



Mate is a traditional drink obtained from the leaves of yerba mate (Ilex paraguariensis A. St.-Hil.), a native plant from South America. Drinking mate is a widespread social practice in Paraguay, Argentina and parts of Brazil, Uruguay, Chile and Bolivia for which different consumption patterns may exist(Reference Cardozo Junior and Morand1). Traditionally, yerba mate leaves and stems are dried, grounded and steeped in hot water to make an herbal tea also called chimarrão. However, some variations in the process and water temperature lead to terere, which is a cold-water beverage, whereas cha mate obtained from the roasted leaves is consumed as a tea substitute or as an instant cold drink. These last years, there is a growing interest for mate in the European and USA markets, mainly due to its richness in a diversity of plant bioactive compounds including polyphenols, methylxanthines and saponins which encourages to consider yerba mate as a functional food(Reference Cardozo Junior and Morand1). In mate, methylxanthines are mainly present as caffeine, saponins as glycosidic derivatives of ursolic and oleanolic acids; however, the major phytochemicals are phenolic acids(Reference Borré, Kaiser and Pavei2,Reference Bravo, Goya and Lecumberri3) . In this respect, the particularity of mate is to be, with coffee, a food source very rich in a diversity of chlorogenic acids which can constitute major contributors to the daily polyphenol intake in heavy consumers(Reference Gebara, Gasparotto-Junior and Santiago4). The composition in chlorogenic acids in mate differentiate from that of coffee by the presence of dicaffeoyl quinic acids that may represent up to half of total chlorogenic acids(Reference Gebara, Gasparotto Junior and Palozi5). In addition to mono chlorogenic acids and dicaffeoyl quinic acids that both represent more than 90 % of total polyphenols, mate also contains some flavonol glycosides(Reference Bravo, Goya and Lecumberri3). A recent study has assessed the bioavailability of yerba mate polyphenols in humans and showed that they are poorly absorbed in the small intestine and extensively metabolised by the gut microbiota prior to their absorption(Reference Gómez-Juaristi, Martínez-López and Sarria6). Hence, the circulating metabolites include some phase 2 derivatives of hydroxycinnamic acids and mainly reduced forms of hydroxycinnamic acids resulting from microbial conversion and their conjugates.
Mate is still little explored compared with other polyphenol-rich beverages like tea or coffee; however, as recently reviewed a growing body of evidence from in vitro and animal studies put forward a diversity of biological effects suggesting a potential interest of mate consumption to prevent cardiometabolic diseases(Reference Cardozo Junior and Morand1,Reference Gan, Zhang and Wang7) . In particular, supplementation studies with mate in diet-induced dysmetabolic murine models have reported anti-inflammatory(Reference Pimentel, Lira and Rosa8), antioxidant and anti-atherogenic(Reference Bravo, Mateos and Sarriá9) effects and also acknowledged the capacity of mate extract to improve blood lipids(Reference Balzan, Hernandes and Reichert10), insulin sensitivity(Reference Hussein, Matsuda and Nakamura11) and endothelial function(Reference Gao, Liu and Qu12). Despite these promising preclinical results, to date clinical evidence is lacking because only few randomised controlled trials with mate have been conducted so far. From these studies, a chronic intake of yerba mate has been shown to improve microcirculation in volunteers with high blood viscosity(Reference Yu, Yue and Liu13), to decrease mass and percentage of body fat in obese subjects(Reference Kim, Oh and Kim14) or to increase the antioxidant capacity in overweight and dyslipidaemic subjects through the elevation of serum levels of paraoxonase-1(Reference Balsan, Pellanda and Sausen15). In men at moderate cardiovascular risk, the sub-chronic consumption of a standardised mate extract tended to improve some cardiometabolic biomarkers such as fasting glucose, HDL-cholesterol or inflammatory markers but significance was not reached(Reference Gebara, Gasparotto Junior and Palozi5).
Molecular mechanisms underlying beneficial effects of mate consumption on cardiometabolic health have been studied mainly on rodent models of obesity-induced metabolic dysregulation, primarily on metabolically active tissues such as liver, adipose tissue and skeletal muscle. In the liver, regular intake of mate modulates NF-kappaB pathway(Reference de Meneses Fujii, Jacob and Yamada16) and PI3K-Akt pathway(Reference Arçari, Santos and Gambero17) but also the expression of genes involved in lipid oxidation and lipogenesis(Reference Gao, Long and Jiang18). Moreover, beneficial effects of regular mate consumption on vascular endothelial function have been attributed in part to the upregulated mRNA expression of hepatic low-density lipoprotein receptor and scavenger receptor B1(Reference Gao, Liu and Qu12). In the white adipose tissue, mate acts through an enhanced expression of uncoupling proteins and increased fatty acid oxidation via AMPK phosphorylation(Reference Pang, Choi and Park19,Reference Choi, Park and Kim20) and decreases the expression of the genes related to chronic low-grade inflammation(Reference Arçari, Bartchewsky and dos Santos21).
Due to the richness of mate in polyphenols, many of its beneficial health effects have been attributed to these compounds(Reference Heck and de Mejia22). Systematic analyses of published nutrigenomic data have identified the key cellular processes and pathways modulated by polyphenols in cellular and animal models of cardiometabolic disease(Reference Ruskovska, Massaro and Carluccio23,Reference Monfoulet, Ruskovska and Ajdžanović24) , and in humans(Reference Ruskovska, Budić-Leto and Corral-Jara25). These include cellular processes such as inflammation, lipid metabolism, and endothelial function and pathways such as MAPK signalling pathway, TNF signalling pathway, PI3K-Akt signalling pathway, focal adhesion or PPAR signalling pathway. These effects derive not only from the modulation of expression of mRNA and miRNA but also probably from the modulation of lncRNA that may be associated with the initiation, development and progression of cardiometabolic diseases(Reference Jiang, Sun-Waterhouse and Chen26).
The majority of the studies that explore nutrigenomic effects of polyphenols and/or polyphenol-rich foods focus on several key genes that are relevant to the health effects of interest. As such, these studies give only a fragmented picture of the genomic impacts of polyphenols, and therefore do not allow a comprehensive understanding of the underlying molecular mechanisms of action. In contrast, studies that apply a holistic approach, namely an analysis of global gene expression using array or RNAseq technologies, provide a global picture of nutrigenomic effects, but their number is still very limited. In particular, the implementation of such a holistic approach has the advantage to allow the analysis not only of protein coding genes but also protein non-coding RNA, such as miRNA and lncRNA, which are known to contribute to the global genomic effects. Therefore, the aim of this study was to explore global genomic modulations in the PBMC of men predisposed to cardiovascular risk and consuming a standardised mate extract. By applying a comprehensive bioinformatic analysis of obtained nutrigenomic data, we aimed to reveal the key biological processes and cellular functions modulated by a regular intake of a polyphenol-rich mate extract and to examine their links with cardiometabolic risk factors.
Materials and methods
Subjects
Thirty-five male volunteers, aged 45–65 years, were recruited for the study as previously reported(Reference Gebara, Gasparotto Junior and Palozi5). All subjects had no more than one of the five criteria associated with metabolic syndrome proposed by the National Cholesterol Education Program’s Adult Treatment Panel III (NCEP-ATP III) and approved by Brazilian scientific societies in the First Brazilian Guideline for Diagnosis and Treatment of Metabolic Syndrome (2005)(Reference Lipsy27,28) . For men, these criteria include waist circumference above 40 inches, blood pressure above 130/85 mmHg, fasting TAG level above 1·70 mmol/l, fasting HDL-cholesterol level less than 1·04 mmol/l and fasting blood sugar above 5·55 mmol/l.
Exclusion criteria included intake of antioxidants or vitamin supplements in the last 3 months prior to the study; smokers or individuals that have quit smoking for less than 3 years; chronic alcoholism; diagnosed chronic diseases (diabetes, hypercholesterolaemia, severe hypertension, mental illness and renal or hepatic disease) and intake of associated medications. This study was conducted according to the guidelines laid down in the Declaration of Helsinki, and all procedures involving human subjects/patients were approved by the Human Ethics Committee of the Paranaense University (CAAE: 22531313.5.0000.0109). All volunteers signed a Consent Form, thirty-five completed the study, but one was excluded from the analysis for not following the guidelines during the protocol. Thirty-four volunteers were evaluated, and nutrigenomic analyses were performed on a subset of the volunteers. This study was registered at www.clinicaltrials.gov under Protocol ID UNP-ILCV-1518 (https://clinicaltrials.gov/ct2/show/NCT02789722).
Description of mate extract used in the study
The volunteers were given a standardised mate extract to be consumed daily. The extract was supplied by the Brazilian company SUSTENTEC (www.sustentec.org.br) and was prepared by infusion of dry mate leaves and drying in a spray-dryer (ratio 5:1 leaves/extract). The phytochemicals content, including chlorogenic acid derivatives, caffeine, and theobromine, in the resulting dry extract was analysed and quantified by HPLC coupled to a diode detector (HPLC-DAD – Varian – PRO STAR Mod. 210) and was provided in the full clinical paper(Reference Gebara, Gasparotto Junior and Palozi5). Based on data obtained from the quantification of caffeoyl quinic acids (CQA) in the extract and considering the minimal daily dose of total CQA ingested (about 510 mg/d) through the consumption of traditional mate as reported by Gebara et al., 2017(Reference Gebara, Gasparotto-Junior and Santiago4), the daily dose of mate extract in the study was set at 2250 mg which provided 581 mg CQA. Di-caffeoylquinics accounted for about 54 % of the CQA present in mate extracts and 46 % were mono-caffeoylquinic acids(Reference Gebara, Gasparotto Junior and Palozi5). This standardised mate extract was used for encapsulation (250 mg dry extract/capsule) and to reach 581 mg CQA/d, nine capsules had to be consumed daily. For placebo, the volunteers took placebo capsule that contained 250 mg of starch only.
Intervention study
A two-arms controlled, randomised, crossover trial was conducted over 4 weeks for comparing effects with mate extract v. placebo. Two weeks of wash-out were applied between treatments. The randomisation was carried out by an independent person outside from the clinical study, who was also responsible for handling the capsules. The total duration of the study per volunteer was 12 weeks, including the inclusion phase (2 weeks), two experimental periods of 4 weeks each separated by a wash-out period of 2 weeks.
The volunteers were evaluated for inclusion, where parameters such as blood cells count, urea, creatinine, hepatic enzymes, lipid parameters, fasting blood glucose and anthropometric and haemodynamic parameters (waist circumference, BMI, pulse and blood pressure) were evaluated. During the entire study period, participants were asked to maintain their lifestyle, and their dietary habits except that they were instructed to completely refrain from consuming mate products (chimarrão, terere) and to limit their total intake of polyphenol-rich beverages (tea, coffee, cocoa, wine, soya milk and fruit juices) to 200 ml/d. Before the beginning of each treatment period, the capsules of mate or placebo were distributed to volunteers who were instructed to consume three capsules every 8 h, from morning to evening (3 × 3 caps/d, providing a total of 2250 mg of dry extract or placebo), for 28 d. The consumption of the capsules was monitored, and a daily record was provided to volunteers at the beginning of each period to report their daily intake of study products. Volunteers had to bring back the record at the end of each period. Adherence to the intervention was assessed by examining the daily records and counting the capsules not consumed.
Isolation of PBMC
Among the volunteers enrolled in the randomised controlled crossover study, twelve volunteers, selected by drawing lots at the beginning of the trial, participated in the present nutrigenomic study. This number of volunteers has been chosen regarding several previous nutrigenomic analyses in clinical trials in which the number of volunteers varied from 3 to 15(Reference Milenkovic, Vanden Berghe and Boby29,Reference Milenkovic, Deval and Dubray30) . Blood samples were collected in the morning, around 7:00 a.m., after overnight fasting at the end of each experimental period of 4 weeks of consumption of either mate extract or placebo using heparin vacutainer tubes (Becton Dickinson, Franklin Lakes, NJ, USA). The tubes were immediately centrifuged for 20 min at 1500 g at room temperature. The cell layer was collected and washed twice with sterile PBS, with centrifugation for 10 min at 300 g between each washing step. The obtained pellet of PBMC was immediately frozen and kept at −80°C until used.
Total RNA extraction
The PBMC were lysed using a lysing buffer solution from the RNeasy Micro Kit (Qiagen). Total RNA extraction was performed using the RNeasy Micro Kit as recommended by the manufacturer. RNA quality was assessed by 1 % agarose gel electrophoresis and quantity determined by measuring absorbance at 260 and 280 nm on NanoDrop ND-1000 spectrophotometer (Thermo Scientific). The total RNA was stored at −80°C until analysed.
Microarray analysis
Fifty ng of total RNA extracted were amplified and fluorescently labelled to produce Cy5 or Cy3 cRNA using the Low Input Quick Amp Labeling two colour kit (Agilent, Santa Clara, CA, USA) in the presence of spike-in two colour control, as recommended by the manufacturer. Following the purification, 825 ng of labelled cRNA were hybridised onto G4845A Human GE 4 × 44K v2 microarray (Agilent, USA) containing 27 958 Entrez Gene RNA sequences, according to the manufacturer’s instructions. Microarrays were scanned with Agilent G2505 scanner (Agilent, USA) and data extracted by Feature Extraction software (Agilent, USA) version 11.0 using linear and Lowess normalisation. Statistical analyses were performed using the free R 2.1 software (https://www.r-project.org)(Reference Dessau and Pipper31). Data were analysed by Student’s t-test to detect differentially expressed genes, and the probability values were adjusted by Benjamini-Hochberg correction for multiple testing at 0·01 to eliminate false positives.
Bioinformatics
Principal component analysis with quantile normalisation was conducted using the bioinformatic tool MetaboAnalyst, version 5.0 (https://www.metaboanalyst.ca, accessed on 14.08.2021)(Reference Xia and Wishart32). Associations between transcripts and study groups were searched using unsupervised hierarchical clustering of the samples and the differentially expressed probes. Rows were centred, and unit variance scaling was applied to rows. Both rows and columns were clustered using correlation distance and average linkage. The clustering results were illustrated as a heatmap of expression signals, which was constructed using the ClustVis tool (https://biit.cs.ut.ee/clustvis, accessed on 08.06.2021)(Reference Metsalu and Vilo33).
Gene ontology enrichment analysis was conducted using the bioinformatic tool ShinyGO, version 0.66 (http://bioinformatics.sdstate.edu/go, accessed on 14.08.2021)(Reference Ge, Jung and Yao34) and applying the following settings: P-value cut-off = 0·05; # of top pathways to show = 50; species – human. Pathway enrichment analyses were conducted using the GeneTrail 3, version 3.2 (https://genetrail.bioinf.uni-sb.de, accessed on 14.08.2021)(Reference Gerstner, Kehl and Lenhof35), as a platform to access Kyoto Encyclopedia of Genes and Genomes (KEGG) and WikiPathways databases, and applying the following settings: over-representation analysis; null hypothesis (for P-value computation) – two-sided; method to adjust P-values – Benjamini–Hochberg; significance level – 0·05; all supported genes taken as a reference.
Protein–protein interactions were analysed with the bioinformatic tool OmicsNet (https://www.omicsnet.ca, accessed on 18.08.2021)(Reference Zhou and Xia36,Reference Zhou and Xia37) that was used as a platform to access the data from the database STRING, applying the following settings: experimental evidence; confidence score – 900; minimum network.
Analysis of potential transcription factors that regulate the differentially expressed protein coding genes was conducted with the bioinformatic tool TRANSFAC and JASPAR PWMs that was accessed through the platform Enrichr (https://maayanlab.cloud/Enrichr, accessed on 14.08.2021)(Reference Chen, Tan and Kou38,Reference Kuleshov, Jones and Rouillard39) .
Potential binding interactions between top transcription factors, their regulatory cell signalling proteins and major circulatory metabolites were examined by molecular docking using the SwissDock docking analysis tool (http://www.swissdock.ch/docking, accessed on 09.10.2021)(Reference Grosdidier, Zoete and Michielin40). Protein 3D structures were obtained from UniProt Data Bank (https://www.uniprot.org, accessed on 09.10.2021)(41) and chemical structures of metabolites from PubChem database (https://pubchem.ncbi.nlm.nih.gov, accessed on 09.10.2021)(Reference Kim, Chen and Cheng42).
miRNA-target enrichment analysis and functional enrichments for target genes of individual miRNA were conducted with the bioinformatic tool Mienturnet (http://userver.bio.uniroma1.it/apps/mienturnet, accessed on 16.08.2021)(Reference Licursi, Conte and Fiscon43), using the data available from the miRTarBase, while setting the threshold for the minimum number of miRNA–target interactions to 1. Targets of lncRNA were identified using the bioinformatic tool LncRRIsearch, version 1.0 (http://rtools.cbrc.jp/LncRRIsearch/index.cgi, accessed on 16.08.2021)(Reference Fukunaga, Iwakiri and Ono44), where we searched for available transcripts, and for each transcript we used top 40 targets with the lowest value for minimum energy of binding.
All interaction networks were constructed with Cytoscape software, version 3.7.2. (https://cytoscape.org)(Reference Shannon, Markiel and Ozier45). Elements that are in common for investigated data sets were retrieved using the following softwares: Venny, version 2.1 (https://bioinfogp.cnb.csic.es/tools/venny, accessed on 19.08.2021) and InteractiVenn (http://www.interactivenn.net, accessed on 23.08.2021)(Reference Heberle, Meirelles and da Silva46). For visualisation of the multi-level multi-omic regulation of the cellular processes, we used a diagram from the KEGG database (https://www.genome.jp/kegg, accessed on 10.10.2021)(Reference Kanehisa and Goto47).
The association of polyphenol modulated genes with human diseases was analysed using the Comparative Toxicogenomics Database (https://ctdbase.org, accessed on 09.10.2021)(Reference Davis, Grondin and Johnson48). To explore correlations between polyphenol modulated genes and genes associated with CMD, we first searched the GEO (https://www.ncbi.nlm.nih.gov/gds, accessed on 09.10.2021) for suitable data sets to obtain gene expression profiles of CMD and drugs commonly used for the treatment of insulin resistance, dyslipidaemia or hypertension. The available data sets were further analysed with GEO2R. Pearson’s correlation coefficients between these gene expression profiles and polyphenol-modulated genes were calculated using the R program (https://www.r-project.org)(Reference Dessau and Pipper31).
Results
Consumption of mate extract modulates expression of genes in PBMC
Following RNA extraction, two samples had to be removed because of low quality of RNA, and following microarray hybridisation and scanning, two additional samples were removed because of low quality of microarray hybridisation. As a result, the whole nutrigenomic analysis was performed on eight samples. Characteristics at screening of the subset study population involved in the nutrigenomic study are presented in Table 1. To get a general insight into the effect of mate extract on global gene expression, we first performed principal component analysis of the microarray data using the bioinformatic tool MetaboAnalyst. These results, presented in Fig. 1(a), show a clear separation of groups (mate v. placebo), suggesting changes in gene expression profile with regular consumption of mate extract. This analysis also shows a significant effect of mate extract on the expression of analysed transcripts. Furthermore, we performed hierarchical cluster analysis of global gene expression profiles of each of the eight volunteers who consumed placebo and mate extract. The obtained heatmap is presented in Fig. 1(b). This comparison of global gene expression profiles identified two distinct groups of profiles that correspond to the profiles of volunteers that consumed mate extract and those that consumed placebo. Moreover, the hierarchical analysis suggests opposite expression profiles in the two groups suggesting again significant impact on the expression of genes in humans after the regular consumption of mate extract.
Table 1. Characteristics of the study population at screening day
(Mean values and standard deviation; range, n 8)
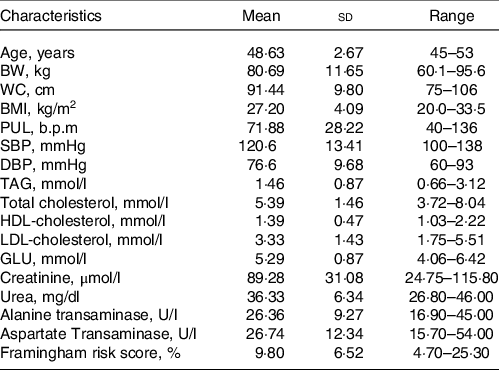
BW, body weight; WC, waist circumference; PUL, pulse; GLU, glucose.
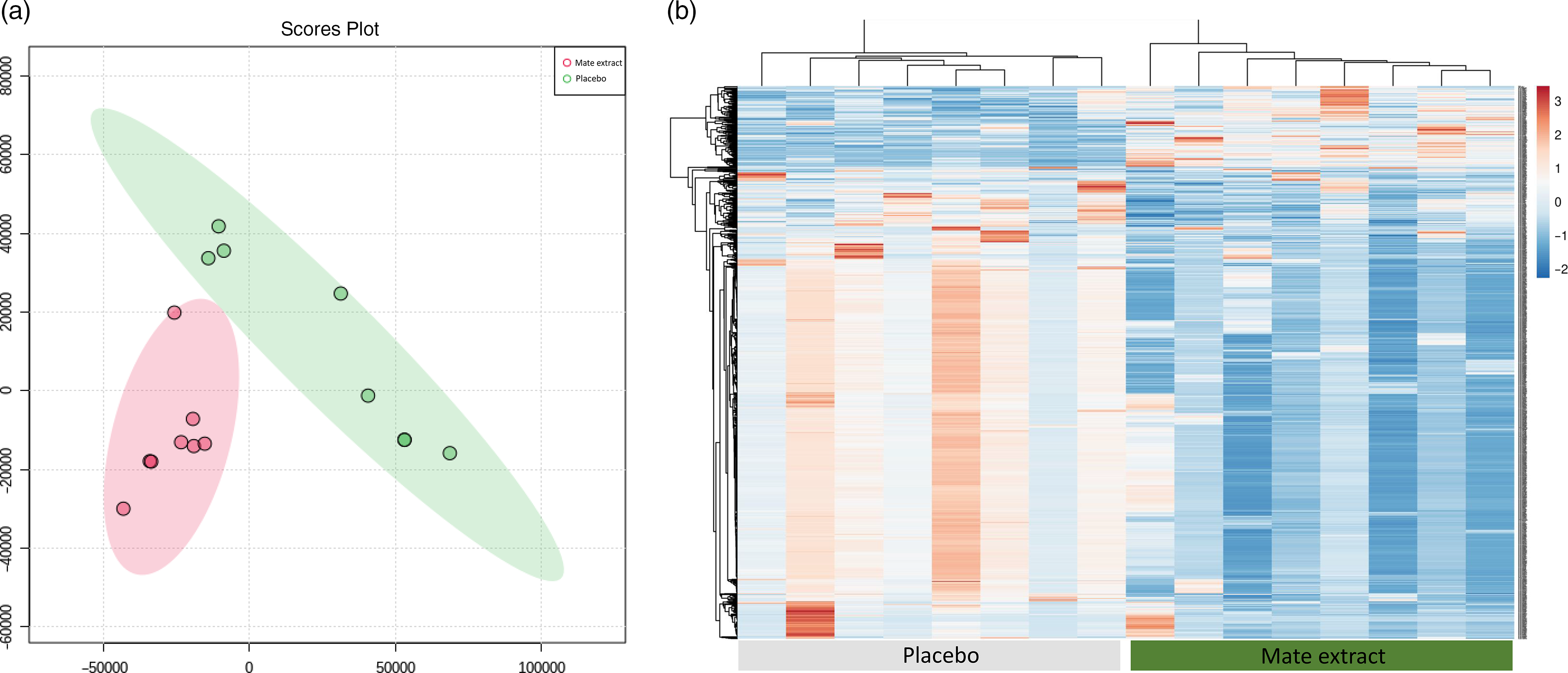
Fig. 1. Modulation of global gene expression in PBMC in men at moderate cardiovascular risk with mate extract. (a) Principal component analysis of gene expression profile with consumption of mate extract or placebo (MetaboAnalyst, with quantile normalization). (b) Heatmap obtained with hierarchical cluster analysis of global gene expression profiles of each of the eight volunteers who consumed placebo and mate extract.
Following the previous analysis suggesting changes in global gene expression profiles, we performed statistical analysis to identify genes whose expression has been significantly affected by consumption of mate extract, which revealed 2635 differentially expressed genes. The fold changes of these genes were observed to vary from −2·92 to 8·06, with variation from −2·92 to −1·06 for down-regulated genes and from 1·07 to 8·06 for up-regulated genes. A closer insight into identified differentially expressed genes showed that there are 2385 differentially expressed protein coding genes, six miRNA, and 244 lncRNA. These results suggest that the consumption of mate extract can significantly affect the expression of genes, not only protein coding but also protein non-coding genes in circulating immune cells. All categories of differentially expressed genes were further subjected to functional and integrative bioinformatic analyses.
Functional analysis of differentially expressed protein coding genes
The intake of mate extract significantly modulated the expression of n 2385 protein coding genes, 95 % of which were down-regulated. To get an insight into the cellular functions of significantly modulated protein coding genes, we first conducted gene ontology enrichment analysis using the bioinformatic tool ShinyGO. The treemap plot of significantly over-represented gene ontologies is presented in Fig. 2(a). This analysis showed that mate extract consumption affected several biological functional categories that include cell adhesion and migration, cell development and differentiation, cell signalling, as well as processes related to the nervous and circulatory system.

Fig. 2. Bioinformatic analyses of differentially expressed protein coding genes. (a) Gene ontology analysis of significantly modulated protein coding genes with consumption of mate extract. (b) Pathway enrichment analyses of protein coding genes modulated with consumption of mate extract. Top fifty pathways related to cellular processes in human immune cells, twenty five from each of the two interrogated databases (KEGG and WikiPathways). In alphabetical order, x-axis represents the number of hits. *from KEGG; **from WikiPathways. (c) Two-D Force Atlas presentation of protein–protein interactions potentially affected with consumption of mate extract, generated in OmicsNet. Proteins that have more than fifteen interactions with other proteins within the network are presented in orange colour.
To get a more precise picture of the cellular functions that are regulated by protein coding genes significantly modulated following mate extract consumption, we conducted pathway enrichment analyses. To this end, we interrogated two databases: KEGG and WikiPathways that we reached using the platform for bioinformatic analyses GeneTrail. The top fifty pathways that are related to cellular processes (top twenty five from each of the interrogated databases) are depicted in Fig. 2(b). These top pathways are involved in cell signalling (cAMP signalling pathway, PI3K-Akt signalling pathway or MAPK signalling pathway), cell motility and interaction (ECM–receptor interaction, focal adhesion, tight junction, regulation of actin cytoskeleton or Ras signalling), inflammation (cytokine–cytokine receptor interaction) or functions of the nervous system (axon guidance, glutamatergic synapse or GABAergic synapse). Interestingly, some of the top pathways are common for both KEGG and WikiPathways, such as MAPK signalling pathway, PIK3-Akt signalling pathway or Ras signalling pathway, as well as the pathways related to Ca signalling, circadian rhythm, focal adhesion, function of gamma-aminobutyric acid or vitamin A metabolism.
The next step was to investigate the potential protein–protein interactions of proteins that are coded by the genes identified as significantly modulated with mate extract. To this end, we used the bioinformatic tool OmicsNet as a platform to access the database STRING. The analysis revealed a network of interactions of identified proteins as presented in Fig. 2(c). This analysis also revealed genes that form nodes in the network that is genes having the highest number of interactions with other genes. The number of interactions reached eighty for epidermal growth factor receptor, or more than fifty for nuclear factor NF-κ-B p105 subunit. The hub proteins of the network, namely those presenting more than fifteen interactions (n 40), are presented in orange colour in Fig. 2(c) and listed in Table 2. Furthermore, using the functionality of the bioinformatic tool OmicsNet, we retrieved the specific interactions of these hub proteins. This analysis showed that approximately 1000 PPI are particularly affected with consumption of mate extract. Using the software Cytoscape, we built this sub-network centred on hub proteins and the proteins they interact with, which is presented in Supplementary Fig. S1. Pathway enrichment analyses of hub proteins conducted in GeneTrail showed that these genes are involved mainly in cell signalling (chemokine signalling pathway, cAMP signalling pathway, MAPK signalling pathway and relaxin signalling pathway), cell motility and interaction (adherens junction, gap junction, focal adhesion, Rap1 signalling pathway and Ras signalling pathway) or fluid shear stress and atherosclerosis. However, pathway enrichment analyses of hub proteins together with the proteins they interact with pinpointed focal adhesion as primarily affected pathway related to cellular processes, followed by Rap1 signalling pathway, Ras signalling pathway and several pathways involved in cell signalling, such as PI3K-Akt signalling pathway, ErbB signalling pathway, cAMP signalling pathway, MAPK signalling pathway or chemokine signalling pathway.
Table 2. Proteins with more than fifteen interactions within the network of protein–protein interactions (PPI)

Potential transcription factors and docking with known major metabolites
Next, we aimed to identify which transcription factors (TF) could have their activity affected by mate extract and be involved in the regulation of the expression of identified significantly modulated protein coding genes. To this end, we interrogated the database TRANSFAC and JASPAR PWM that we accessed through the platform Enrichr. We identified nineteen TF significantly associated with the protein coding genes modulated with regular consumption of mate extract (Table 3), the network of which is presented in Supplementary Fig. S2. The identification of potential transcription factors involved in the regulation of gene expression by mate extract suggests that circulating polyphenol metabolites that are present in the circulation after the intake of this extract could interact with transcription factors and/or cell signalling proteins regulating their activity. Therefore, we aimed to identify the capacity of major metabolites of mate phenolic acids to interact and bind to these proteins using a 3D docking tool. Based on a previous human study(Reference Gómez-Juaristi, Martínez-López and Sarria6) that has identified the phase 2 and microbial-derived metabolites present in plasma after mate intake, we have assessed the binding capacity of two major metabolites, namely caffeic acid-3-sulphate for phase 2 metabolites and dihydro-ferulic acid for microbial-derived metabolites. We observed that caffeic acid-3-sulphate (phase 2 metabolite) could interact with HNF1A (binding capacity of –7·61 kcal/mol), MYB (binding capacity of –7·15 kcal/mol) and MEF2A (binding capacity of –7·2 kcal/mol) (Fig. 3(a), Table 4), whereas dihydro-ferulic acid (gut microbiome metabolite) presents a slightly lower binding capacity to these transcription factors. We observed that this metabolite has a binding capacity of –6·88 kcal/mol with HNF1A, binding capacity of –6·9 kcal/mol with MYB and binding capacity of –6·45 kcal/mol with MEF2A.
Table 3. Transcription factors that potentially modulate differentially expressed protein coding genes with consumption of mate extract

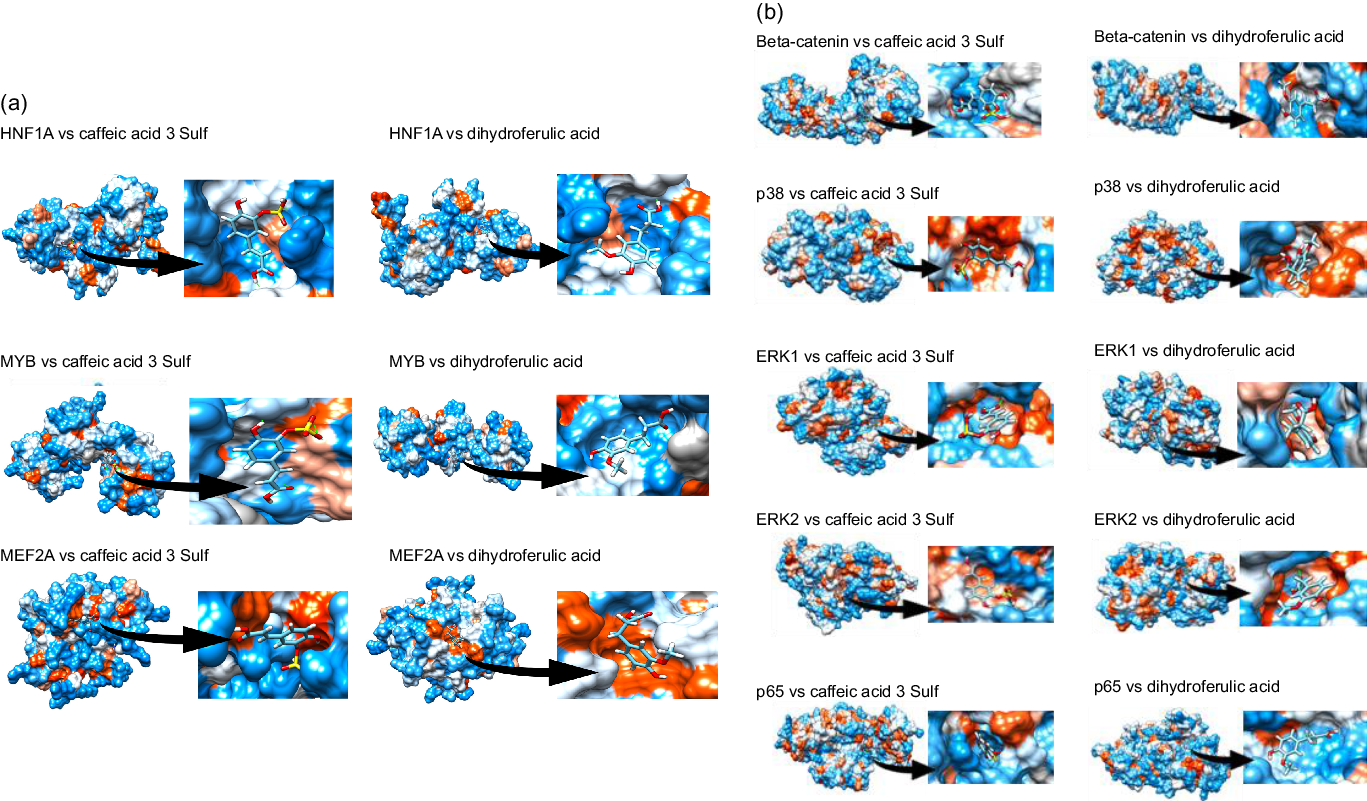
Fig. 3. Docking of major circulatory metabolites with top transcription factors (TF) (a), and cell signalling proteins regulating their activity (b).
Table 4. Binding capacity of major mate circulating metabolites with top transcription factors (TF) and cell signalling proteins

Together with transcription factors, we also aimed to identify potential binding capacity of these two major mate metabolites to cell signalling proteins regulating the activity of identified TF. In silico analysis showed that caffeic acid-3-sulphate presents a high binding capacity with β-catenin (cell signalling protein interacting with TCF4), p65 (cell signalling protein interacting with HNF1A and MYB), ERK1/2 (cell signalling proteins interacting with GATA6) but also with p38 which interacts with MEF2A (Fig. 3(b)). Regarding the gut microbiome-derived metabolite tested, dihydro-ferulic acid, its binding capacity to these cells signalling proteins varied from −6·79 to −7·02 kcal/mol (Table 4). These in silico analyses suggest that the circulating metabolites of phenolic acids present in mate extract can not only bind to transcription factors but also to cell signalling proteins. Interactions between metabolites and cell signalling proteins could induce modifications of their kinase activity, which can modulate the activity of downstream cell signalling proteins and consequently transcription factors, changes that result in the observed modulations of gene expression.
miRNA – identification of their targets and functional analyses
We observed that the regular intake of mate extract caused a significant modulation in the expression of six miRNA: hsa-miR-100-5p, hsa-miR-1-3p, hsa-miR-205-5p, hsa-miR-3654, hsa-miR-7-5p and hsa-miR-99a-5p, all of them being down-regulated. Since miRNA are small non-coding RNA with a role in post-transcriptional modulation of the expression of protein coding genes, our first analysis aimed to identify their target mRNA. To this end, we interrogated the miRTarBase that was accessed using the bioinformatic tool Mienturnet. For our set of six differentially expressed miRNA, a total number of 981 statistically significant potential target mRNA (P < 0·05) were identified. Using the software Cytoscape, the network of interactions between these miRNA and their target mRNA was built, which is depicted in Fig. 4(a). To get insight into the function of target genes of miRNA whose expression was modulated by mate extract, we conducted pathway enrichment analyses. First, interrogation of the Mienturnet database revealed significantly over-represented pathways, from both KEGG and WikiPathways databases, that are associated with each of the miRNA identified as differentially expressed by mate extract consumption (Fig. 4(b) and (c)). Among the pathways identified as potentially affected by these miRNA are cell signalling pathways (such as MAPK signalling, PI3K-Akt signalling and VEGFR signalling), or pathways regulating cellular functions such as cell adhesion (adherens junction pathway, focal adhesion), cellular senescence or cancer development. Second, we also interrogated KEGG and WikiPathways using identified potential target genes through the platform GeneTrail. Top fifty pathways, namely twenty-five top pathways from each of the interrogated databases, are depicted in Fig. 4(d). Among them, pathways, such as ErbB signalling pathway, MAPK signalling pathway, PI3K-Akt signalling pathway, Ras signalling pathway, chemokine signalling pathway, focal adhesion, regulation of actin cytoskeleton and oxidative phosphorylation are common for both KEGG and WikiPathways.
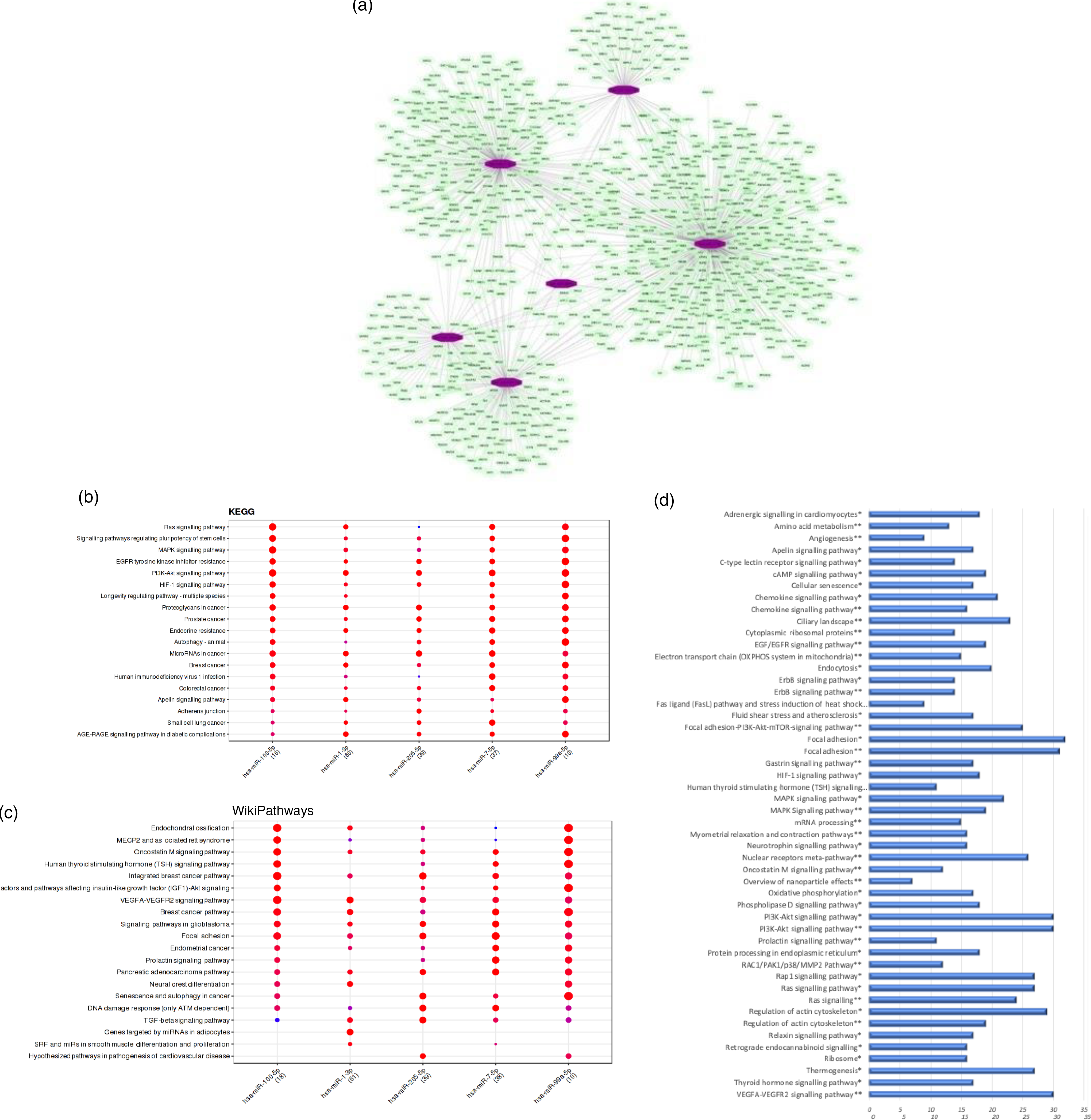
Fig. 4. Bioinformatic analyses of differentially expressed miRNA and their target mRNA. (a) Network of miRNA significantly modulated with consumption of mate extract and their target mRNA. (b) KEGG pathways associated with miRNA modulated with consumption of mate extract. (c) WikiPathways associated with miRNA modulated with consumption of mate extract. (d) Pathway enrichment analyses of target genes of miRNA modulated with consumption of mate extract. Top fifty pathways related to cellular processes in human immune cells, twenty five from each of the two interrogated databases (KEGG and WikiPathways). In alphabetical order; x-axis represents the number of hits. *from KEGG; **from WikiPathways.
Long non-coding RNA – identification of their targets and functional analysis
Mate extract also modulated the expression of 244 lncRNA, the majority of which (97 %) were down-regulated. As lncRNA target various RNA, our first goal was to identify potential targets of differentially expressed lncRNA. Using the bioinformatic tool LncRRIsearch, we were able to identify 112 available lncRNA transcripts and 803 targets of the lncRNA that are modulated by mate extract, a network of which is presented in Supplementary Fig. S3. Pathway enrichment analysis was conducted for identified target genes using GeneTrail, as a platform to interrogate KEGG and WikiPathways databases. Top fifty pathways that are related to cellular processes (top twenty five from each of the interrogated databases) are depicted in Fig. 5. Most of these pathways are involved in cell signalling, but there are also pathways involved in cell motility and interaction (tight junction, Ras signalling pathway), neurofunction (cholinergic synapse, dopaminergic synapse, GABAergic synapse, long-term depression and long-term potentiation), oxidative stress and antioxidant defense (phytochemical activity on NRF2 transcriptional activation) or apoptosis.
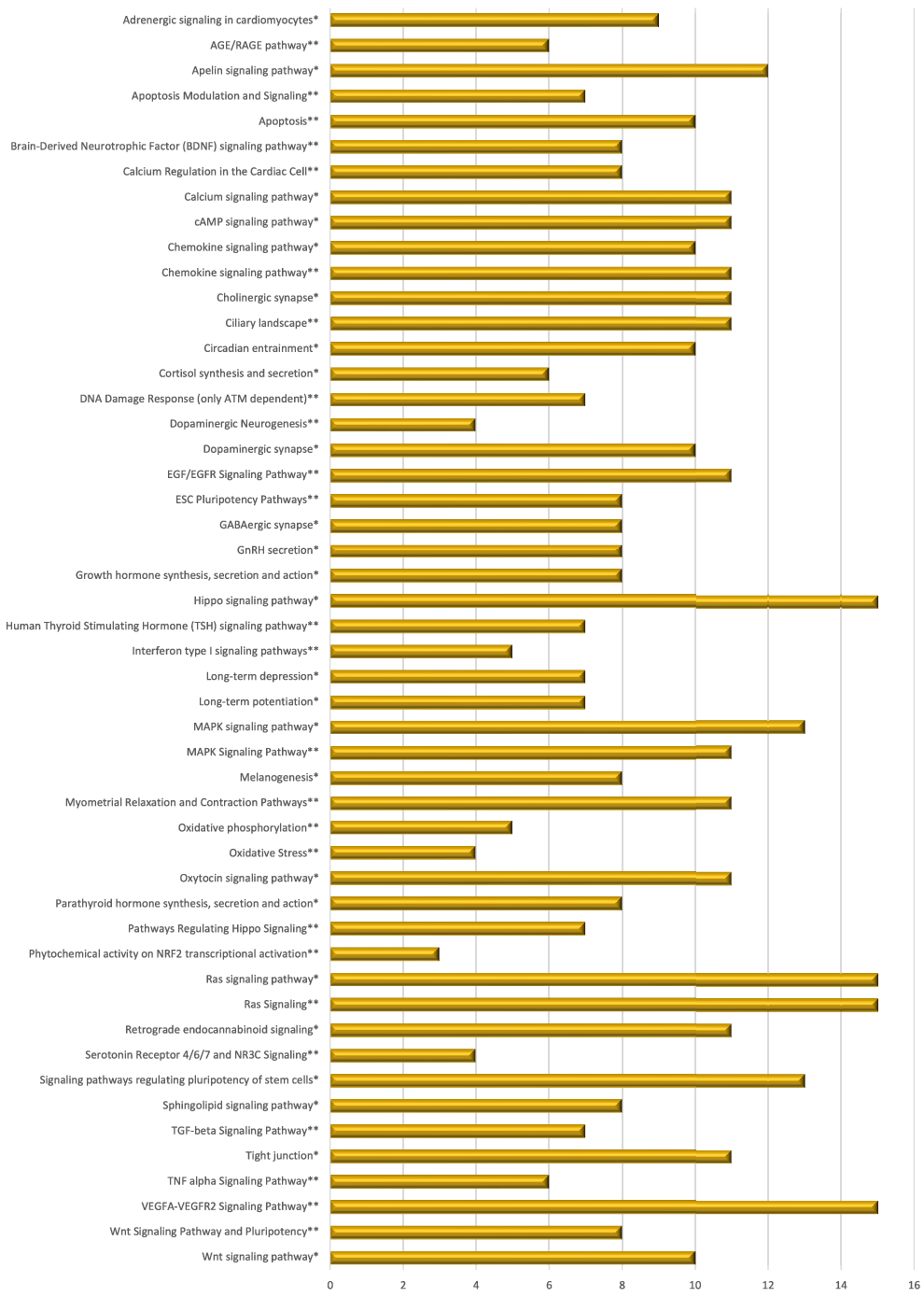
Fig. 5. Pathway enrichment analyses of target genes of lncRNA modulated with consumption of mate extract. Top fifty pathways related to cellular processes in human immune cells, twenty five from each of the two interrogated databases (KEGG and WikiPathways). In alphabetical order; x-axis represents the number of hits. *from KEGG; **from WikiPathways.
Integrative analysis of multi-omic data
Our next step was to conduct a comprehensive comparative and integrative analysis of observed nutrigenomic modulations with regular consumption of mate extract. First, we compared differentially expressed protein coding genes to miRNA and lncRNA targets. This analysis showed a relatively small percentage of overlapping elements, indicating a considerable diversity of molecular targets of mate extract (Fig. 6(a)). Much higher percentage of overlap was observed when analysing top pathways related to cellular processes significantly associated with differentially expressed protein coding genes, miRNA targets, and lncRNA targets (Fig. 6(b)). The central pathways in this diagram include cAMP signalling pathway, MAPK signalling pathway, Ras signalling pathway, adrenergic signalling in cardiomyocytes, retrograde endocannabinoid signalling and myometrial relaxation and contraction pathways. This analysis clearly indicates multi-level and multi-omic regulation of key cellular functions affected with regular mate consumption.

Fig. 6. Comparative analysis of observed nutrigenomic modulations with consumption of mate extract. (a) Analysis of overlap of differentially expressed protein coding genes and targets of protein non-coding RNA (miRNA and lncRNA). (b) Analysis of overlap of top pathways significantly associated with differentially expressed protein coding genes and targets of protein non-coding RNA (miRNA and lncRNA).
We further built and visualised an integrative network of differentially expressed protein coding genes, TF that potentially regulate their expression, miRNA and lncRNA whose expression was modulated by mate extract and their targets, which is presented in Fig. 7. This network consists of numerous and diverse interactions between molecular targets of mate extract. Some of the genes in the network are subjected to multi-level regulation that includes regulation through the activity of putative TF, and/or regulation by protein non-coding RNA. Of note, three of the genes in the network, that is CACNG7, FAM221A and UBE2Z, not only have their expression modulated by mate extract but are also regulated by specific mate extract affected miRNA and lncRNA. To get an insight into cellular functions of all molecular targets of consumption of mate extract, which include differentially expressed protein coding genes, miRNA- and lncRNA targets together (n 3960), we conducted integrative pathway enrichment analyses using GeneTrail as a platform to access KEGG and WikiPathways databases. Top fifty pathways related to cellular processes (top twenty five from each of the interrogated databases), organised in four groups (cell signalling, cell motility and interaction, neurofunction and other cellular processes), are presented in Fig. 8(a). This multigenomic analysis showed that genes of the integrated network are involved in the processes regulating cellular signalling, cell-cell adhesion and neurological-related pathways. The network of these top pathways, i.e. central integrative network of pathways related to cellular processes, which includes the genes (hits) that belong to each of the pathways is presented in Fig. 8(b). Notably, several pathways, such as chemokine signalling pathway, MAPK signalling pathway, PI3K-Akt signalling pathway, Ras signalling pathway, focal adhesion and regulation of actin cytoskeleton, were among top pathways in both interrogated databases, which points out the key cellular processes affected with regular consumption of mate extract. For a detailed in-depth analysis of the multi-level multi-omic regulation of the cellular processes affected with consumption of mate extract, we focused on one of the pathways that emerged consistently from our previous bioinformatics analyses, that is focal adhesion. For visualisation of the regulation of the elements of the focal adhesion pathway, we used the diagram of this pathway from the KEGG database. The multi-level multi-omic regulation of the genes that are part of this pathway is depicted in Fig. 8(c), which presents differentially expressed protein coding genes resulting from consumption of mate extract, regulation by different transcription factors, as well as with identified miRNA and several lncRNA.
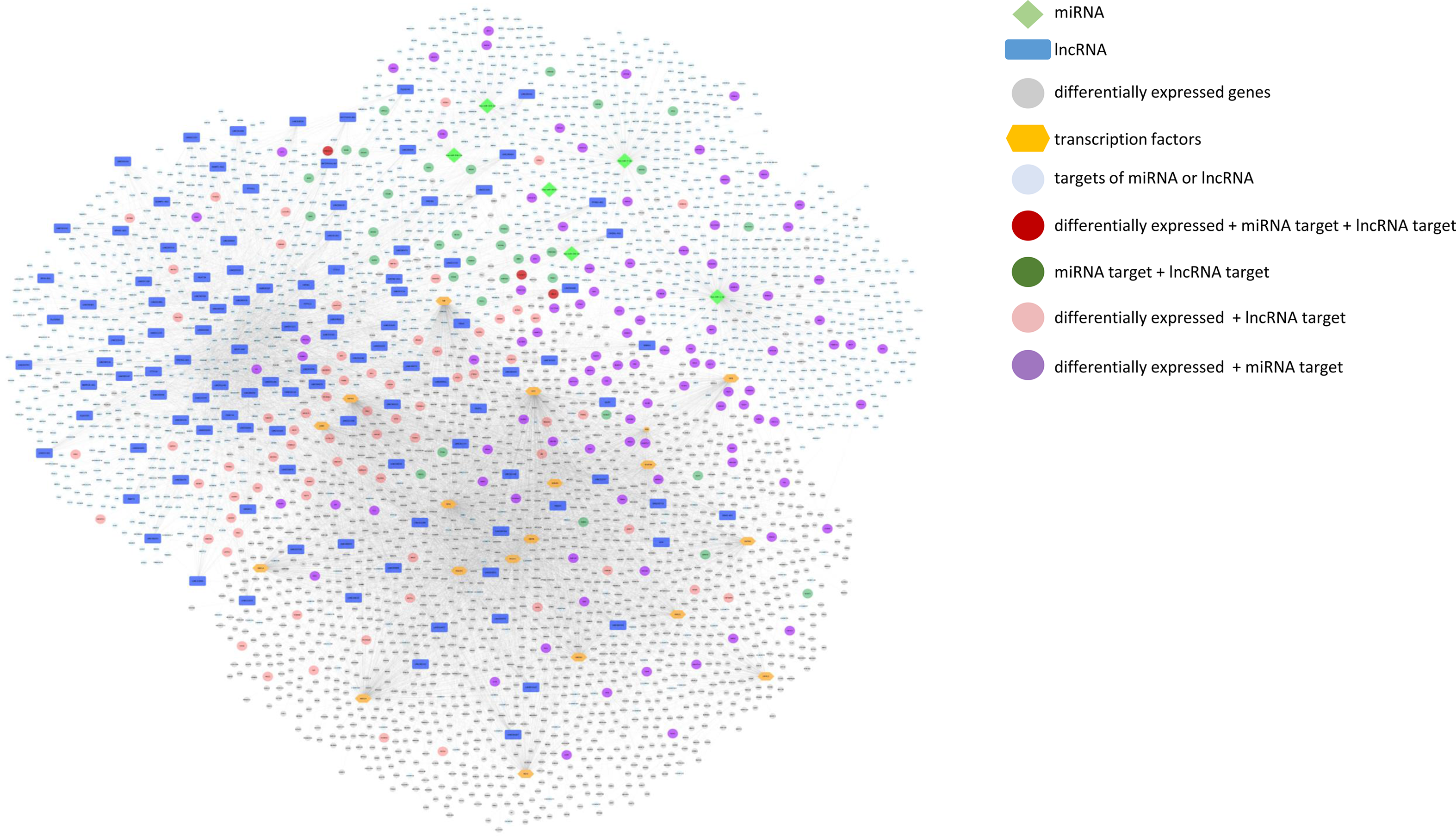
Fig. 7. Integrative network of differentially expressed genes and transcription factors (TF) that potentially regulate their expression, miRNA and lncRNA, and their targets (grey circle: differentially expressed genes; blue squares: lncRNA; light green diamond shape: miRNA; orange hexagonal: TF; light blue: targets of miRNA or lncRNA; dark red: differentially expressed + miRNA target + lncRNA target; dark green: miRNA target + lncRNA target; light red: differentially expressed + lncRNA target; violet: differentially expressed + miRNA target).

Fig. 8. Integrative analyses of multigenomic modifications observed in response to consumption of mate extract. (a) Integrative pathway enrichment analyses of differentially expressed protein coding genes, miRNA- and lncRNA targets. Top fifty pathways related to cellular processes in human immune cells, twenty five from each of the two interrogated databases (KEGG and WikiPathways); y-axis represents the number of hits. *from KEGG; **from WikiPathways. (b) Central integrative network of pathways related to cellular processes. (c) Focal adhesion as an example of multi-level multi-omic regulation of cellular processes with consumption of mate extract.
Correlation with diseases
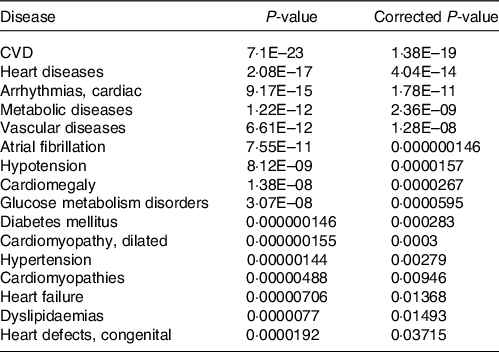
Together with the analysis of cellular pathways and functions potentially affected by identified changes in the expression of genes following consumption of mate extract, we aimed to identify associations with known human diseases. To this end, we used the Comparative Toxicogenomics Database, a database that interrelates differentially expressed genes with diseases, revealing their potential role in prevention or development of these disorders. This analysis showed that identified differentially expressed genes are associated with CVD, including heart diseases, vascular diseases or hypertension but also metabolic diseases, including glucose metabolism disorders, diabetes or dyslipidaemia (Table 5).
Table 5. Associations of gene expression profile of consumption of mate extract with known human diseases
To identify if changes in gene expression profiles (up- or down-regulation) are correlated with development or prevention of cardiovascular and metabolic diseases, we analysed the correlations between modulated genes by mate extract and gene expression profiles identified in the patients with these diseases which are available in the GEO database. Interestingly, we observed that the gene expression profile we obtained following consumption of mate extract is inversely correlated with gene expression profile in patients with metabolic syndrome (GSE124534) and type 2 diabetes (GSE23561) (Fig. 9). We also observed inverse correlations between mate-modulated genes and genes presenting changes in expression in hypertensive patients (GSE75672) and patients with coronary artery disease (GSE23561) (Fig. 9). Altogether, these results suggest that mate extract can have a positive effect on cardiometabolic risk factors by modulating gene expression profiles.

Fig. 9. Correlations between gene expression profile of consumption of mate extract and gene expression profiles identified in the patients with cardiometabolic diseases.
Discussion
Our study is the first to analyse through a systems biology approach, the multigenomic effects of polyphenol-rich mate extract in PBMC in humans, with the objective to better understand by which molecular mechanisms mate polyphenols could influence human health and to determine if the observed genomic modulations can be associated with development of some specific diseases. Another originality of this study is to take advantage of the holistic approach in sample analysis and to use a variety of bioinformatic tools for data analysis and interpretation in an integrative way. The investigations were conducted in PBMC because they are easily accessible cells in humans and their transcriptomic profiles may reflect diet-related diseases and therefore health status prior to development of disease state as well as nutritional status and responses(Reference Afman, Milenkovic and Roche49). This nutrigenomic analysis was performed in male volunteers only. Our objective was to select a study population representative of that consuming traditional mate beverages and also reflecting the increased prevalence of adverse metabolic risk factors in the Brazilian adult population, such as overweight/obesity, dyslipidaemia and hypertension(Reference Conde and Monteiro50,Reference Schmidt, Duncan and Azevedo e Silva51,Reference Picon, Fuchs and Moreira52) . As recently established from analysis of the existing literature, the interindividual variability in the response to polyphenols intake obviously exists(Reference Gibney, Milenkovic and Combet53,Reference Milenkovic, Morand and Cassidy54) , and this variability has often led to inconclusive results in clinical trials aiming to demonstrate the health effects of specific polyphenols. A range of factors, such as age, sex, genetic background, or health status, are suspected to be involved in this between-subject variability, however their respective contributions are far to be demonstrated for now. For these reasons, we focused the inclusion of volunteers on men only, middle-aged (70 % of Brazilian men consuming traditional mate were reported to be >30 years old)(Reference Gebara, Gasparotto-Junior and Santiago4), presenting at most one of the five criteria of metabolic syndrome, and without medication. The polyphenol-rich mate extract used in this study contains different bioactives, among which is also caffeine. Therefore, the observed nutrigenomic changes are prompted by the simultaneous action of all these bioactives. In agreement with data from our study, few previous studies have shown that caffeine and caffeine-rich beverages have the capacity to regulate the expression of large number of genes(Reference Barnung, Nøst and Ulven55,Reference Takahashi, Saito and Jia56) .
In order to give a biological perspective of the observed genomic modulations, we conducted pathway enrichment analyses at three levels, that is differentially expressed protein coding genes, miRNA- and lncRNA targets, as well as integrated multigenomic analysis covering all of the above (Fig. 10). In these analyses, we observed several pathways related to cellular processes that appear consistently as most strongly affected by mate extract. These pathways are involved in cell signalling, such as cAMP signalling pathway, PI3K-Akt signalling pathway or MAPK signalling pathway, pathways that are ubiquitously represented and play key roles in numerous and diverse cellular processes, including cellular processes associated with cardiometabolic risk factors. For example, PI3K-Akt signalling pathway and MAPK signalling pathway are associated with obesity, insulin resistance or polycystic ovary syndrome, all of which are implicative for type 2 diabetes(Reference Huang, Liu and Guo57,Reference Diamanti-Kandarakis and Dunaif58) , while cAMP signalling pathway is involved in both central and peripheral regulation of energy homeostasis(Reference Ravnskjaer, Madiraju and Montminy59), cardiac fibrosis(Reference Delaunay, Osman and Kaiser60) or leukocyte transendothelial migration(Reference Lorenowicz, Fernandez-Borja and Hordijk61). Some of the beneficial health properties attributed to mate tea have been related to its high content of polyphenols(Reference Bravo, Goya and Lecumberri3). Hydroxycinnamic acids, also known as hydroxycinnamates and more collectively as chlorogenic acids, constitute the main polyphenols in yerba mate(Reference Gómez-Juaristi, Martínez-López and Sarria6). Randomised controlled trials have shown that chronic intake of hydroxycinnamic acid-rich foods or supplements (extracts or pure compounds) has beneficial effects on cardiometabolic risk factors in humans(Reference Martínez-López, Sarriá and Mateos62,Reference Roshan, Nikpayam and Sedaghat63,Reference Bumrungpert, Lilitchan and Tuntipopipat64) . Of note, it has been shown that these effects are more pronounced in populations at increased cardiometabolic risk(Reference Martini, Chiavaroli and González-Sarrías65). Genomic modulations underlying the cardiometabolic health properties of hydroxycinnamic acids have also been studied. For example, it has been shown that the protective effects of ferulic acid on vascular endothelium are accompanied with decreased expression of monocyte chemoattractant protein-1 and TNF-α mRNA in rat abdominal aorta(Reference Yin, Qi and Song66). Concordantly, ferulic acid induces an increase in mRNA expression of cardiac antioxidant enzymes in rats, namely superoxide dismutase, glutathione peroxidase and catalase(Reference Yeh, Ching and Yen67), while sinapic acid prevents deregulated expression of cardiovascular genes such as transforming growth factor-b (TGF-β), β-myosin heavy chain and endothelial nitric oxide synthase in a rodent model of hypertension(Reference Silambarasan, Manivannan and Krishna Priya68). In addition, it has been shown that cardioprotective effects of sodium ferulate in rats involve modulated expression of genes such as atrial natriuretic factor, protein kinase C-β, Raf-1 proto-oncogene serine/threonine kinase, extracellular signal-regulated kinase-1/2 and mitogen-activated protein kinase phosphatase-1(Reference Luo, Chen and Yang69). The genomic effects of hydroxycinnamic acids have also been investigated in the context of the neurological manifestations of cardiometabolic diseases. It has been observed that dietary supplementation with decaffeinated green coffee improves diet-induced insulin resistance and brain mitochondrial energy metabolism in mice, accompanied with modulation of a number of genes in the brain that are implicated in cellular energy metabolism(Reference Ho, Varghese and Wang70).
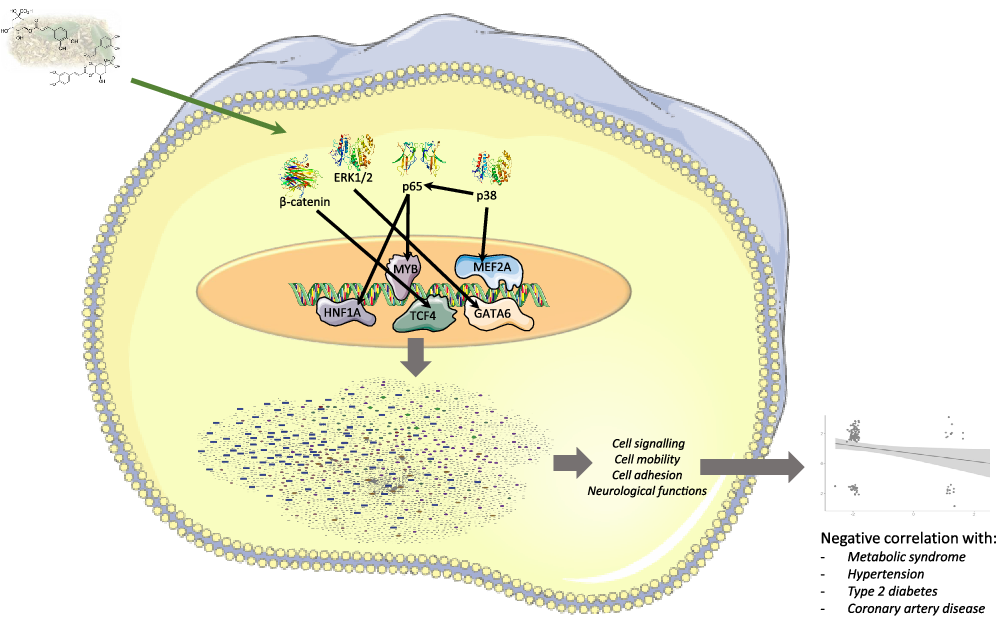
Fig. 10. Summary figure representing multi-omic results and genomic modifications induced in human immune cells by mate extract consumption.
Another group of cellular pathways that are strongly affected by mate extract are the pathways involved in cell motility and interactions. Of these, following integrated pathway analysis of protein coding and protein non-coding RNA, the focal adhesion pathway clearly stands out by appearing among the top cellular pathways in almost every analysis, followed by several related pathways such as tight junction, regulation of actin cytoskeleton or Ras signalling pathway, but also adherens junction, gap junction and fluid shear stress and atherosclerosis. The synergistic activity of these pathways is strongly indicative of regulation of vascular function and atherosclerosis, which is also supported by the results of gene ontology analysis showing that the circulatory system is strongly affected by mate extract. In addition, recent studies have shown that platelets are involved not only in the late stages of atherosclerosis and plaque rupture(Reference Libby71) but also platelet–endothelial interactions occur early in atherogenesis, in part caused by endothelial von Willebrand factor (VWF), which is inflammation mediated(Reference Chen and Chung72,Reference Shim, Liu and Atkinson73) . The concept of inflammation and inflammatory response also applies to other cardiometabolic risk factors and diseases such as obesity, insulin resistance and type 2 diabetes(Reference Libby71). Accordingly, our analyses show that chemokine signalling pathway and cytokine–cytokine receptor interaction are significantly affected with regular consumption of mate extract. In addition, changes in the expression of some of the major genes involved in the above discussed pathways, such as down-regulation of C-C Motif Chemokine Ligand 7 (CCL7; FC = -1·94), TNF Superfamily Member 11 (TNFSF11; FC = -1·91), Nuclear Factor κ B Subunit 1 (NFKB1; FC = -1·19) and VWF (FC = -2·04), or upregulation of Forkhead Box O3 (FOXO3; FC = 1·27), suggest a decrease in risk of development of cardiometabolic diseases. CCL7, also known as Monocyte Chemotactic Protein 3, has been shown to be involved in the pathophysiology of atherosclerosis, at least through its induction by oxidised low-density lipoprotein (oxLDL)(Reference Jang, Kim and Jeoung74), or its effect of direct induction of coronary artery smooth muscle cells proliferation(Reference Maddaluno, Di Lauro and Di Pascale75). Studies have also shown that CCL7 is involved in the chronic inflammation of obese white adipose tissue as a major causative factor of insulin resistance and type 2 diabetes. Treatment with naringenin, a polyphenol from the class of flavanones, significantly reduced the expression of CCL7 in the white adipose tissue of mice fed high-fat diet, suggesting protective effects of naringenin in obesity-related diseases(Reference Tsuhako, Yoshida and Sugita76). On the other hand, there is evidence that activation of the transcription factor NF-kappaB and downstream inflammatory signalling pathways lead to hepatic insulin resistance, β-cell dysfunction and ultimately type 2 diabetes. Studies have shown that TNFSF11 (also known as Receptor Activator of Nuclear Factor κ B Ligand – RANKL), a member of the tumour necrosis factor superfamily, acts as a potent stimulator of NF-kappaB. TNFSF11 is expressed in various cells and tissues, including liver and pancreatic β-cells, and its high serum concentrations independently predict manifestation of type 2 diabetes(Reference Kiechl, Wittmann and Giaccari77). In the cardiovascular system, TNFSF11 is involved not only in both intimal and medial vascular calcification but also in the calcification of heart valves(Reference Wu, Rementer and Giachelli78). Therefore, down-regulation of TNFSF11 mRNA, especially when accompanied with down-regulation of VWF, as it is the case in our study, is indicative of beneficial effects on cardiovascular health. VWF is among the key mediators of early atherogenesis, advanced atherosclerosis and atherothrombosis. A recent study showed that lifestyle intervention in individuals with impaired glucose tolerance or impaired fasting plasma glucose improves their prothrombotic state, including a decrease of VWF(Reference Hörber, Lehmann and Fritsche79). In contrast to the above genes, the expression of FOXO3 mRNA in PBMC is increased by regular consumption of mate extract. FOXO3 transcription factor plays pivotal role in the biological processes of stress response(Reference Morris, Willcox and Donlon80), and as such its activity strongly impacts age-related diseases, including cardiometabolic diseases and sustains longevity(Reference Sanese, Forte and Disciglio81,Reference Bao, Song and Hong82) . Therefore, FOXO3 emerges as a promising target in management of age-related diseases(Reference Jimenez, Silva and Calissi83). It has been shown that FOXO3 can be activated by metformin(Reference Hartwig, Loebel and Steiner84) or curcumin(Reference Zingg, Hasan and Cowan85), which leads to reduced ROS levels in the immune cells and protection against oxidative damage. Altogether, the results of our pathway enrichment analyses show that regular consumption of polyphenol-rich mate extract significantly affects cellular processes that play critical roles in cardiometabolic health in humans. These results are corroborated with our analysis of associations of nutrigenomic effects of mate extract with known human diseases using the Comparative Toxicogenomics Database, and further verified with our global gene expression correlation analysis that demonstrated inverse and statistically significant correlations with gene expression profiles of patients with metabolic syndrome, type 2 diabetes, hypertension and coronary artery disease.
Furthermore, through an in-depth analysis, data from the literature and molecular docking, we identified potential TF which activity could be modulated by mate consumption and regulate the expression of identified differentially expressed genes. Among identified TF are particularly those that are closely related to the pathophysiology of cardiometabolic diseases, such as MEF2A, MYB and HNF1A. MEF2A is a member of the family of myocyte-specific enhancer factor 2 (MEF2) TF, which includes MEF2A, B, C and D. These proteins play central roles in the differentiation, morphogenesis and maintenance of several tissue types(Reference Pon and Marra86) but are also implicated in cellular processes related to cardiometabolic health. MEF2A, together with MEF2D, has been shown to mediate the effects of nuclear receptor corepressor 1 (NCoR1) in cardiomyocyte hypertrophy(Reference Li, Sun and Chen87). Also, MEF2A is a major TF involved in GLUT4 transcription in the soleus muscle(Reference Takeda, Yamamoto and Takemura88), which defines its role in maintaining insulin sensitivity. In addition, a recent study pinpoints the role of MEF2 family of TF as a critical upstream regulator of several transcription factors that play roles in promoting an anti-inflammatory and antithrombotic atheroprotective endothelium(Reference Lu, Martino and Gerlach89). Consistent with the results of the above studies, it has been shown that ferulic acid can increase the expression of MEF2C mRNA in mouse C2C12 myotubes(Reference Chen, Guo and Jia90). As for the MYB, it has been shown that it exacerbates experimental atherosclerosis directly through its effects on B lymphocytes, limiting the levels of atheroprotective oxLDL-specific IgM antibodies(Reference Shikatani, Besla and Ensan91), while several human studies associate HNF1A polymorphisms with circulating CRP concentrations and CVD(Reference Reiner, Barber and Guan92,Reference Hsu, Ko and Teng93,Reference Hong, Kim and Suh94) . Importantly, in a subgroup representative of our entire study population, significant decreases in plasma levels of CRP and IL-6 were observed(Reference Gebara, Gasparotto Junior and Palozi5), suggestive for cardiometabolic protective effects of mate extract and putative mediating role of HNF1A. Our in silico docking analyses suggest that polyphenol metabolites can interact with these transcription factors and cell signalling proteins. These interactions could affect their activity and consequently result in changes in the expression of genes, as observed in our study. These results could be corroborated with few previous studies showing that interaction between MEF2A and molecules like acetamide or carboxamide can affect MEF2 activity and modulate its interaction with DNA(Reference Zia, Imran and Rashid95). Similarly, binding of ligands to p38 cell signalling proteins has been shown to modulate kinase activity and potentially affects the activity of downstream TF(Reference Huang96). These analyses reveal novel molecular mechanisms of actions of mate polyphenols underlying their health properties.
We also observed that regular consumption of mate extract caused a significant down-regulation of six miRNA in PBMC in humans. MiRNA represent a class of small non-coding RNA that function as post-transcriptional regulators of gene expression, and as such are involved in the regulation of various biological processes and linked with the development of diseases(Reference Saliminejad, Khorram Khorshid and Soleymani Fard97), including cardiometabolic diseases(Reference Włodarski, Strycharz and Wróblewski98). Previously it has been shown that polyphenols can modulate the expression of miRNA in experimental models of cardiometabolic diseases(Reference Milenkovic, Berghe and Morand99,Reference Krga, Tamaian and Mercier100,Reference Milenkovic, Deval and Gouranton101) and in humans(Reference Milenkovic, Deval and Dubray30,Reference Rodriguez-Mateos, Istas and Boschek102) . All miRNA that we have identified as differentially expressed with regular consumption of mate extract, except hsa-miR-3654, have already been investigated in the context of cardiometabolic diseases. A recent multi-omics study pinpointed plasma miR-100-5p as one of the four major miRNA associated with the pathogenesis of risk factors for type 2 diabetes and coronary heart disease(Reference Mens, Maas and Klap103). Likewise, a previous occurrence of either gestational hypertension, preeclampsia or gestational diabetes mellitus has been associated with the upregulation of several whole blood miRNA, including miR-100-5p and miR-1-3p(Reference Hromadnikova, Kotlabova and Dvorakova104), while miR-205-5p is upregulated in the lung of patients with idiopathic pulmonary arterial hypertension(Reference Li, Zhang and Xu105) and is associated with unstable atherogenesis and pro-inflammatory macrophage phenotype in a rodent model susceptible to atherosclerosis plaque formation(Reference Meng, Yin and Yu106). Increased serum expressions of the other two miRNA that we identified as down-regulated by mate extract, that is miR-99a-5p and miR-7-5p, have been reported in patients with atrial fibrillation and in hypertensive patients with left ventricular hypertrophy, respectively(Reference Natsume, Oaku and Takahashi107,Reference Kaneto, Nascimento and Moreira108) . Accordingly, it has been shown that regular exercise significantly modulates mean serum expression of several miRNA related to CVD, including down-regulation of miR-7-5p(Reference Barber, Zellars and Barringhaus109).
In addition to the differentially modulated protein coding genes and miRNA, our microarray analysis also revealed a number of lncRNA transcripts that are significantly modulated with regular consumption of mate extract. LncRNA are recently discovered non-coding RNA that are more than 200 bp long single-stranded transcripts, representing the widest, the most heterogeneous and the most complex class of non-coding RNA(Reference Ma, Bajic and Zhang110). By definition, lncRNA do not have protein coding potential, but some of them can translate to small peptides. As they can bind to DNA, RNA and/or proteins, they exhibit an extensive diversity in their mechanisms of action, and as such can regulate gene expression on transcriptional and post-transcriptional levels(Reference Quinn and Chang111,Reference Dykes and Emanueli112) . LncRNA are poorly characterised, but nevertheless there is evidence that they play key roles in the development and normal physiology. In addition, deregulation of specific lncRNA has been shown to be involved in pathophysiology of various diseases, including cardiometabolic diseases(Reference Wijesinghe, Nicholson and Tsintzas113,Reference Uchida and Dimmeler114) . More recently, it has also been shown that diets, such as lipid-rich diet or high glyacemic diets, can modulate lncRNA expression in brain vascular cells(Reference Nuthikattu, Milenkovic and Norman115,Reference Nuthikattu, Milenkovic and Rutledge116,Reference Nuthikattu, Milenkovic and Rutledge117) . Our microarray data show that some of the lncRNA involved in the processes of atherogenesis and/or insulin resistance can have their expression modulated following regular consumption of mate extract. These results suggest that lncRNA present novel molecular targets of mate, and their expression is suggestive for its protective cardiometabolic properties. For example, genome-wide associated studies revealed that single nucleotide polymorphisms in ANRIL (also known as CDKN2B-AS1), a 3·8 kb long lncRNA, are strongly associated with coronary artery disease genetic susceptibility in humans(Reference Xu, Fang and He118,Reference Cheng, Cai and Chen119) . Studies have also shown that ANRIL expression in atherosclerotic plaques, whole blood or PBMC correlates well with atherosclerosis severity(Reference Fasolo, Di Gregoli and Maegdefessel120), and that ANRIL plasma expression is increased in patients with in-stent restenosis, indicating its potential clinical use as optimal prognostic factor(Reference Wang, Su and Liu121). Besides, it has been shown that the expression of ANRIL, HOTAIR and several other lncRNA is increased in PBMC from type 2 diabetes patients and that they positively correlate with poor glycemic control, insulin resistance and systemic inflammation(Reference Sathishkumar, Prabu and Mohan122), while HOXA cluster lncRNA is elevated in PBMC from patients with carotid artery atherosclerosis and positively correlates with inflammatory factors such as VCAM1 and CCL2(Reference Zhu, Liu and Yu123). Another lncRNA, namely GATA6-AS, which is highly expressed in endothelial cells, is involved in the regulation of endothelial gene expression and angiogenesis. This lncRNA is upregulated in HUVEC in hypoxic conditions and exerts its activity by binding with nuclear enzyme lysyl oxidase-like 2 and acting as negative regulator of its function(Reference Neumann, Jaé and Knau124). Altogether, these findings are indicative of significant involvement and complex roles of these lncRNA in the pathophysiology of cardiometabolic diseases. Despite this evidence, further research is needed to better understand the roles of specific lncRNA and the potential protective effect of regular consumption of mate tea.
Some study limitations should be acknowledged. Our study focused only on male volunteers, for two main raisons: this study population presents predisposition to cardiovascular and metabolic diseases and also reduces potential heterogeneity in the response between men and women, as it has been observed for other polyphenols. Also, our nutrigenomic analysis was performed only at a unique time point, that is at the end of the study period, because in previous studies nutrigenomic analysis at the end of the study proved to be sufficient to detect significant changes in the expression of genes and be able to identify underlying mechanisms of actions(Reference Milenkovic, Deval and Dubray30,Reference Rodriguez-Mateos, Istas and Boschek102,Reference Milenkovic, Rodriguez-Mateos and Lucosz125) .
In conclusion, our study provides for the first-time evidence that mate consumption can modulate expression of genes in immune cells that are involved in regulation of cell motility, interaction or inflammation. Modulation of expression of both protein coding and non-coding genes is suggestive of multi-omic modes of action of mate bioactives involved in the health properties of this drink. The observed changes in gene expression in this nutri(epi)genomic study might be a link to the protective cardiometabolic effects of mate and other inflammatory-related diseases. Future studies therefore need to consider this mode of action, use multi-omic approach and go into deeper analysis including not only genomic but also studies of health effects.
Acknowledgements
We thank all research volunteers for participation in this study.
This research was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq-Brazil) 200200/2012-7.
D. M. conceptualised the paper. T. R. and D. M. conducted bioinformatic analyses. C. I. B., K. S. G. and E. L. C. G. conducted the clinical study. T. R., D. M. and C. M. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
The authors declare no conflict of interest.
Supplementary material
For supplementary material/s referred to in this article, please visit https://doi.org/10.1017/S0007114522001027

















