


Overweightness and obesity have become globally common conditions that can significantly reduce quality of life and life expectancy(Reference Peeters, Barendregt and Willekens1). Alongside overweight, the prevalence of metabolic syndrome is increasing(Reference Mikkola, Keinanen-Kiukaanniemi and Laakso2). Increased concentrations of blood lipids and glucose in overweight subjects are risk factors for the development of CVD and diabetes(Reference Esteghamati, Khalilzadeh and Anvari3). It is well documented that obesity is a systemic inflammatory condition, characterised by increased plasma concentrations of C-reactive protein (CRP) and inflammatory cytokines(Reference Yudkin, Stehouwer and Emeis4, Reference Dandona, Aljada and Bandyopadhyay5). Furthermore, it has been demonstrated that there is a correlation between weight, or waist circumference, adiposity and the blood-inflammatory status(Reference Bahceci, Gokalp and Bahceci6, Reference Festa, D'Agostino and Williams7).
Changes in gastrointestinal (GI) functions in overweight subjects have not been studied extensively. Intestinal glucose and amino acid transporters are typically adapted to the luminal nutrient concentrations(Reference Diamond and Karasov8). Excessive luminal nutrient load may exceed the absorptive capacity of the transporters(Reference Diamond9), allowing increased availability of nutrients for colonic microbes. Thus, nutrients not available in the colon of healthy normal weight subjects may have an impact on the composition of the microbial community and the amount of energy harvested from the diet. Recently, a new role for intestinal microbes and the production of SCFA(Reference Ley, Turnbaugh and Klein10, Reference Turnbaugh, Ley and Mahowald11) in the development of obesity has been suggested. Specifically, body weight-based differences between the balance of the relative proportions of the two major intestinal bacterial phyla, Firmicutes and Bacteroidetes, have been described(Reference Ley, Turnbaugh and Klein10).
The aim of the present study was to characterise the intestinal immunological and microbiological environment in obese and normal weight subjects in more detail, and furthermore, to correlate intestinal biomarkers with well-described blood biomarkers for obesity.
Materials and methods
Study subjects
In total, forty subjects were recruited to take part in the present study in the area of Kuopio, Eastern Finland. The main inclusion criteria of the study subjects were for them to be aged between 20 and 55 years and to have a BMI of 20–25 kg/m2 for the normal weight group and a BMI of 30–37 kg/m2 for the obese group. In addition, the subjects had to be accustomed to consuming a mixed diet with moderately low-fibre content (a typical fibre intake of less than 19 g/d for men and 17 g/d for women). The exclusion criteria were critical illness, inflammatory bowel disease, celiac disease or malignancy in the GI tract. Use of anti-obesity drugs, laxatives and weight control products was prohibited. Furthermore, regular (daily) use of probiotics, fibre supplements or bran as well as the regular and/or abundant use of non-steroidal anti-inflammatory drugs was not allowed. The use of antibiotics was prohibited for 3 months before and also during the study. Subjects with a tendency towards constipation (defecation frequency < 3/week) and subjects with recent (within 1 month) severe acute diarrhoea were excluded. In addition, alcohol or drug abuses were exclusion criteria. The present study was conducted according to the guidelines laid down in the Declaration of Helsinki, and all procedures involving human subjects were approved by the Research Ethics Committee, Hospital District of Northern Savo. The purpose of the study was explained to the participants, and written informed consent was obtained from all subjects.
Study design and background information
In telephone screening, potential study subjects were interviewed on their consumption of fibre containing food items using an internet-based questionnaire (www.leipatiedotus.fi)(Reference Lyly, Soini and Rauramo12). The questionnaire calculated the daily fibre intake in grams per day. The cross-sectional study consisted of two study visits. On the first visit, the demographic data and other background information on the study subjects were collected on a structured form (background demographics and diet in Table 1). Furthermore, anthropometric measurements were taken and subjects were instructed how to collect the faecal samples. Body weight was measured twice on the first study visit on a digital scale (Scale Seca 707, Vogel & Falke GmpH & Co., Hamburg, Germany) and the mean was used in the results. Body height was measured with a Seca telescoping measuring rod, type 221, to the nearest crossed millimetre. The waist circumference was measured at a level midway between the superior aspect of the iliac crest and the lower lateral margin of the ribs. The circumference was measured twice and the second measurement was used as the final result. Typically, study subjects are more relaxed during the second measurement and thus the last measurement is considered more reliable. No overnight fasting was requested before the anthropometric measurements. During the 1-week study period, subjects recorded in a diary all changes in their medication and health status as well as their bowel function. On the second study visit, subjects returned the faecal samples and blood samples were taken.
Table 1 Sex, age, life style, dietary restrictions, intake of dietary fibre, bowel function and use of food supplements of the study subjects (n 40)
(Mean values with their standard errors)

* At least half an hour, two to three times a week.
Sample collection
All blood samples were extracted after a 10- to 12-h overnight fast during the second study visit. The samples were collected to serum tubes for S-insulin, S-hs-CRP, S-leptin and S-adiponectin analysis; to citrate-containing tubes for B-glucose analysis; to gel tubes for total cholesterol, fS-LDL and fS-HDL and fS-TAG analysis; and to EDTA-containing tubes for P-IL-6, P-TNF-α, P-ghrelin, P-peptide tyrosine–tyrosine (PYY) and P-orexin analysis. The samples were analysed as fresh (B-glucose, S-insulin, total cholesterol, LDL- and HDL-cholesterol and TAG) or stored at − 70°C (S-leptin, S-adiponectin, S-hs-CRP, P-IL-6, P-TNF-α, P-ghrelin, P-PYY and P-orexin) until analysis. Three spot samples of faeces were collected in a 60 ml specimen tube with an applicator. All faecal samples were placed immediately after collection into a home freezer or were freshly transferred, with a maximum of 1·5 h delay, for freezing in the study unit. Both frozen and fresh samples were transferred to the study unit in a polystyrene box with frozen cool bags. Thereafter, the samples were stored at − 20°C until analysed.
Blood measurements
Blood lipids, sugar and insulin
Serum total TAG, total-, LDL- and HDL-cholesterol were analysed by enzymatic photometric assay (Konelab 60i Clinical Chemistry Analyzer, Thermo Electron Corp., Vantaa, Finland) using commercial reagents (Konelab cholesterol, LDL-cholesterol, HDL-cholesterol and TAG, Thermo Electron Corp.) and plasma glucose by using commercial reagents (Konelab glucose, Thermo Electron Corp.). Also serum insulin was analysed by TR-FIA using commercial kits (AutoDELFIATMo insulin kit, Perkin-Elmer, Boston, MA, USA).
Satiety-related peptides
Serum leptin and ghrelin were analysed by RIA (Multigamma 1261-001, Perkin-Elmer/Wallac Oy, Turku, Finland) using commercial kits (Human Leptin RIA Kit, cat. no HL-81K, Linco Research Inc., St Charles, MO, USA and Ghrelin (Total) RIA Kit, cat. no GHRT-89HK, Linco Research Inc., respectively). Serum adiponectin was analysed by an immunoenzymometric assay using commercial kits (Human Adiponectin/Acrp30 Immunoassay, R&D Systems Inc., Minneapolis, MN, USA). PYY was analysed by RIA (Multigamma 1261-001, Perkin-Elmer/Wallac Oy) using commercial kits (PYY (Total) RIA Kit, cat. no. PYYT-66HK, Linco Research Inc.). Plasma orexin-A concentrations were analysed with RIA using commercial kits (Orexin-A (Human, Rat, Mouse) RIA Kit; Cat no RK-003-30, Phoenix Pharmaceuticals, Inc., Belmont, CA, USA).
Inflammatory biomarkers
IL-6 and TNF-α were analysed with solid phase ELISA method using commercial kits (Quantikine® HS/Human IL-6 immunoassay and Quantikine® HS/Human TNF-α Immunoassay, R&D Systems Inc., Minneapolis, MN, USA). Hypersensitive CRP was analysed with turbidimetry (Hitachi 912, Roche, GmbH, Germany) using Tina-quant CRP (latex) high sensitive assay (Product number 1972855, Roche Diagnostics).
Faecal measurements
DM, pH, ions and energy
Immediately after melting, the frozen faecal samples were weighed for various physico-chemical analyses. For DM determination, approximately 1 g faecal sample was weighed, dried at 105°C for 16 h, cooled down in a desiccator to room temperature, reweighed and the DM content (%) calculated. For pH measurements, 1 g faecal sample was mixed with 1 ml distilled water and the pH measured immediately (SevenEasy pH, Mettler Toledo GmbH, 8603 Schwerzenbach, Switzerland). For Na+ and K+ analyses, 1 g faecal sample was washed and then dissolved in HCl. The solution was diluted with water, and the K content of the solution was determined by an inductively coupled plasma emission spectrometer at wavelength 766·4 nm and the Na content at wavelength 589·5 nm. The energy contents of the faecal samples were determined by an adiabatic bomb calorimeter.
Protein and fat
The faecal protein measurement was based on total n (N × 6·25 = protein) as analysed by the Kjeldahl method. Determination of the total faecal fat was modified from the American Organization of Analytical Chemists 963.15 method (1973; Fat in Cacao Products, Soxthlet Extraction Method). In short, the total amount of petrol diethyl ether-soluble fat was determined after hydrolysis gravimetrically. Fatty acids were analysed as their methyl esters (fatty acid methyl esters) by GC(Reference Cristie13).
Phenolic and acidic compounds
The method of analysis for phenolic and acidic compounds in human faeces was based on the method by Knust et al. (Reference Knust, Erben and Spiegelhalder14). Briefly, a faecal matrix was initially extracted with a phosphate buffer, and the extract was then acidified and re-extracted with a diethyl ether. Individual phenolic and acidic compounds were identified as their silyl derivatives by GC–MS and semi-quantitated using an internal surrogate standard (salicylic acid). Of the forty-six identified compounds, twenty belonged to di- and hydroxy acids, eleven belonged to phenolics, ten belonged to fatty acids and four belonged to sterols. The results were expressed as a sum of the compounds belonging to each subgroup. Moreover, the individual compounds (succinic acid, lactic acid, 3-OH-benzenepropanoic acid and cholesterol) with the highest concentrations in the subgroups were selected for more detailed statistical analysis.
Soluble and insoluble carbohydrates
To separate the soluble and insoluble carbohydrates from faecal samples, 1 g faeces was weighed into a centrifuge tube and 9 ml PBS was added (the PBS consisted of 8·5 g NaCl, 1·21 g K2HPO4, 0·34 g KH2PO4, 0·5 g NaN3 (to prevent bacterial growth), 1 mm phenylmethyl sulphonium fluoride (to prevent serine proteases), 2 mm iodoacetamide (to inhibit cystein-containing enzymes), 10 mm EDTA (to inhibit metalloproteases) and 1 litre H2O) and stored on ice while weighing other tubes. The samples were shaken vigorously for 1 h at 4°C and then centrifuged for 30 min at 4°C at 15 000 g. The clear supernatants were used for soluble carbohydrate analysis, and the pellet was used for the analysis of insoluble polysaccharides. The samples were stored at − 20°C until analysed.
The extraction of soluble carbohydrates from faecal supernatants was modified from the method described by Miller & Hoskins(Reference Miller and Hoskins15). The faecal supernatants were centrifuged in a 35 ml tube for 30 min at 4°C at 15 000 g to remove any particulate matter. The clear faecal supernatant was (via filter through a 0·45 μm filter) transferred to another centrifuge tube and 25 ml ice-cold ( − 18°C) ethanol was added to a final percentage of 70–80 %. The samples were mixed and allowed to stand at 4°C for 1 h. After centrifugation at 4°C for 20 min at 10 000 g, the supernatants were discarded and the pellets were dissolved in 1 ml water. The amount of soluble carbohydrates is the sum of hydrolysed hexoses and amino sugars. To hydrolyse hexoses in the soluble carbohydrate, a fraction of 0·1 ml of the faecal suspension or the hexose standard solution (fucose, arabinose, rhamnose, galactose, xylose, glucose and mannose) was pipetted into a 2 ml microfuge vial, and 0·1 ml 4 m trifluoroacetic acid was added. The samples were incubated at 100°C for 3 h and evaporated to dryness. The samples were dissolved in 1 ml water and filtered through a 0·45 μm membrane filter. The separation and analyses of hexoses were made using high pH ion exchange chromatography (HPLC) and a pulsed electrochemical detector. The analytical column was CarboPac PA1 (4 mm × 250 mm), and the separation was done using a gradient elution with a mobile phase that consisted of a mixture of water and 0·2 m NaOH. To hydrolyse amino sugars in a soluble carbohydrate, a fraction of 0·1 ml of the faecal suspension or the standard solution (galactose amine and glucose amine) was pipetted into a 2 ml microfuge vial, and 0·1 ml 8 m HCl was added. The samples were incubated at 100°C for 5 h and evaporated to dryness. The samples were dissolved in 2 ml water and filtered through a 0·45 μm membrane filter. The separation and analyses of glucose amine and galactose amine were made by HPLC–pulsed electrochemical detector as described earlier.
To analyse the insoluble carbohydrates in the faecal samples, 2 ml of 60 % ice-cold H2SO4 was added to the solid pellet obtained after separation of the soluble and insoluble faecal fractions (see above). A glass rod was used to disperse the pellet thoroughly. The samples were allowed to stand at room temperature (20–25°C) for 1 h, then 22 ml water was added to the samples and they were kept at 100°C for 5 h. The samples were allowed to cool and then diluted to 50 ml with water and filtered through a 0·45 μm membrane filter. The samples were further diluted 1/10 with 20 mm NaOH. The amino sugars and hexoses were analysed by HPLC–pulsed electrochemical detector as described earlier.
Ammonia, SCFA and biogenic amines
Ammonia, SCFA and branched-chain fatty acids (BCFA) concentrations in faecal samples were measured according to a method of Ouwehand et al. (Reference Ouwehand, Tiihonen and Saarinen16). The biogenic amines in faeces were analysed using the method described by Saarinen(Reference Saarinen17).
Microbial analyses
Total microbial counts
The total bacterial cell counts in digesta samples were determined by flow cytometry (FACSCalibur, Becton Dickinson, Franklin Lakes, NJ, USA) as previously described(Reference Apajalahti, Kettunen and Kettunen18). The bacterial fractions were recovered by suspending faecal samples in a 50 mm sodium phosphate buffer (pH 9·0), followed by centrifugation (48 000 g, 30 min, 22°C) and washing. The cell samples were diluted, fixed and stained with the fluorescent nucleic acid-binding dye SYTO 24 (Molecular Probes, Leiden, The Netherlands). The results were expressed as the quantity of bacteria/g fresh digesta weight.
DNA extraction and PCR
DNA was extracted from the washed bacteria using the method described by Apajalahti et al. (Reference Apajalahti, Sarkilahti and Maki19), whereby bacteria were subjected to five freeze-thaw cycles and subsequently treated with lysozyme (17·5 mg/ml, 4 h, 37°C) and proteinase K (0·1 mg/ml, 1 h, 37°C). The recovered bacterial DNA was used to quantify total bifidobacteria, lactobacilli, Clostridium group XIVab, Clostridium perfringens, Bacteroides and sulphate-reducing bacteria using primers and probes as described in Table 2.
Table 2 Primers and probes used to quantify selected members the faecal microbiota

Quantitative real-time PCR was performed using 1 μg isolated bacterial genomic DNA. A 25-μl amplification reaction consisted of 1 × TaqMan Universal Master Mix (Applied Biosystems, Forster City, CA, USA) with 300 nm of both reverse and forward primers, and 200 nm TaqMan probe (Applied Biosystems). All assays were run on an ABI Prism 7000 Sequence Detection System (Applied Biosystems) using the instrument's default settings for thermal cycling and fluorescence measurements. For standard curves, bacterial genomic DNA from C. perfringens (ATCC 13 124) and in-house isolated genomic DNA from Desulfovibrio intestinalis (DSM 11 275) were applied. Based on the genomic sizes, the weight of one copy of each of the C. perfringens and D. intestinalis genome was calculated, and the amount of chromosomes in 1 pg was estimated. Standard amplification curves were constructed by using 1, 10, 100 and 1000 pg bacterial genomic DNA as a template. The results are expressed as the quantity of bacteria/g faeces.
Immunological analyses
The concentrations of IgA, TNF-α, calprotectin and PGE2 were measured from the soluble fraction of the faeces as previously described(Reference Ouwehand, Tiihonen and Saarinen16). Briefly, the frozen samples were thawed and extracted with bovine serum albumin and stored at − 20°C before analysis. Concentrations of IgA, TNF-α and PGE2 were determined with an ELISA in accordance with the respective manufacturer's instructions (E80-102, Bethyl Laboratories, Inc., Montgomery, TX, USA; Biosource Europe S.A., Nivelles, Belgium; Cayman Chemical Company, Ltd, Ann Arbor, MI, USA), and the results were expressed as μg or pg per gram fresh digesta weight.
Statistical analyses
The basic statistics (mean and standard error) were applied to the data. The comparison of normal weight and obese group means was carried out using the two-sample t test, and a P-value lower than 0·05 was considered as significant. For addressing associations between inflammatory blood biomarker and intestinal biomarkers, Pearson correlation coefficients were calculated by combining the data from both groups. The association between the parameters was considered significant if the P-value was below 0·05.
Results
A number of different biomarkers both from blood and faecal samples were measured from twenty normal weight and twenty obese subjects. The waist circumference was on average 82 and 108 cm, and the BMI was 23 and 33 kg/m2, in the normal weight and obese subjects, respectively (Table 1).
Blood biomarkers
Concentrations of blood insulin and TAG were higher (P < 0·001 and P = 0·026, respectively), whereas concentrations of HDL-cholesterol were lower in the obese subjects (P = 0·006). Blood glucose, total cholesterol and LDL-cholesterol were similar in both groups. The inflammatory biomarkers, CRP and IL-6, were higher in the obese subjects (P = 0·055 and 0·024), whereas concentrations of TNF-α were similar in both groups. Of the measured satiety hormones, concentrations of leptin were higher and concentrations of ghrelin were lower in the obese subjects (P < 0·0001 and P = 0·03, respectively). Concentrations of adiponectin, PYY and orexin were similar in both groups (Table 3).
Table 3 Blood biomarkers in the obese and normal weight subjects
(Mean values with their standard errors)

CRP, C-reactive protein; PYY, peptide tyrosine–tyrosine.
* The sample size used for calculating the descriptive statistics. n is smaller than twenty in some cases due to missing data. Two-sample t test was used to compare the mean values between the two groups.
† A P-value < 0·05 was considered to indicate significant difference between the groups.
Faecal measurements
Chemical composition of the faecal samples was studied with focus on the available nutrients and energy (Table 4). The remaining measured energy in the faecal samples was similar in both groups, as well as the other measured physico-chemical properties (faecal weight, DM content, pH, and concentrations of Na+ and K+), with the exception of concentrations of ammonia and carbohydrates that showed a trend for higher levels in the obese subjects (P = 0·15 and 0·14, respectively). The residual concentrations of macronutrients, proteins and fats were similar in both groups.
Table 4 Chemical composition of faeces from the normal weight and obese subjects
(Mean values with their standard errors)

fw, fresh digesta weight.
* The sample size used for calculating the descriptive statistics including the number of samples below the detection limits (given in parenthesis). For calculation of the mean values, the data below detection limits were set to half of the limit value. Two-sample t test was used to compare the mean values between the two groups.
† Detection limit 0·01 g/kg fw.
‡ Detection limit 0·01 g/kg fw.
While the total concentration of bacteria was similar, the microbial composition was different in the obese and normal weight subjects. The level of sulphate-reducing bacteria (P = 0·05) and Bacteroides (P = 0·07) appeared higher in the normal weight subjects than in the obese subjects (Table 5). Of the microbial metabolites, the sum of SCFA was similar in both groups. However, the concentrations of lactic acid and valeric acid were different (P = 0·08 and 0·04) between the study groups (Table 5). The sum of BCFA (P = 0·03) was higher in the obese subjects, as were the two main components, 2-methylbutyric acid and isovaleric acid (P = 0·02 and 0·03, respectively). However, neither the sum of biogenic amines nor any of the single biogenic amines differed significantly between the groups. The sum of phenolic concentrations was significantly higher in the obese subjects (P = 0·02). In more detail, the 3-OH-benzenepropanoic acid concentrations were elevated in the obese subjects (P = 0·06).
Table 5 Faecal microbes and microbial metabolites in the normal and obese subjects
(Mean values with their standard errors)

fw, fresh digesta weight; BCFA, branched-chain fatty acid.
* The sample size used for calculating the descriptive statistics including the number of samples below the detection limits (given in parenthesis). For calculation of the mean values, the data below detection limits were set to half of the limit value. Two-sample t test was used to compare the mean values between the two groups.
† Detection limits: 1·2 × 102 cells/g.
‡ Detection limits: 5·1 × 101 cells/g.
§ A P-value < 0·05 was considered to indicate significant difference between the groups.
∥ Detection limits: 0·1 mmol/kg.
¶ Detection limits: 30 nmol/kg.
** Detection limits: 15 nmol g/kg.
†† Detection limits: 10 mg/kg.
‡‡ Detection limits: 0·4 μg/kg.
§§ Detection limits: 0 pg/g.
No differences were detected in the concentrations of faecal immune markers, IgA, TNF-α, calprotectin and PGE2 (Table 5).
Correlations between blood inflammatory biomarkers and intestinal biomarkers
It was of special interest to study the correlations between inflammatory blood biomarkers and the intestinal biomarkers. Serum CRP and IL-6 appeared to be correlated, especially with some of the microbial metabolites, i.e. concentrations of phenolics (P = 0·04 and 0·01) and di- and hydroxy acids (P = 0·04 and 0·04), whereas serum TNF-α was associated with the total number of bacteria and the faecal concentrations of sterols (P = 0·006 and P < 0·001). An inverse correlation between the numbers of faecal Bacteroides and waist circumference (P = 0·05) as well as weight (P = 0·09) was noted. In addition, an inverse correlation between the total faecal microbial counts and serum CRP (P = 0·04) was found (Table 6).
Table 6 Pearson correlation coefficients (r) and P-values for testing whether the correlation is zero or not for faecal and clinical measurements combining data from both the groups
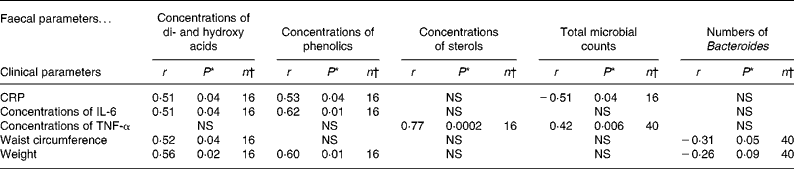
CRP, C-reactive protein.
* A P-value < 0·05 indicates significant association.
† Values refer to the number of observations for which measurements on both parameters are available.
Discussion
The obese subjects included in the present study were found to represent typical characteristics regarding their fasting satiety-regulating hormones, metabolic disturbances and inflammatory biomarkers. In accordance with previous findings, the plasma TAG concentrations were higher and HDL-cholesterol was lower in obese subjects than in normal weight subjects(Reference Couillard, Bergeron and Prud'homme20). Moreover, the increased insulin concentrations in the study subjects may indicate insulin resistance. The elevated plasma leptin levels in the present study are also in line with previous findings(Reference Considine21) indicating leptin resistance in obese subjects(Reference Considine21). Circulating ghrelin, which has the opposite effect to leptin, is, however, decreased as reported also by Tschop et al. (Reference Tschop, Weyer and Tataranni22). Adipose tissue has been described as an origin for pro-inflammatory cytokines such as IL-6 and TNF-α(Reference Compher and Badellino23). It has been proposed that hypoxia caused by enlarged adipocyte size may underlie an inflammatory response, increasing IL-6 and TNF-α production and decreasing secretion of anti-inflammatory adiponectin(Reference Trayhurn, Wang and Wood24). Increased serum IL-6 concentrations are reported to be associated with visceral adiposity, whereas serum TNF-α showed more associations with total body fatness(Reference Couillard, Bergeron and Prud'homme20, Reference Cartier, Lemieux and Almeras25). The present study also indicated a correlation between serum IL-6 and waist circumference, while serum TNF-α correlated with faecal sterol concentrations. The latter phenomenon possibly indicating of a feedback signalling from the stressed adipocytes, indicated by secretion of TNF-α, to the intestinal tissue to reduce absorption of sterols forms the diet, thus perhaps resulting in increased concentrations present in the faeces. Alternatively, in rodent model, inflammation has shown to impair reverse cholesterol transport to faeces(Reference McGillicuddy, de la Llera and Hinkle26). High-fat feeding has shown to increase gut permeability of bacterial lipopolysaccharides to plasma, which in turn triggers low-grade inflammation and obesity-associated disorders(Reference Cani, Amar and Iglesias27, Reference Cani, Bibiloni and Knauf28). Anti-inflammatory adiponectin concentrations are typically decreased in obesity(Reference Weyer, Funahashi and Tanaka29); however, in the present study, the decrease was not statistically significant. It is of interest that, despite the systemic inflammatory status, the measured faecal immunological biomarkers were not affected by obesity. Previously, it has been described that concentrations of faecal PGE2 may be affected by age(Reference Tiihonen, Tynkkynen and Ouwehand30), and furthermore, that faecal IgA concentrations may be elevated during the allergy season in subjects allergic to birch pollen(Reference Ouwehand, Nermes and Collado31). Both calprotectin and TNF-α are used to monitor the state of intestinal inflammation, and antibodies specific to TNF-α are also used as treatment to suppress the intestinal inflammation, in patients with inflammatory bowel disease and ulcerative colitis(Reference Foell, Wittkowski and Roth32–Reference Yadav and Liu34).
Only relatively small changes were detected in the faecal microbial composition between the obese and normal weight groups. However, the fermentation pattern in obese subjects is more pronounced in protein than carbohydrate fermentation as suggested by increased concentrations of BCFA, phenolics and a tendency for increased ammonium concentrations(Reference Cummings and Macfarlane35). Surprisingly, in spite of the indirect indications of elevated amounts of protein fermentation, less sulphate-reducing bacteria were detected in the obese subjects. In accordance with previous observations, the levels of Bacteroides were decreased in the obese subjects(Reference Ley, Turnbaugh and Klein10). Interestingly, the waist circumference was inversely correlated with the numbers of Bacteroides, underlining the possible role of Bacteroides in the absence of adiposity. Increased numbers of faecal Bacteroidetes have also been detected in elderly over 70 years of age than in young adults(Reference Makivuokko, Tiihonen and Tynkkynen36). In a recently published retrospective study on children at risk of developing allergy, less bifidobacteria and more Staphylococcus aureus were found in the faeces of children that later became overweight(Reference Kalliomaki, Collado and Salminen37). In the present study, the levels of S. aureus were not determined, but the levels of bifidobacteria were similar in both the normal weight and obese subjects. Furthermore, other associations between chronic conditions such as irritable bowel syndrome and intestinal microbiota have also been reported(Reference Malinen, Rinttila and Kajander38).
The elevated concentrations of many residual microbial metabolites, both originating from carbohydrate and protein fermentation, in the faeces suggest a different fermentation profile in the colon of the obese subjects. Previously, it has been described that faecal SCFA are found in higher levels in obese subjects than in lean subjects(Reference Schwiertz, Taras and Schafer39), while, in the present study, the difference was statistically significant only in the faecal concentrations of BCFA. Recently, Samuel et al. (Reference Samuel, Shaito and Motoike40) have indicated SCFA as signalling molecules for enteroendocrine cells to produce PYY and thus decreasing gut motility and further increasing SCFA absorption and energy extraction from the gut lumen. Microbial metabolites may also regulate food intake differently in obese and in lean subjects, as has been indicated by rat studies(Reference Bray41). Many metabolites resulting from putrefaction, such as phenols(Reference Hughes and Rowland42), have been linked with health risks, such as colon cancer(Reference Rafter43), whereas metabolites, such as butyrate, may be chemopreventive in colon carcinogenesis(Reference Scheppach and Weiler44). Obesity has been identified as a risk factor for the development of colon cancer(Reference Larsson and Wolk45); the higher faecal levels of BCFA and phenols could potentially be a partial explanation for these epidemiological observations. Concentrations of faecal butyrate were not found to be decreased in the obese subjects in the present study. In summary, the changed microbial metabolite profile in the faeces of obese subjects may reflect the adaptation of the microbial community to the different nutrients available.
In the present study, faecal phenolics and lactic acid were found to be associated with the blood inflammatory biomarkers, CRP and IL-6. It remains to be determined whether or not these coinciding changes are somehow causally linked to each other. l-lactate is generally a signal of anaerobic metabolism in tissues and thus may induce inflammatory response in adipose tissues. Lactic acid is also produced by many different GI microbes and is typically utilised by other intestinal microbes(Reference Duncan, Louis and Flint46) and does not accumulate in the faeces of healthy subjects. If, however, lactate is accumulated in the lower GI tract due to an imbalance in the lactate-producing and lactate-utilising bacteria, it can be absorbed into the blood circulation either by passive diffusion (d-lactate) or via the monocarboxylate transporter (l-lactate)(Reference Ritzhaupt, Wood and Ellis47). Therefore, lactic acid originating from intestinal fermentation can have also systemic effects, e.g. with muscle cells(Reference Lombardi, Fabris and Bassetto48). Unfortunately, plasma lactate levels were not analysed in the present study. In obesity, elevated blood lactate concentrations are suggested to originate from increased lactate production by adipocytes(Reference DiGirolamo, Newby and Lovejoy49). However, the postprandial rise in circulating lactate may be partly explained by the intestinal lactate production. Chronic hyperlactaemia has been suggested to have a role in the development of insulin resistance in muscle cells(Reference Lombardi, Fabris and Bassetto48). Excessive amounts of lactic acid in the blood circulation have been shown to cause neurotoxicity and cardiac arrhythmia(Reference Chan, Slater and Hasbargen50, Reference Vella and Farrugia51). Also dietary or microbe-derived phenolics can be absorbed from the GI tract, but their uptake can be incomplete(Reference Jenner, Rafter and Halliwell52). In addition to microbial metabolites, structural microbe-derived products, such as lipopolysaccharides, have been recently suggested to affect the development of metabolic diseases in animal models(Reference Reigstad, Lunden and Felin53).
Overall, the changes in the microbial fermentation patterns, including increased faecal phenolics and lactic acid concentrations, described in the obese subjects most probably have an impact on host physiology including the systemic inflammatory condition. Future studies should focus on studying whether these obesity-associated intestinal parameters could be a potential therapeutic target for decreasing obesity-associated systemic effects.
Acknowledgements
The present research received no external funding, apart from the support of Danisco A/S. The authors are indebted to the participants of the study for their cooperation and assistance. The authors thank Markku Saarinen, Antti Kaipainen, Martti Marjamaa, Päivi Nurminen, Heli Putaala, Brita Mäki, Kirsi Stenström, Eeva Linblom and Tiina Laamanen for the various laboratory analyses. Essi Sarkkinen, Tarja Niskanen and Henna Karvonen from Foodfiles Ltd are thanked for the practical arrangements; and Akra-Numero Research and Consultancy Centre, India, for the statistical analysis. All authors contributed to the preparation of the manuscript. K. T. contributed to the planning of the study, data management, coordination of the manuscript preparation and data interpretation. A. C. O. contributed to the microbial analyses, manuscript preparation and data interpretation. N. R. coordinated the planning of the study and contributed to manuscript preparation and data interpretation. There is no conflict of interest.






