral agents that target important aspects of M-directed assembly and budding can be envisioned. For example, inhibition of Newcastle disease virus replication by targeting two distinct sites of the M gene using interfering RNA has been recently reported (Ref. Reference Yin109). In addition, for simian virus 5, proteasome inhibitors and expression of dominant-negative VPS4(E228Q) ATPase blocked budding, probably because of the involvement of the ubiquitin–proteasome pathway in budding (Ref. Reference Schmitt110). For NiV, a recent study showed that ubiquitin-regulated nuclear–cytoplasmic trafficking of NiV-M is important for viral budding (Ref. Reference Wang111). Therefore, compounds that block M-ubiquitinating enzymes by depleting free ubiquitin in the cell (proteasome inhibitors), or that preferentially block nuclear import or export of NiV-M, could be potential antihenipavirus candidates (Fig. 2). Indeed, bortezomib, an FDA-approved proteasome inhibitor used for treating multiple myeloma, reduced viral titres significantly at an IC50 of 2.7 nm, 100-fold less than the achievable plasma concentration in humans (Ref. Reference Wang111). Thus, this FDA-approved agent has the potential for being evaluated as an off-label use for henipavirus treatment. Understanding the cellular components that have important roles in viral assembly and release should also aid the discovery of novel drugs to target these steps of the life cycle of emerging paramyxoviruses.
Molecular mechanisms and antiviral strategies targeting the P, V and C proteins
IFNs are part of the innate immune system and constitute one of the first lines of defence against viral pathogens in mammals (Ref. Reference Weber, Kochs and Haller112) in the early virus–host battle that determines the establishment of an infection (Ref. Reference Yokota, Okabayashi and Fujii113). The P gene encodes for the P, C, V and W proteins, and in the subfamily Paramyxovirinae the P gene products generally have anti-IFN activities (see Ref. Reference Eaton28). In part, P gene antiviral activities are due to their effects in limiting the extent of viral genome replication, because aberrant transcripts activate the retinoic acid inducible gene I (RIG-I; DDX58) RNA helicase pathway, which activates IFN production (Ref. Reference Magoffin, Mackenzie and Wang114). For example, the simian virus 5 P protein (Ref. Reference Dillon and Parks115), Sendai C protein (Ref. Reference Cadd116), measles C protein (Ref. Reference Reutter117), J virus and Beilong virus C proteins (Ref. Reference Magoffin, Mackenzie and Wang114), HPIV-3 C protein (Ref. Reference Magoffin, Mackenzie and Wang114), and henipavirus C, V and W proteins (Ref. Reference Sleeman118) have all been shown to inhibit viral genome replication. Recently, a study in golden hamsters showed that V and C proteins play key roles in NiV pathogenicity (Ref. Reference Dutch151). In addition, all the henipavirus P gene proteins have been shown to inhibit IFN signalling pathways (reviewed in Refs Reference Fontana, Bankamp and Rota119, Reference Ramachandran and Horvath120).
Because restoring IFN responses has been successful in the treatment of cancer, autoimmune and infectious diseases (Refs Reference Foster and Mathurin121, Reference Maher122, Reference Pfeffer123), this type of approach might also be suitable against emerging paramyxovirus infections. One study showed that the IFN inducer poly(I)–poly(C12U) (Ampligen®, a mismatched double-stranded RNA) prevented death from NiV infection in a hamster model (Ref. Reference Georges-Courbot124). Ampligen was also observed to be effective against SARS-coronavirus infection in a mouse model (Ref. Reference Barnard125), and has shown positive effects in HIV-infected patients (Ref. Reference Thompson126). Congruent with these studies is the finding that NiV and HeV replicate more efficiently in Vero cells, which are defective in IFN responses, compared with other cell lines (Ref. Reference Aljofan127). Therefore, stimulation of IFN production seems to be a promising treatment for henipavirus infections.
Broad-spectrum and other antiviral strategies
Most current antiviral drugs target differences between viral agents and hosts, such as specific viral protein moieties important for viral entry, replication, assembly, budding and so on, conferring specificity for the infected cells. However, targeting specific viral protein moieties is not always the best solution, because viral resistance by mutagenesis is very common when targeting single or even multiple viral proteins (Refs Reference Phillips128, Reference Pillay129). Thus strategies that target nonprotein determinants of important steps in the viral life cycle, particularly for a broad assortment of viruses, are highly desirable. For example, broad-spectrum compounds that target the viral membrane fluidity required for viral entry or exit, or RNA replication, have recently been explored.
LJ001, a viral membrane inhibitor
Recently, a high-throughput screening assay based on NiV/vesicular stomatitis virus (VSV)-pseudotype viral entry inhibition identified a small molecule that intercalates into and irreversibly damages viral membranes, but not cellular membranes, at low micromolar concentrations (Ref. Reference Wolf130). Studies with lipid biosynthesis inhibitors indicated that LJ001 exploits the differences between static viral membranes and biogenic cellular membranes with reparative capacity. LJ001, a rhodanine derivative, was effective against numerous enveloped viruses, but not against nonenveloped viruses, and showed no overt toxicity in vitro or in vivo, with an SI of >100. LJ001 inactivated virions while leaving envelope proteins functionally intact, inhibiting a post-binding but pre-fusion step (Ref. Reference Wolf130). Thus, LJ001 might represent a new class of broad-spectrum antivirals that target physiological rather than physical differences between viral and cellular lipid membranes. A potential mechanism of action would be disruption of the proper balance between saturated and unsaturated phospholipids that is required for the positive to negative membrane curvature transitions during the fusion process (reviewed in Ref. Reference McMahon and Gallop131). Elucidating the exact mechanism by which LJ001 damages membranes will shed light on whether differences between viral and cellular membranes can be exploited by other chemotypes, and help refine medicinal chemistry efforts to improve bioavailability and in vivo efficacy.
Cationic compounds
In another study, a high-throughput screen based on live virus infection identified three compounds unsuitable for internal administration, but possibly suitable for topical applications (Ref. Reference Aljofan132). These three compounds – gliotoxin, Gentian Violet and Brilliant Green – have been previously used as antibacterial and antifungal agents, and showed antiviral activity against NiV, HeV, VSV and HPIV-3. Additionally, gliotoxin inhibited influenza A, suggesting a broad-spectrum activity for this compound. Although the mode of action of these cationic compounds is not known, it has been proposed that they directly bind to and inhibit viral membranes (Ref. Reference Aljofan132).
Calcium influx inhibitors
In a recent study that tested licensed pharmaceuticals against henipavirus replication in vitro, calcium chelators and compounds that released intracellular calcium stores, as well as calcium channel and calmodulin antagonists, inhibited henipavirus replication at the micromolar range (Ref. Reference Aljofan133). However, the mechanism that links calcium influx to henipavirus replication is unknown, and in vivo assays have not been reported.
Ribavirin
Ribavirin is a broad-spectrum antiviral used particularly for HRSV and hepatitis C, and it is also used for RNA viruses for which there is no other available treatment (Refs Reference Olszewska and Openshaw134, Reference Snell135). It is a purine nucleoside analogue, and although its exact mechanism of inhibition of viral replication is not completely understood, it is known that ribavirin interferes with RNA metabolism, which is required for virus replication (Ref. Reference Parker136). For the emerging paramyxoviruses, various results with ribavirin have been reported. In the first NiV outbreak in Malaysia in 1998–1999, a 36% reduction in mortality in humans was reported (Ref. Reference Chong137). In addition, several studies have reported the inhibition of henipavirus replication by ribavirin in vitro (Refs Reference Porotto74, Reference Freiberg76, Reference Georges-Courbot124, Reference Aljofan138, Reference Wright, Crameri and Eaton139). However, in vivo studies carried out in animal models have not yielded promising results with ribavirin (Refs Reference Freiberg76, Reference Georges-Courbot124). The inability of ribavirin to cross the blood–brain barrier might account for its inadequacy in in vivo studies. It has been previously shown that ribavirin is effective in the brain only when administered intracranially (not by intraperitoneal injection) in a hamster model (Ref. Reference Honda140). In the Malaysian epidemic, the effect of ribavirin in late-onset NiV encephalitis was not reported (Refs Reference Chong137, Reference Honda140). In addition, the complex molecular mechanisms of inhibition of viral replication by ribavirin, such as induction of error catastrophe (excessive RNA mutations) and depletion of intracellular GTP pools, might not allow the rapid design of more potent analogues (reviewed in Ref. Reference Leyssen, De Clercq and Neyts141).
Chloroquine
Chloroquine (9-aminoquinoline) is used for the treatment of pathogens that require endosome acidification, such as malaria and pH-dependent viruses. Because the henipaviruses require endosomal cleavage of their F protein, it was not surprising that chloroquine was found to be a potential inhibitor of NiV infection in vitro (Refs Reference Porotto74, Reference Pallister75, Reference Freiberg76). However, oral administration of chloroquine did not protect ferrets from lethal NiV infection (Ref. Reference Pallister75) even though effective serum chloroquine concentrations were achieved, and peritoneal administration of chloroquine alone or in combination with ribavirin did not protect hamsters from lethal NiV or HeV challenge (Refs Reference Pallister75, Reference Freiberg76). As with ribavirin, the lack of in vivo success with chloroquine might be due to its inability to cross the blood–brain barrier or inadequate tissue distribution (Ref. Reference Koreeda142), and to its effects on the immune system that might not favour the host (Ref. Reference Savarino143). In vitro versus in vivo discrepancies in choroquine treatment results have also been reported for influenza, SARS, HIV and chikungunya viruses (Ref. Reference Savarino143).
siRNA
An alternative way of inhibiting viral gene expression is by the use of small interfering RNA (siRNA) (Ref. Reference Castanotto and Rossi144). In one recent study, siRNA molecules directed against the L and N genes were tested against minigenome and live henipavirus replication in vitro (Ref. Reference Mungall145). Whereas some siRNA had effects on both minigenome and live virus replication, some had effects only on minigenome replication and some on neither. In addition, siRNA targeting more-conserved genome sequences, for instance in P, V or W, has been proposed (Ref. Reference Mungall145). Although somewhat promising, one disadvantage of this approach is the need for gene-therapy-based siRNA delivery methods, which might not be readily available.
Inhibitors of macropinocytosis
A recent report indicates that NiV can enter cells by macropinocytosis (Ref. Reference Pernet146). This type of entry pathway for NiV necessitates phosphorylation of the cytoplasmic domain of ephrinB2, after NiV-G attachment. Although it is not known whether this is a major pathway utilised for NiV entry, drugs that affect macropinocytosis, with the exception of chloroquine, affected NiV entry, but not cell–cell fusion (Ref. Reference Pernet146). Two of the strongest inhibitors of NiV entry were latrunculin A and the amiloride analogue 5-(N-ethyl-N-isopropyl)amiloride (EIPA). Although the first one is probably hazardous in vivo, EIPA is a commonly used antihypertensive agent, and can be evaluated for its in vivo efficacy in animal models of henipavirus infection.
Favipiravir (T-705)
Favipiravir is a compound with promising broad-spectrum antiviral activities. Host enzymes metabolise its precursor into a ribofuranosyltriphosphate derivative that selectively inhibits viral RNA-dependent RNA polymerases, for reasons not fully understood (reviewed in Ref. Reference Furuta147). Importantly, it does not inhibit host DNA or RNA synthesis, and is not cytotoxic to mammalian cells. In vivo experiments with T-705 against influenza virus, arenavirus, bunyaviruses, West Nile virus, yellow fever virus and foot-and-mouth disease virus have shown one or more of the following results: protection from death, reduction of viral loads and limitation of symptoms. In addition, protective effects of T-705 were observed when it was administered 1–7 days after virus inoculation (see Ref. Reference Furuta147). Although these pathogens were not paramyxoviruses, in vitro susceptibility of HRSV to T-705 has been observed (Ref. Reference Furuta148), suggesting that favipiravir might serve as an antiviral against emerging paramyxoviruses.
Future of antiviral strategies
The various antiviral strategies discussed in this review are summarised in Table 1. In general, a better understanding of the structures and functions of viral and host proteins involved in the viral life cycle (Fig. 2) will aid in the development of new antiviral therapeutics. In addition, animal model experiments that examine the potential antivirals arising from the in vitro studies described above are important – for example, because not all compounds can successfully cross the blood–brain barrier. Because the emerging virus entry mechanisms have been explored in greater detail than the assembly and budding mechanisms, further progress in the elucidation of these late (and other) steps of the viral life cycle is imperative. Prompt antiviral discovery and characterisation against emerging paramyxoviruses should be facilitated by the use of pseudotyped and reverse genetics viral systems.
Table 1. Effect of antiviral agents on emerging paramyxovirus infections

a100% protection in vivo at 100–112 μg.
bHuman mAb m102.4: protection of 1/3 pre-infused and 3/3 post-infused ferrets at a dose of 50 mg.
cProtection of 5/6 animals, at a dose of 3 mg/kg once a day.
dSurvival increased by 1–3 days, at a dose of 25–100 mg/kg.
eNo protection at 50–150 mg/kg.
Abbreviations: EIPA, 5-(N-ethyl-N-isopropyl)-amiloride; HeV, Hendra virus; HR2, heptad repeat 2; IFN, interferon; mAb, monoclonal antibody; NiV, Nipah virus; PEG, polyethylene glycol; HRSV, human respiratory syncytial virus; siRNA, small interfering RNA.
Acknowledgements and funding
This work was supported by grants AI060694, AI069317, AI065359 (Pacific Southwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases), AI082100 and AI07495 from the US National Institutes of Health. We apologise to investigators whose work was not discussed because of space limitations. We thank Frederic Vigant and Arnold Park for their assistance with figures and editorial comments, respectively. We also sincerely thank our peer reviewers for their valuable comments and suggestions.




