


The essential dietary micronutrient Zn is a structural component of, and/or catalytic cofactor for, several hundred proteins, so plays critical roles in a diverse array of cellular processes(Reference Vallee and Falchuk1). Zn is required for the catalytic activity of RNA polymerases and as a component of Zn-finger domains of numerous transcription factors(Reference Cousins2–Reference Mackay and Crossley4), suggesting that alterations in Zn status can have potentially complex effects on gene expression. This view is supported by transcriptomic studies that document pleiotropic gene-regulatory effects of changes in Zn availability(Reference Beck, Li and Bao5–Reference Yamada, Suzuki and Koizumi14).
The small intestine is the primary site of Zn homeostasis in mammals(Reference Krebs15), so a number of transcriptomic investigations have focused on the regulatory effects of dietary Zn supply on small-intestinal gene expression in rodent models(Reference Blanchard, Moore and Green6, Reference Pfaffl, Gerstmayer and Bosio11, Reference Blanchard and Cousins16, Reference Blanchard and Cousins17). By transferring nutrients between mother and fetus, the function of the placenta is analogous to that of the intestine. Observational studies in human subjects have suggested links between poor maternal Zn status and measures of pregnancy outcome(Reference Tamura and Goldenberg18). Despite this evidence, no large-scale analyses of the effects of Zn availability on placental gene expression have been performed to date. An understanding of gene-regulatory responses to changes in Zn availability in the human intestine and placenta will provide valuable information on the processes modulated by altered Zn nutrition and its potential impact on human health, particularly during fetal development.
We employed oligonucleotide microarrays to compare the effects of Zn on gene expression in the well-characterised human intestinal cell line Caco-2 and the human placental cell line JAR. We suggested previously that the regulation of expression of Zn transporter genes by Zn may occur over different concentration ranges in intestinal and placental cells, appropriate to the normal concentration of total extracellular Zn encountered by the two different tissues in vivo (Reference Ford19). Whilst enterocytes in the intestine are exposed to the Zn concentration of the intestinal lumen, which is very variable and likely to be in the order of 100 μm following a meal or nutrient supplement(Reference Cragg, Christie and Phillips20), placental trophoblasts are exposed to much lower and less variable serum Zn concentrations of approximately 12–15 μm(Reference Hotz, Peerson and Brown21). In view of this hypothesis our experimental protocol was designed to allow us to establish if both cell types display parallel transcriptomic response profiles to Zn occurring at physiologically relevant concentrations. In contrast, our analysis demonstrates that a different repertoire of genes responds to changes in Zn availability in intestinal cells compared with placental cells. We also provide further evidence that Zn affects gene transcription differently in the Caco-2 compared with the JAR cell line by demonstrating differential regulation by Zn of both MT2A promoter activity and Zn-sensitive transcription factor expression. We suggest that the latter may be responsible, at least in part, for the differential gene responses to altered Zn supply in intestinal and placental cells. Interestingly, in relation to reports of nutrient interactions between Zn and other metals, our microarray analyses identified a number of Fe- and Cu-related genes as targets for regulation by Zn in intestinal or placental cells.
Methods
Cell culture
Caco-2 and JAR cells were cultured as described previously(Reference Cragg, Christie and Phillips20). Absence of mycoplasma contamination was confirmed using the Venor GeM detection kit (Cambio Ltd, Cambridge, Cambs, UK). Zn was added to confluent JAR monolayers or differentiated Caco-2 monolayers in six replicate 75 cm2 flasks. Either 7 d (JAR) or 14 d (Caco-2) post-seeding, culture medium was replaced with 15 ml basal medium (about 3 μm-Zn, assessed by atomic absorption spectroscopy) or medium supplemented to 12 μm or 100 μm with ZnCl2; cells were then cultured for 12 or 24 h.
Preparation of RNA samples for microarray analysis
Total RNA was extracted from cells using TRIzol reagent (Invitrogen Corp., Carlsbad, CA, USA). Traces of genomic DNA were digested by incubation with RNase-free DNaseI (Roche, Basel, Switzerland), then RNA was extracted using phenol–chloroform. RNA from the six replicate flasks was pooled, and then concentration was determined using a NanoDrop™ spectrophotometer (NanoDrop Technologies, Thermo Fisher Scientific Inc., Pittsburgh, PA, USA). RNA quality was confirmed by denaturing agarose gel electrophoresis and also on an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA), accepting an RNA Integrity Number (RIN)>9.
Preparation of Arabidopsis thaliana external control cRNA
Total RNA extracted from Arabidopsis thaliana leaves was reverse transcribed to cDNA using SuperScript III RNase H- enzyme (Invitrogen Corp.). PCR amplification was performed over thirty cycles (95°C for 30 s; 55°C for 30 s, 72°C for 90 s), using ThermoStart DNA polymerase (ABgene Ltd, Epsom, Surrey, UK) (forward primer sequence: GCATCAACGCGAAAACAGTTGG; reverse primer sequence: CTTATACTCCCTTCGATGTACC). The cDNA fragment was subcloned into the pCR2.1TOPO vector (Invitrogen Corp.) to give the plasmid pCR2.1TOPO-ara_control. The construct was linearised 3′ of the insert using BamHI, then complementary RNA was transcribed from the T7 promoter using the mMessage mMachine kit (Applied Biosystems/Ambion, Austin, TX, USA). A poly(A)-tail was added to the cRNA transcript using the Ambion poly(A)-tailing kit.
MWG custom microarrays
Human low-density custom glass microarray slides were obtained from MWG Biotech Ltd (Ebersberg, Germany) and were spotted with duplicate arrays of 50-mer oligonucleotides. Each duplicate array incorporated 190 experimental human genes, nine human housekeeping genes and two A. thaliana control genes. One of the A. thaliana genes was utilised as an external control gene, which was spiked into array labelling reactions, whilst the other was used as a negative control gene to measure background levels of fluorescence on array slides.
Preparation of labelled cDNA for microarray hybridisation
Labelled cDNA samples for microarray analyses were prepared using the CyScribe amino-allyl post-labelling kit (Amersham/GE Healthcare). Each experimental and reference sample was labelled in duplicate, once with Cy3 and once with Cy5. Reverse transcription reactions contained 20 μg total RNA plus 2·8 ng synthetic poly(A)-tailed A. thaliana external control RNA (equivalent to 1000 spike copies/cell). Measurements of cDNA yield and CyDye incorporation were by spectrophotometry. Cy3 and Cy5 incorporation was measured using absorbance settings of 550 and 649 nm respectively.
Hybridisation and scanning protocols
Equal amounts (40 pmol) of Cy3- and Cy5-labelled experimental and reference cDNA were hybridised to array slides. Two independent hybridisations were performed for each reference v. experimental comparison, each with genes spotted in duplicate, using a dye-swap approach. Hybridisations were carried out using an HS4800 Automatic Hyb Chamber (Tecan, Lyon, France). Microarray slides were pre-hybridised in buffer containing 3 × saline–sodium citrate (SSC), 0·15 % SDS, for a total of 90 s at 42°C. Hybridisation was performed at 42°C for 16 h with low agitation. Slides were washed at 30°C for 30 s with 2 × SSC, 0·1 % SDS, followed by 1 × SSC, then 0·5 × SSC. Scanning was performed with a GenePix 4000B confocal laser scanner (Molecular Devices, Sunnyvale, CA, USA), using laser excitation at 532 nm for Cy3 and 635 nm for Cy5. Photo-multiplier tube gain was optimised for each slide so that detection of low and high intensity signals was balanced and saturation tolerance was set at 0·005 % to avoid pixel saturation. GenePix Pro 6.0 software (Molecular Devices) was used to extract raw data from the images.
Microarray data normalisation and analysis
Gene expression levels were normalised and analysed using GeneSpring GX 7.3 software (Agilent Technologies, Inc.). A cut-off was applied to discard data from weak signals; present calls were determined as signals that had intensity values in either the Cy3 or Cy5 channel greater than the mean intensity of the A. thaliana negative control spots plus two times the standard deviation. Expression ratios (i.e. Cy5:Cy3, or Cy3:Cy5 for dye swaps) for each gene were normalised using factors calculated from the median intensity ratios of the A. thaliana external control gene and a panel of nine housekeeping genes. To estimate the expected variance of ratios of non-regulated genes, 95 % CI were calculated using the mean and standard deviation of the normalised set of housekeeping genes. Genes were considered up- or down-regulated (i.e. differentially expressed between reference and experimental samples) if they had: (i) an average ratio, calculated from the four replicates, outside of the 95 % CI, or (ii) ratios outside of the 95 % CI in at least three out of four replicates. Statistical analysis was by Student's t test with a Benjamini–Hochberg multiple testing correction. Genes with P < 0·05 were determined to be significantly regulated by the experimental Zn treatment. All statistical analyses were performed using log-transformed data. Microarray data were annotated to MIAME (minimum information about a microarray experiment) standard and have been deposited in the ArrayExpress database (www.ebi.ac.uk/arrayexpress; EMBL – European Bioinformatics Institute, Cambridge, Cambs, UK) with the accession number E-MEXP-1064.
Real-time reverse transcriptase polymerase chain reaction for validation of microarray results
Reverse transcription was carried out with SuperScript III RNase H- RT (Invitrogen), using random hexamer primers and 2·5 μg DNase-treated RNA. Real-time PCR was performed using SYBR-Green I Master Mix (Roche) with the Lightcycler 480 (Roche) for fluorescence detection. Cycling conditions were as follows: denaturation at 95°C for 5 min, then forty-five cycles of 95°C for 10 s; 53°C or 60°C for 15 s; 72°C for 6 s. Primer sequences are listed in Supplementary Table 1. Expression levels were normalised to expression levels of glyceraldehyde-3-phosphate dehydrogenase. Primer specificity was assured by a single melting curve peak. Statistical analysis of data (n 3) was by one-way ANOVA followed by Dunnett's post test.
Generation of reporter plasmid constructs
The − 358 to +40 region of the MT2A promoter was subcloned into the vector pBlueTOPO (Invitrogen Corp.), upstream of the β-galactosidase reporter gene, as described previously(Reference Helston, Phillips and McKay22).
Reporter gene assay
Transfection and measurement of β-galactosidase reporter gene activity in Caco-2 and JAR cells were as described previously for Caco-2 cells(Reference Helston, Phillips and McKay22). The protein concentration of cell lysates was determined using the Bradford assay (Bio-Rad, Hercules, CA, USA).
Results
Comparison of global gene responses to zinc in intestinal and placental cell lines
We employed dual-label (Cy3, Cy5), low-density oligonucleotide microarrays to examine changes in gene expression levels between experimental cells grown under Zn-supplemented conditions (12 μm or 100 μm extracellular Zn) and reference cells grown in basal culture medium (3 μm extracellular Zn), for either 12 or 24 h. Microarray slides incorporated two A. thaliana control genes, one of which was used as a negative control (to measure background levels of fluorescence on array slides), whilst the other was utilised as an external spike-in control gene (to correct for effects of systematic variations in the microarray data). Poly(A)-tailed cRNA corresponding to the A. thaliana external control gene was spiked in equal quantities into experimental and reference microarray sample labelling reactions and expression ratios of each gene were normalised to expression ratios of nine housekeeping genes spotted on the arrays and the Arabidopsis external control. Only genes classified as present (signal intensities in reference or experimental samples greater than the mean of the negative control spots plus 2 sd) were included for analysis. Data from four replicate spots were used to calculate the average expression ratio of each gene under each experimental condition. Identification of differentially expressed genes was based on 95 % CI, calculated two-sided from the log-transformed mean expression ratio (μ) of the panel of nine housekeeping genes (μ ± 1·96 × sd). Lists of genes identified as differentially expressed in Caco-2 and JAR cells under each experimental condition are provided in Tables 1 and 2. All information relating to these microarray experiments, including a complete dataset of expression ratios for all genes and experiments, can be viewed at www.ebi.ac.uk/arrayexpress (accession number: E-MEXP-1064).
Table 1 Genes identified as differentially expressed in Caco-2 cells by DNA microarray hybridisation following exposure to 12 μm or 100 μm extracellular zinc concentrations for 12 or 24 h

* RNA samples extracted from Caco-2 cells exposed for the indicated time period to the indicated extracellular Zn concentration.
† GenBank accession number.
‡ Name of encoded protein.
§ Magnitude of changes observed by array, increased (+) or decreased ( − ) relative to expression levels in control cells (3 μm-Zn).
∥ Each gene n 4.
¶ By Student's t test.
Table 2 Genes identified as differentially expressed in JAR cells by DNA microarray hybridisation following exposure to 12 μm or 100 μm extracellular zinc concentrations for 12 or 24 h

* RNA samples extracted from JAR cells exposed for the indicated time period to the indicated extracellular Zn concentration.
† GenBank accession number.
‡ Name of encoded protein.
§ Magnitude of changes observed by array, increased (+) or decreased ( − ) relative to expression levels in control cells (3 μm-Zn).
∥ Each gene n 4.
¶ By Student's t test.
In the 12 h experiments, supplementation of Caco-2 and JAR cells with 100 μm-ZnCl2 led to differential expression (outside the 95 % CI) of seven and three genes respectively, whilst supplementation with 12 μm-ZnCl2 led to no detectable changes in the JAR cell line. Gene responses to treatment with 12 μm-ZnCl2 for 12 h were not measured in the Caco-2 cell line because insufficient microarray slides were available. In the 24 h experiments, following exposure to 100 μm-ZnCl2, eleven genes in the Caco-2 cell line and fourteen genes in the JAR cell line were identified as differentially expressed, whilst exposure to 12 μm-ZnCl2 led to detectable changes in the expression of only six genes in the Caco-2 cell line and two genes in the JAR cell line. Interestingly, with the exception of the metallothionein genes (MT1H, MT2A, MT3), the lists of genes identified as differentially expressed in Caco-2 and JAR cells did not overlap. In general the observed changes in gene expression in Caco-2 and JAR cells treated with Zn concentrations of 12 μm and 100 μm were small and all were less than 2-fold. The exceptions to this generalisation were the MT genes (isoforms MT1H, MT2A and MT3), which were up-regulated at 100 μm-Zn by approximately 5–6-fold in Caco-2 cells and 7–8-fold in JAR cells.
A number of genes related to transport or metabolism of Cu and Fe were identified as differentially expressed in either Caco-2 or JAR cells. Following exposure to 100 μm-Zn for 24 h, the expression of hephaestin mRNA was down-regulated in Caco-2 cells, whilst ceruloplasmin expression was up-regulated in JAR cells under the same conditions. In addition, expression levels of transferrin, Fe-responsive element binding protein 2 (IREB2) and the Cu transporter hCTR2 were all increased in JAR cells exposed to 100 μm-Zn for 24 h, whilst the Cu transporter ATP7B was down-regulated in these cells following 24 h exposure to 12 μm extracellular Zn.
Validation of changes in gene expression by real-time reverse transcriptase polymerase chain reaction
We selected six of the candidate genes identified from microarray analyses of Caco-2 and JAR cells for verification by real-time RT-PCR (Supplementary Table 1). These genes comprised the following, all related to the transport and/or metabolism of dietary essential metals: Zn transporter ZnT1; multi-copper ferroxidases hephaestin and ceruloplasmin; Cu transporters hCTR2 and ATP7B; Fe-regulated transcription factor IREB2. Gene expression levels were normalised to the expression of glyceraldehyde-3-phosphate dehydrogenase. Real-time PCR results confirmed the up-regulation of both ZnT1 and IREB2 in JAR cells exposed to 100 μm-Zn for 24 h, detected via microarray analysis, and also demonstrated a significant increase in the expression of both genes at 12 μm-Zn (Table 3). The down-regulation of hephaestin mRNA expression in Caco-2 cells exposed to 100 μm-Zn for 24 h, detected by microarray analysis, was also confirmed by real-time PCR. For the remaining genes, results of microarray and real-time PCR analyses did not correlate. Our published data demonstrate a robust up-regulation of MT2A at increased extracellular Zn concentrations (100 μm and 150 μm) in Caco-2 and JAR cells(Reference Ford19, Reference Cragg, Christie and Phillips20, Reference Jackson, Helston and McKay23), so we did not repeat confirmation of metallothionein responses to Zn in the present study.
Table 3 Comparison of fold-changes in gene expression induced by zinc treatment in Caco-2 and JAR cells as detected by DNA microarray hybridisation or real-time PCR
(Mean values with their standard errors)

− , Reduced expression; ZnT, Zn transporter; +, increased expression; CTR, Cu transporter; IREB2, Fe-responsive element binding protein 2.
* Statistically significant (P < 0·05).
† GenBank accession number.
‡ Name of encoded protein.
§ Sample analysed, indicating cell line, Zn concentration and exposure time.
∥ Fold-changes, compared with expression levels in cells exposed to 3 μm extracellular Zn for the corresponding time-point, for n 4 (microarray) or n 3 (real-time PCR).
¶ Statistical analysis for microarray data was by Student's t test followed by Benjamini–Hochberg multiple testing correction (microarray), using an expression ratio of 1 as the control value.
†† Statistical analysis for real-time PCR data was by one-way ANOVA followed by Dunnett's post-test.
Profile of zinc regulation of the human MT2A promoter in Caco-2 and JAR cell lines
To investigate differences between the regulation profile of a well-characterised Zn-responsive gene in human intestinal and placental cells, we examined the effect of a range of extracellular Zn concentrations on transcriptional activity of the highly Zn-responsive human MT2A promoter in both Caco-2 and JAR cell lines. A β-galactosidase reporter construct incorporating the MT2A promoter was transfected transiently into Caco-2 and JAR cells and cells were exposed for 24 h to culture medium containing 3–150 μm-Zn as ZnCl2. The MT2A promoter–reporter construct showed concentration-dependent transcriptional activation by Zn in both Caco-2 and JAR cells, but the profile of concentration-dependence differed between the two cell lines (Fig. 1). In Caco-2 cells, the MT2A promoter was fairly refractory to Zn concentrations below 75 μm, whilst promoter activity in JAR cells was responsive to much lower Zn concentrations. For this comparison, reporter gene activity in each cell line was normalised to the corresponding value measured at 3 μm-Zn, to exclude any influence of transfection efficiency. A low level of β-galactosidase activity (approximately 10 % of the activity measured at 3 μm-Zn) measured in both Caco-2 and JAR cells transfected with a corresponding negative control reporter construct, containing no promoter insert(Reference Jackson, Helston and McKay23), was unchanged by extracellular Zn concentration. This observation excludes the possibility that the differential effect of Zn on β-galactosidase activity observed in the two cell lines could be attributed to any effect on endogenous gene expression or to any promoter-independent effect on reporter gene activity. These observations are consistent with the different extracellular Zn concentrations sensed by the two corresponding tissues in vivo. It is notable that the transcriptional activation of the MT2A promoter detected using our reporter gene approach was in excellent agreement with the pattern and magnitude of the response of the MT2A gene to Zn indicated by the microarray analysis (Fig. 1). Microarray analysis detected an increase in MT2A mRNA expression in Caco-2 cells at 100 μm-Zn but not at 12 μm-Zn (compared with 3 μm-Zn). In JAR cells the MT2A mRNA was increased at both 12 μm- and 100 μm-Zn.
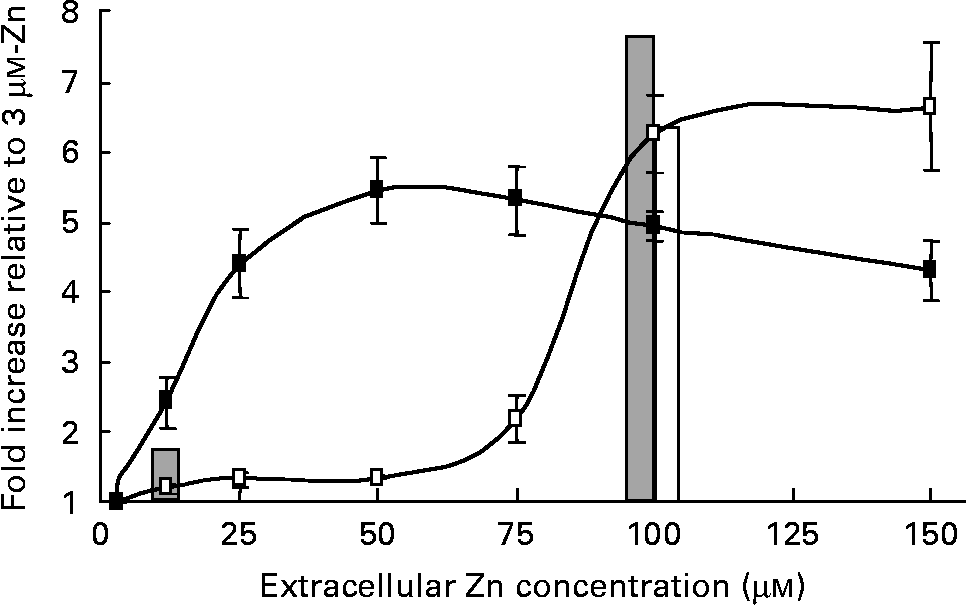
Fig. 1 Concentration dependence of transcriptional activation of the human MT2A promoter by Zn in Caco-2 (–□–) and JAR (–■–) cells. Cells were exposed to culture medium containing 3–150 μm-Zn as ZnCl2 for 24 h. Promoter activity was detected as activity in cell lysates of a β-galactosidase reporter gene immediately downstream of the MT2A promoter ( − 358 to +40) in the vector pBlue-TOPO (Invitrogen), assayed using the substrate chlorophenol red-β-d-galactopyranoside. Data are normalised to activity at 3 μm-Zn. Values are means (n 9–30), with standard errors represented by vertical bars. The bars show the fold-increase in MT2A mRNA at 12 μm- and 100 μm-Zn in Caco-2 (□) and JAR (![]() ) cells, compared with 3 μm-Zn, at 24 h detected by DNA microarray hybridisation.
) cells, compared with 3 μm-Zn, at 24 h detected by DNA microarray hybridisation.
Regulation of metal response element binding transcription factor (MTF)-1 and MTF-2 mRNA by zinc in Caco-2 and JAR cells
Differential sensitivity of Zn-responsive transcription factors may in part explain differences in gene responses to Zn in Caco-2 and JAR cell lines reported in the present and other studies(Reference Ford19, Reference Cragg, Christie and Phillips20). We sought to investigate this proposal by examining the effects of Zn on transcript levels of the Zn-responsive metal response element binding transcription factor (MTF)-1, and its recently identified homologue MTF-2. The responses of MTF-1 and MTF-2 to increased extracellular Zn concentration were measured by real-time RT-PCR (Fig. 2). MTF-1 and MTF-2 mRNA were unchanged in both cell lines at 12 μm- compared with 3 μm-Zn, but were reduced in Caco-2 cells only, and not in JAR cells, at 100 μm-Zn. Therefore, the abundance of both transcription factor mRNA were responsive to the extracellular Zn concentration in the Caco-2 cell line, but not the JAR cell line.

Fig. 2 The effect of extracellular Zn concentration on metal response element binding transcription factor (MTF)-1 (a, c) and MTF-2 (b, d) mRNA expression in Caco-2 (a, b) and JAR (c, d) cells. Cells were cultured for 24 h at the Zn concentrations indicated (added as ZnCl2) then total RNA was extracted and levels of MTF-1 and MTF-2 mRNA were measured by real-time RT-PCR, using SYBR green fluorescence and the Lightcycler 2·0. Data are expressed relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels measured in the same samples. Values are means (n 6), with standard errors represented by vertical bars. Statistical analysis was by one-way ANOVA followed by Bonferroni's multiple comparisons test. *** Mean value was significantly different from that at 3 μm-Zn (P < 0·001). ††† Mean value was significantly different from that at 12 μm-Zn (P < 0.001).
Discussion
We used the high-throughput capacity of DNA microarrays to examine gene regulation by Zn in intestine and placenta, using the human intestinal cell line Caco-2 and the human placental cell line JAR as appropriate models, and examined gene responses to extracellular Zn concentrations of 12 μm and 100 μm, representative of the Zn concentrations typically encountered by the placenta and intestine in vivo. In general, magnitudes of gene responses were modest and, with the exception of the metallothionein genes, all changes were less than 2-fold. These observations are consistent with results of other microarray studies, which report moderate gene-regulatory effects of both elevated and reduced Zn availability(Reference Blanchard, Moore and Green6, Reference Kindermann, Doring and Fuchs8). The use of a fold change cut-off is commonly employed as a method for classification of regulated genes in microarray experiments, including several examples of studies using DNA microarrays to identify Zn-regulated genes(Reference Beck, Li and Bao5, Reference Kindermann, Doring and Fuchs8–Reference Moore, Blanchard and McCormack10, Reference tom Dieck, Doring and Roth13). The choice of which fold change cut-off to use is somewhat arbitrary and a recent report highlighted the fact that using arbitrary threshold criteria can preclude changes with functional importance from consideration, as genes with little inherent variation, but expression ratios just below the cut-off, would be disregarded unnecessarily(Reference Blanchard, Moore and Green6). In view of this argument, we chose to use an alternative approach to identify genes differentially expressed, whereby the threshold cut-off was selected based on the variability of the data, allowing identification of small but significant changes in gene expression. The variance of the normalised set of housekeeping genes was used to generate an estimate of expected variance for the dataset, leading to predicted 95 % CI, and genes with expression ratios outside these CI were selected as differentially expressed. This approach has been used in other low-density microarray studies(Reference de Longueville, Atienzar and Marcq24, Reference de Longueville, Surry and Meneses-Lorente25) and measurement of variability of the data has been used as a technique to identify Zn-regulated genes in rat liver and jejunum(Reference Pfaffl, Gerstmayer and Bosio11). Statistical analysis of large datasets also requires consideration of the problems associated with multiple hypothesis testing, which results in the generation of false positives (type I errors). To control for false discovery rate we applied a Benjamini–Hochberg multiple testing correction (MTC) to statistical analyses of our microarray data. Although MTC limits identification of false positives, we recognise the possibility that some differentially expressed genes in the array experiments were missed due to the application of this correction.
The Zn supply to enterocytes and placental trophoblasts in vivo is subject to fluctuation, albeit to a different extent in each tissue. Placental trophoblasts are exposed to the Zn concentration of the blood, which can experience some degree of flux but is generally maintained within a fairly narrow range (12–15 μm) and rarely exceeds 20 μm. In contrast, enterocytes are typically exposed to much higher and more variable Zn concentrations, potentially exceeding 100 μm following a meal or nutrient supplement. In view of the analogous roles of the intestine and placenta in nutrient transfer, we hypothesised that alterations in Zn supply would induce parallel changes in gene expression in Caco-2 and JAR cells. However, given that each tissue senses Zn over different concentration ranges, we predicted that response profiles to Zn in intestinal and placental cells would be analogous, but would occur at different, physiologically relevant Zn concentrations.
In contrast to these predictions, our array analyses demonstrated that approximately equal numbers of genes were regulated by Zn in both Caco-2 and JAR cells under the same conditions, and that fewer genes were regulated at 12 μm- than at 100 μm-Zn in both cell lines. Additionally, we found a lack of correlation between Zn-responsive genes in Caco-2 and in JAR cells at the two Zn concentrations employed in the present study. Our observations suggest that, despite the analogous roles of the intestine and placenta in nutrient transfer, Zn may modulate different transcription factors and/or regulatory pathways, leading to the regulation of different subsets of genes, in placental and intestinal cells. We propose a number of possible explanations for this finding. One possibility is that exposure to the same concentration of extracellular Zn does not result in comparable changes in intracellular Zn in the two cell lines, as a result of differences in the balance between Zn uptake, efflux and compartmentalisation by specific transport proteins. In support of this concept, we previously reported differential effects of Zn on the expression of Zn uptake and efflux transporters in human intestinal and placental cell lines(Reference Ford19, Reference Cragg, Christie and Phillips20). Tissue- or cell type-specific gene regulation by Zn may also be due to the existence of transcription factors and regulatory elements that are expressed or function only in certain cell types. The best-characterised Zn-responsive transcription factor in mammals is MTF-1, which mediates Zn-induced transcriptional activation by binding to metal response elements in gene proximal promoter regions(Reference Andrews26). MTF-1 is involved in Zn-induced transcriptional activation of human metallothionein 2A(Reference Koizumi, Suzuki and Ogra27) as well as mouse metallothionein 1/2(Reference Heuchel, Radtke and Georgiev28) and the Zn transporter ZnT1(Reference Langmade, Ravindra and Daniels29). MTF-2 was identified recently as a putative homologue of MTF-1(Reference Cousins, Blanchard and Popp7) and, although its function remains elusive, MTF-2 and MTF-1 mRNA were decreased and increased respectively in Zn-deficient THP-1 cells(Reference Cousins, Blanchard and Popp7), suggesting potential reciprocal roles in Zn-regulated gene expression. In the present study we demonstrate that the expression of both MTF-1 and MTF-2 mRNA is decreased in response to elevated extracellular Zn (100 μm) in Caco-2 cells only, and not in JAR cells. We suggest that the differential sensitivity to Zn of these two transcription factors may explain to some extent the differences between gene regulation in Caco-2 and JAR cells. Since a number of other reports document tissue- or cell type-specific regulation of gene expression by Zn, including variations in the direction and magnitude of change(Reference Ford19, Reference Cragg, Christie and Phillips20, Reference Liuzzi, Blanchard and Cousins30–Reference Pfaffl and Windisch32), it is possible that the differential sensitivity of MTF-1 and MTF-2 extends to other tissues and cell types.
In addition we present evidence that cell type-specific responses of specific genes to changes in Zn availability are a consequence of differential sensitivity of the Zn-sensing mechanism(s) in each cell type. Using a reporter gene approach, in the present study we demonstrated that transcriptional activation of the highly Zn-responsive human MT2A promoter occurs over different Zn concentration ranges in Caco-2 and JAR cells, suggesting that there may be different ‘set points’ for the Zn-sensor in the intestine and placenta.
The results of our microarray analysis offer several opportunities for future investigation of the physiological importance and mechanisms of regulation of specific genes by Zn. For example, a number of genes related to transport or metabolism of Cu and Fe were identified as Zn-regulated genes in either Caco-2 or JAR cells. These included the multi-copper ferroxidases hephaestin and ceruloplasmin, the Cu transporters hCTR2 and ATP7B, the Fe-binding protein transferrin and the Fe-regulated mRNA stability factor IREB2. Regulation of these genes by Zn is of considerable interest as previous studies have reported adverse effects of elevated Zn availability on Cu and Fe status in both animal models(Reference Cox, Schlicker and Chu33) and human subjects(Reference Fischer, Giroux and L'Abbe34–Reference Donangelo, Woodhouse and King36) but the mechanisms through which these effects are mediated are not well understood. It should be noted that the regulatory effects of Zn on the mRNA levels of these genes, as detected by microarray analysis, were confirmed by real-time PCR for hephaestin and IREB2 only. Therefore, further confirmation will be required before the physiological relevance of the remaining genes can be assessed.
Hephaestin, a basolateral membrane ferroxidase responsible for oxidation of Fe to the ferrous state, is highly expressed in the gut and is required for the release of Fe from the enterocyte, as illustrated by the anaemic phenotype of sex-linked anaemia (sla) mice that have a mutant form of hephaestin(Reference Vulpe, Kuo and Murphy37). Human studies show an inhibitory effect of high doses of Zn on intestinal Fe absorption(Reference Whittaker38–Reference Olivares, Pizarro and Ruz40). The observed down-regulation of hephaestin transcript levels in Zn-supplemented (100 μm) Caco-2 intestinal cells may be a mechanistic explanation for these effects. In support of this view, a previous study reported negative effects of Zn supplementation on both hephaestin protein expression in the intestine and retention of an oral dose of 59Fe in rat pups given supplemental Zn orally from postnatal day 2(Reference Kelleher and Lonnerdal41).
Microarray analyses detected up-regulation of the mRNA stability factor IREB2 following treatment of JAR cells with 100 μm extracellular Zn for 24 h and a similar effect was detected at 12 μm-Zn by real-time RT-PCR. These data indicate that, in placental cells, IREB2 is particularly sensitive to changes in the extracellular Zn supply. A previous study demonstrated down-regulation of IREB2 expression in human mononuclear THP-1 cells treated with 40 μm-ZnSO4(Reference Cousins, Blanchard and Popp7), suggesting that the pattern of regulation by Zn may be cell type specific. IREB2 is expressed in human placenta(Reference Bradley, Leibold and Harris42) and may be involved in the post-transcriptional control of placental transferrin receptor 1, which is responsible for Fe uptake at the villous membrane of the placenta by receptor-mediated endocytosis(Reference McArdle, Danzeisen and Fosset43, Reference Verrijt, Kroos and Huijskes-Heins44). Elevated Zn concentrations can affect placental Fe transport, as shown by the increased fetal plasma Fe and placental Fe content when pregnant rats were fed Zn-supplemented diets(Reference Barone, Ebesh and Harper45). The observed up-regulation of IREB2 expression in JAR cells at elevated extracellular Zn concentrations may be explained by the potential role of IREB2 in the regulation of placental Fe uptake pathways.
The observed apparent differences in gene regulation derived from the microarray analysis and kinetic real-time RT-PCR may be a consequence of the relatively small changes in expression observed, the use of different normalisation methods or the interrogation of different regions of the sequence using the two different techniques, which may lead to the detection of splice-variant-specific effects with only one or other approach. Interestingly, in confirmation of our findings, a recent study of the effect of dietary Zn deficiency on gene expression in rat hepatocytes reported that the direction of regulation of ceruloplasmin detected by array data was opposite to that observed with Northern blotting(Reference tom Dieck, Doring and Roth13). Limitations with respect to microarray sensitivity or interrogation of different regions of the coding sequence using microarray hybridisation compared with RT-PCR may also account for the fact that we detected no Zn-dependent change in expression of ZnT1 and ZnT5 mRNA in Caco-2 cells, in contrast to our previous observations made in human intestinal mucosa, where we measured reduced ZnT1 mRNA levels in response to oral Zn supplementation(Reference Cragg, Phillips and Piper46), and in Caco-2 cells, where we measured increased levels of ZnT1 and ZnT5 mRNA at the same concentration of extracellular Zn (100 μm)(Reference Cragg, Christie and Phillips20), but over a longer (3 or 7 d) period of exposure, which may, itself, be an alternative explanation. Similarly, a time-dependent effect may explain the observation that microarray analysis in the present study detected an increase in ZnT1 mRNA in the placental cell line JAR after exposure to 100 μm-Zn for 24 h, whilst we previously detected no change in ZnT1 expression in JAR cells exposed to 100 μm-Zn for 7 d(Reference Ford19).
Two other Zn-regulated genes identified by microarray analyses merit further comment. The first of these is liver fatty acid-binding protein, which was down-regulated by 1·4-fold in Caco-2 cells treated with 12 μm-Zn for 24 h. Previous microarray analyses showed that mRNA levels of a number of genes involved in lipid metabolism, including liver fatty acid-binding protein, were modulated by low Zn status in rats(Reference tom Dieck, Doring and Fuchs12, Reference tom Dieck, Doring and Roth13). Regulation of liver fatty acid-binding protein expression by Zn supply may in part provide a mechanistic explanation for reports that Zn-deficient rats develop fatty livers, characterised by increased TAG accumulation(Reference Eder and Kirchgessner47, Reference Eder and Kirchgessner48). The second gene is the Na+/H+ exchanger NHE1, which was up-regulated by 1·5-fold in Caco-2 cells exposed to 100 μm-Zn for 24 h. Consistent with this finding, previous studies demonstrated that elevated extracellular Zn concentrations (>50 μm) activated Na+/H+ exchange in HT-29 human colonic cells and enhanced recovery from acidic pH(Reference Azriel-Tamir, Sharir and Schwartz49).
In summary, the data presented in the present study demonstrate that human intestinal and placental cells display differential transcriptomic responses to changes in Zn availability. These effects may in part be attributed to transcription factors whose activities are dependent on whether the intracellular environment is intestinal or placental. The response profile of the human MT2A promoter to Zn indicates that there may be a different set-point for the Zn sensor in these two tissues. Data from microarray analyses suggest that different genes respond to changes in Zn availability in intestinal cells compared with placental cells and a number of these genes represent targets for future research into nutrient interactions between Zn and other essential metals. An understanding of these interactions is essential for assessing the potential risks to nutritional status posed by dietary Zn supplementation.
Acknowledgements
The present study was supported by the United Kingdom Biotechnology and Biological Sciences Research Council Grant 13D/18 271 (to D. F. and J. C. M.) and by a research studentship from the Medical Research Council (to K. A. J.). We would also like to acknowledge financial support from the Rank Prize Funds.
K. A. J. was responsible for all experimental and analytical work, other than that attributed to other authors as follows, and for the initial preparation of the manuscript. R. A. V. was responsible for the identification of genes to include on the microarrays and for preliminary optimisation of microarray hybridisation procedures. J. A. M. carried out and analysed experiments presented as Fig. 2. D. C. S. provided advice and support concerning microarray analysis. J. C. M. provided support and guidance throughout the study, including the initial study design, and provided advice on preparation of the manuscript. D. F. provided support and guidance throughout the study, including the initial study design, and contributed to the preparation of the manuscript.
The authors have no conflicts of interest to declare.
The supplementary material (Supplementary Table 1) mentioned in this article can be found at http://journals.cambridge.org/bjn







