


Seeing is believing and the development of high-resolution microscopes originally provided the most conclusive evidence for the existence of bacteria or animalcules (tiny animals) as they were first described.Reference Lane1 Microscopy continues to be a central tool in modern bacterial biophysics and, when combined with quantitative image analysis tools, microscopes can provide unambiguous quantitative data to answer many of the questions related to bacterial behaviour.
A simple inverted optical microscope is shown in Figure 1.1. It follows a simple 4f geometry, where f is the focal length of the condenser and objective lenses, and in practice, additional improvements are standard on laboratory microscopes e.g. Köhler illumination (to illuminate specimens in a uniform manner), phase contrast (to provide additional contrast for thin or transparent specimens), fluorescence optics (to allow imaging of fluorescent samples, Figure 13.8) and confocal pin holes (to improve background rejection, Figure 13.7).Reference Mertz2

Figure 1.1 A simple inverted optical microscope in a 4f configuration (f is the focal length of both the condenser and the objective lenses).Reference Mertz2 An E. coli cell is shown, and the flagella would be invisible without a dedicated contrast mechanism (fluorescence or phase contrast is needed).
Once objects are identified in a microscopy image (the process of segmentation), analysing their dynamics by linking objects in consecutive images provides a rich source of additional information i.e. tracks are created that can be statistically analysed. Tracks can describe the motion of whole cells on the microscale, single molecules on the nanoscale or organelles on intermediate length scales e.g. the swimming behaviour of Escherichia coli (of micrometre length scales), the motion of proteins attached to a membrane (of nanometre length scales) or the transverse fluctuations of the endoplasmic reticulum in eukaryotic cells (of 10–1 000 nm length scales).Reference Perkins, Allan and Waigh3
1.1 How to Track Cells
Experimentally, tracking single cells is less demanding than single molecules due to their larger size, so it is a good place to start.Reference Dubay, Acres, Riebeles and Nadeau4 For strains of readily culturable bacteria, cells can be imaged with standard microscopy techniques using absorption contrast i.e. no complicated sample preparation is required, such as staining techniques. To track cells, first a sequence of well-resolved microscopy images need to be acquired. Standard types of imaging modality that can be used to create movies of dispersed bacteria or early stages of biofilms include bright-field microscopy (very high speeds are possible i.e. ~105 frames per second), fluorescence microscopy (specific labelling is possible, but the technique is relatively slow due to the low photon yield of fluorescent processes) and confocal microscopy (allows three-dimensional [3D] imaging, but the scanning process of image acquisition often makes it slow). State-of-the-art super-resolution fluorescence microscopy techniques include lattice sheet microscopy that can achieve ~50 nm resolution at video rates (~50 frames per second)Reference Chen5 and MINIFLUX (a variety of stimulated emission depletion microscopy, STED, Figure 13.10) that can achieve ~1 nm resolution at 1 000 frames per second.Reference Wolff, Scheiderer, Engelhardt, Maththias and Hell6 The super-resolution techniques tend to be technically challenging (Section 13.2.4), and bright-field microscopy is much easier for beginner microscopists.
Ideally, movies of cells should be as long as possible, in terms of the number of frames, to maximise the amount of information available in the resultant tracks. Track length can be limited by the depth of focus of the microscope ( sectioning), field of view of the microscope (sampling in
sectioning), field of view of the microscope (sampling in 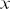 and
and 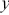 ), excessive particle speeds, the available memory on the camera (particularly an issue with ultrafast cameras), photobleaching of fluorescent labels and phototoxicity that damages the cells.
), excessive particle speeds, the available memory on the camera (particularly an issue with ultrafast cameras), photobleaching of fluorescent labels and phototoxicity that damages the cells.
Once a movie of the cells has been made, the next challenge is to segment the images of cells using image analysis software. Gaussian trackers can be used to locate the positions of compact symmetrical bacterial cells that may be reasonably approximated by Gaussian functions, but more complicated cellular shapes need more sophisticated forms of segmentation, such as neural networks (NNs) or snakes algorithmsReference Szeliski9 (Figures 1.2 and 1.4). Tracks are then made by connecting the centres of the segmented cells together in consecutive frames to form a linked list. Software searches for the closest positions of cells in consecutive images to link the cell centres together. If particles move substantial distances between consecutive images or the particle concentrations are too high, it can be an impossible task to unambiguously identify which particle contributes to which track. Particle positions can typically be measured with subdiffraction-limit resolution (often an order of magnitude improvement is possible on the diffraction limit) and sub-camera pixel resolution, because the weighted mean of measurements of the optical centre of mass of a particle is used i.e. averages over many pixels are calculated. Thus cell positions can be routinely tracked with ~10 nm resolution at ~104 frames per second using standard optical microscopes combined with fast complementary metal oxide semiconductor cameras (Figure 1.3).Reference Waigh10 Higher resolutions have been achieved with quantum metrology using squeezed light.Reference Xu, Zhang, Huang, Ma, Liu, Yonezawa, Zhang and Xiao11, Reference Taylor, Janousek, Daria, Knittel, Hage, Bachor and Bowen12

(a) A Gaussian tracker segments objectsReference Rogers, Waigh, Zhao and Lu7 in a human epithelial cell.Reference Rogers, Waigh, Zhao and Lu7 Endosomes (red, ~100 nm in size) are identified within a frame from a bright-field microscope (inset).

(b) A convolutional neural network (CNN) segments Bacillus subtilis cells (red circles) immersed in a suspension of Brownian particles (red dots).Reference Helgadottir, Argua and Volpe8

Figure 1.3 (a) An image of an early-stage Staphylococcus aureus biofilm from bright-field optical microscopy.Reference Hart, Waigh, Lu and Roberts13, Reference Rogers, van der Walle and Waigh14 (b) Zoomed in region where single bacterial cells (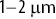 in size) can be observed. (c) A track of a single S. aureus cell made using Gaussian tracker software (Figure 1.2a). (d) Mean square displacements (MSD) as a function of time interval of hundreds of single S. aureus cells in the biofilm calculated from the tracks. S. aureus is immotile, so the cells’ motions are due to thermal forces modulated by the viscoelasticity of the biofilm.
in size) can be observed. (c) A track of a single S. aureus cell made using Gaussian tracker software (Figure 1.2a). (d) Mean square displacements (MSD) as a function of time interval of hundreds of single S. aureus cells in the biofilm calculated from the tracks. S. aureus is immotile, so the cells’ motions are due to thermal forces modulated by the viscoelasticity of the biofilm.
1.2 How to Track Single Molecules
Single molecules can now be routinely tracked both in vitro and in vivo inside single cells, although it was seen as a dramatic advance when single molecules were first imaged in condensed phases (Nobel Prize 2014).Reference Moerner and Kardor15, Reference Leake16 Many people were surprised that it was possible to discriminate single molecules in condensed phases against backgrounds containing vast numbers of molecules of the order of Avogadro’s number (6 × 1023) with a sufficient signal-to-noise ratio (SNR). Single-molecule imaging techniques are primarily based on fluorescence microscopy, and this requires specific labelling of the molecules of interest with fluorophores. An emission filter based on the wavelength shift of emitted photons from a fluorophore compared with the wavelength of the excitation light source (the Stokes shift) can be used to discriminate single fluorescent molecules against the background of a huge number of non-fluorescent molecules.
Large catalogues are available for commercial fluorophores that can label biomolecules with varying degrees of specificity, such as proteins, nucleic acids, carbohydrates and lipids. The specificity of the labels needs to be determined in a biological experiment to be certain of what is labelled using careful control experiments due to the large number of factors that affect fluorophore binding. An elegant solution for labelling proteins is to genetically modify them to add an extra fluorescent protein domain to their structure. This can be very effective for in vivo studies, but green fluorescent proteins (GFPs) can suffer from fast photobleaching (synthetic fluorophores often are much more photostable), bulky GFPs can perturb protein functionality (control experiments are needed), and there are time lags introduced by the GFP transcription that can limit studies of fast intracellular dynamics.
For molecular imaging, the choice of segmentation algorithm is determined in part by the geometry of the molecule. Extended molecules with extensive labelling (e.g. a large DNA molecule in which all the base pairs are fluorescently labelled) require snakes algorithms (Figure 1.4), whereas molecules with point-like labelling often use Gaussian trackers (Figure 1.2a).Reference Szeliski9 AI techniques (e.g. convolutional neural networks [CNNs]) can be more flexible in the types of molecular geometry they can analyseReference Newby, Schaefer, Lee, Forest and Lai19 but will suffer from poor SNRs if they are not properly constrained (Figure 1.2b). Often it is best to constrain artificial intelligence (AI) algorithms using simple physical models e.g. the probabilities of particle displacements can be constrained on the basis that particles will not teleport between different locations, which is a Bayesian approach. Current AI techniques often require extensive data sets to perform the training procedure i.e. they involve supervised learning (Chapter 14).

(a) Time dependence of the tracked contours of the ER tubules from fluorescence microscopy.

(b) Mean position of the ER tubule, where the variances of the transverse motions are highlighted. The tracked contours of ER tubules indicate active motion due to motor proteins.

(c) Peptide fibre positions in a gel from fluorescence microscopy.Reference Cox, Xu, Waigh and Lu17, Reference Cox, Cao, Xu, Waigh and Lu18
Figure 1.4 Snakes algorithms allow tracking of extended objects, such as the endoplasmic reticulum (ER) in human cellsReference Perkins, Allan and Waigh3 or a peptide fibre in a gel.Reference Cox, Xu, Waigh and Lu17
The choice of algorithm to link the positions of segmented particles together into a track also has a variety of optionsReference Chenouard20 e.g. nearest neighbour linking or multi-track optimisation are possible. Particular care is required when particles closely approach one another (they can easily switch labels), and tracks can become fragmented due to low SNRs (they can be stitched together, but often with limited success). Our experience is that the segmentation algorithm plays a more important role than the linking algorithm in the quality of the final tracks, but both are important.
Bayesian tracking techniques (Chapter 14) can be very useful to remove the noise on tracks e.g. Kalman filtering,Reference Wu, Agarwal, Hess, Khargonekar and Tseng21, Reference Murphy22 and the methodology has been extensively developed with satellite imaging. However, care must be taken that this noise is random and Markovian (independent noise fluctuations occur with no memory), since it is an assumption used in Kalman filtering. It is particularly an issue when considering non-Markovian processes e.g. the motility of microorganisms or the intracellular motion of molecules are frequently non-Markovian.Reference Waigh and Korabel23
1.3 The Statistics of Structures
The static images of molecules and cells from microscopy experiments can provide a range of useful information e.g. calculating their sizes, conformations and relative organisation. Standard freeware software allows the segmentation of bacteria in microscopy images and can provide quantitative descriptors of cell shape.Reference Dacret, Quardokus and Brun24 Sophisticated software has also been developed to segment bacterial biofilms and quantify their structures in three dimensions.Reference Hartmann25
Different statistical tools are needed to quantify the relative positions of bacteria or the molecules associated with them. The Ripley K function 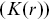 quantifies the intuitive notion of whether particles have been placed at random across a surface or they are clustered together or dispersed.Reference Holmes and Huber27 It is defined so that
quantifies the intuitive notion of whether particles have been placed at random across a surface or they are clustered together or dispersed.Reference Holmes and Huber27 It is defined so that 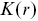 is the expected number of additional points within a distance
is the expected number of additional points within a distance  of a given point. This is a useful tool to understand the distributions of bacteria as they adsorb to surfaces. An alternative measure is given by the pair correlation function
of a given point. This is a useful tool to understand the distributions of bacteria as they adsorb to surfaces. An alternative measure is given by the pair correlation function 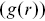 , which is widely used in condensed matter physics, particularly liquid-state theory and models of colloidal matter.Reference Allen and Tildesley28
, which is widely used in condensed matter physics, particularly liquid-state theory and models of colloidal matter.Reference Allen and Tildesley28 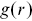 is defined as
is defined as
 (1.1)
(1.1)
where  is again the distance from a test point (Figure 1.5). With a stationary Poisson distribution of points,
is again the distance from a test point (Figure 1.5). With a stationary Poisson distribution of points, 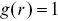 i.e. a complete random arrangement with no correlations.
i.e. a complete random arrangement with no correlations. 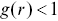 indicates an anti-correlation between points (dispersion), whereas
indicates an anti-correlation between points (dispersion), whereas 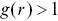 indicates clustering.Reference Holmes and Huber27
indicates clustering.Reference Holmes and Huber27 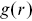 can be related to the interparticle potential if Boltzmann statistics are assumed for systems in thermal equilibriumReference Allen and Tildesley28, Reference Hansen and McDonald29 and has been extensively developed in liquid-state theory. In anisotropic systems,
can be related to the interparticle potential if Boltzmann statistics are assumed for systems in thermal equilibriumReference Allen and Tildesley28, Reference Hansen and McDonald29 and has been extensively developed in liquid-state theory. In anisotropic systems, 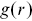 needs to be generalisedReference Kopera and Retsch26 e.g. correlations along separate lattice directions should be averaged separately to maintain the additional information needed to quantify the degree of anisotropy, such as with liquid crystalline materials.
needs to be generalisedReference Kopera and Retsch26 e.g. correlations along separate lattice directions should be averaged separately to maintain the additional information needed to quantify the degree of anisotropy, such as with liquid crystalline materials.

(a) The radial distribution function 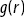 for a random lattice of pointsReference Kopera and Retsch26 (blue). The red curve is from a naïve numerical calculation.
for a random lattice of pointsReference Kopera and Retsch26 (blue). The red curve is from a naïve numerical calculation.

(b) Segmented positions of S. aureus bacteria in a biofilm (coloured with a measure of the linear viscoelasticity via the creep compliance, 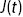 , Chapter 10).
, Chapter 10).

(c) the Ripley K function of the S. aureus bacteria in the biofilmReference Hart, Waigh, Lu and Roberts13 shown in (b) ( is the distance from a test point,
is the distance from a test point, 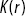 is the Ripley K function and
is the Ripley K function and 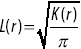 is the black line). It shows considerable clumping of the S. aureus.
is the black line). It shows considerable clumping of the S. aureus.
1.4 How to Analyse Particle Tracking Data
Statistical tools for handling tracking data can be very powerful. They play a central role in modern biological physics, since microscopy methods can provide high-resolution time series of images of living cells, biofilms and single molecules. Robust statistics are needed to test hypotheses on the behaviour of particles e.g. how they move, react, sense and oscillate. Furthermore, the analysis of particle tracking data can be conveniently extended to include tools from machine learning, since they have a common statistical basis, greatly increasing the possibilities for pattern recognition and large-scale automationReference Murphy22 (Chapter 14).
Tracks of individual bacteria provide a rich source of information on their behaviour e.g. their motility, chemosensing and interactions. Statistical tools need to be applied to the tracks to make sense of them. A wide variety of ad hoc bespoke statistical parameters could be defined e.g. a bacterium is motile if its average velocity over 1 s is 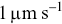 , but they are often unsatisfactory. To choose between alternative possible statistical parameters, standard mathematically elegant methods are preferable, since they provide better prospects for quantitative comparison with both analytical models and simulations. They can also be more robust to varying experimental conditions and thus generalise more easily.
, but they are often unsatisfactory. To choose between alternative possible statistical parameters, standard mathematically elegant methods are preferable, since they provide better prospects for quantitative comparison with both analytical models and simulations. They can also be more robust to varying experimental conditions and thus generalise more easily.
The transport of bacterial cells and molecules in the cells is often anomalous e.g. the central limit theorem breaks down and the probability density functions of their displacements are non-Gaussian. Mathematical models have been developed to describe anomalous transport (Chapter 2), although the relative merits of competing models are still debated.Reference Metzler and Klafter30 A recent innovation is to train NNs on anomalous transport models since NNs can then provide the dynamic segmentation of tracks (Chapter 14).Reference Han, Korabel, Chen, Johnston, Gavrilova, Allan, Fedotov and Waigh31 Particle tracks represent a special case of time series analysis, which find wide-ranging applications inside and outside biology e.g. forecasting the stock market or diagnosing heart disease based on electrocardiograms.Reference Nielsen32 There is thus a huge literature, and a wide range of mathematical tools have been developed.
A central tool for quantifying stochastic motion of particles is the mean square displacement (MSD, Chapter 2). For a random walk, the MSD 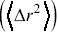 has a simple scaling dependence on time,
has a simple scaling dependence on time, 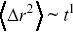 e.g. during Brownian motion (Figure 1.6). Furthermore, scaling of the MSDs is used to define anomalous transport,
e.g. during Brownian motion (Figure 1.6). Furthermore, scaling of the MSDs is used to define anomalous transport, 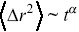 , where
, where 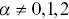 , and this is the type of stochastic motion most commonly observed for cellular motility and the motion of larger molecules inside cells.Reference Waigh and Korabel23, Reference Hofling and Franosch33 Average displacements
, and this is the type of stochastic motion most commonly observed for cellular motility and the motion of larger molecules inside cells.Reference Waigh and Korabel23, Reference Hofling and Franosch33 Average displacements 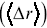 of particles are often not a useful measure of stochastic transport, since for symmetric stochastic processes, they average to zero,
of particles are often not a useful measure of stochastic transport, since for symmetric stochastic processes, they average to zero, 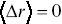 . Higher moments of the displacement probability distribution based on the third and fourth moments
. Higher moments of the displacement probability distribution based on the third and fourth moments 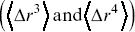 are useful for quantifying the skew and the degree of peakedness (the kurtosis), respectively. Moments of probability distributions of the displacements can provide average quantities to describe stochastic motility, which are reasonably robust to noise, but probability distribution functions (pdfs) contain additional information. Mathematically, the moment distribution is insufficient to unambiguously determine a pdf.Reference Sornette34
are useful for quantifying the skew and the degree of peakedness (the kurtosis), respectively. Moments of probability distributions of the displacements can provide average quantities to describe stochastic motility, which are reasonably robust to noise, but probability distribution functions (pdfs) contain additional information. Mathematically, the moment distribution is insufficient to unambiguously determine a pdf.Reference Sornette34

Figure 1.6 MSD as a function of time interval showing sub-diffusive, diffusive (Brownian), super-diffusive, ballistic and super-ballistic scaling behaviours. Note that super-ballistic scaling is rarely observed in low Reynolds number systems (they are overdamped), although it is possible in turbulent flows.
For stationary statistical processes,Reference Ibe35 often MSDs are averaged over time, and the MSD is then considered as a function of time interval 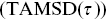 i.e.
i.e.
 (1.2)
(1.2)
where 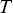 is the duration of the track and
is the duration of the track and  is the time.
is the time. 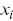 ,
, 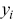 and
and 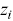 are the Cartesian coordinates of the particle
are the Cartesian coordinates of the particle  . The calculation of time-averaged MSDs (TAMSDs) can provide a major improvement in the SNR at short time intervals in experiments. If there are
. The calculation of time-averaged MSDs (TAMSDs) can provide a major improvement in the SNR at short time intervals in experiments. If there are 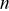 steps in a track, the error bars scale as
steps in a track, the error bars scale as 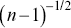 for the shortest time interval of the TAMSD,
for the shortest time interval of the TAMSD, 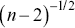 for the next shortest time interval and so on. Ensemble averaging of MSDs (EMSDs) over different particles is also possible to improve the SNR i.e. the MSDs are averaged over
for the next shortest time interval and so on. Ensemble averaging of MSDs (EMSDs) over different particles is also possible to improve the SNR i.e. the MSDs are averaged over  in Equation (1.2).
in Equation (1.2).
There is a general theorem by Birkhoff from dynamical systems theoryReference Birkhoff36, Reference Korabel, Taloni, Pagnini, Allan, Fedotov and Waigh37 that states 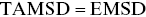 for an ergodic process, and it can be used as a diagnostic for ergodicity breaking e.g. whether glassy behaviour occurs in the tracks. MSDs can be calculated in one, two and three dimensions, and their analysis conveniently generalises to different dimensionalities e.g. to describe the motion of a motor along a DNA chain (one-dimensional [1D]), a particle in the plane of focus of a conventional optical microscope (two-dimensional [2D]) or a particle in a confocal microscope (3D). Stochastic aging (SA) is a separate issue to ergodicity and is often observed in biology e.g. stochastic processes are not stationary and evolve with time during the growth of a cell.Reference Korabel, Taloni, Pagnini, Allan, Fedotov and Waigh37 SA can be diagnosed by delaying tracks by different aging times (i.e. chop off the start of the data corresponding to the aging time) and then comparing the resultant MSDs. Aging and glassy phenomena have direct implications in medicine e.g. scarring during wound healing that involves non-ergodic glassy fibrous composites of collagen.
for an ergodic process, and it can be used as a diagnostic for ergodicity breaking e.g. whether glassy behaviour occurs in the tracks. MSDs can be calculated in one, two and three dimensions, and their analysis conveniently generalises to different dimensionalities e.g. to describe the motion of a motor along a DNA chain (one-dimensional [1D]), a particle in the plane of focus of a conventional optical microscope (two-dimensional [2D]) or a particle in a confocal microscope (3D). Stochastic aging (SA) is a separate issue to ergodicity and is often observed in biology e.g. stochastic processes are not stationary and evolve with time during the growth of a cell.Reference Korabel, Taloni, Pagnini, Allan, Fedotov and Waigh37 SA can be diagnosed by delaying tracks by different aging times (i.e. chop off the start of the data corresponding to the aging time) and then comparing the resultant MSDs. Aging and glassy phenomena have direct implications in medicine e.g. scarring during wound healing that involves non-ergodic glassy fibrous composites of collagen.
Velocities need to be handled carefully with stochastic processes, since with random walks they depend on the time scale at which they are measured.Reference Berg38 Often instantaneous velocities are defined in experiments as 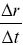 (
(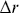 is the displacement of a particle over a time interval,
is the displacement of a particle over a time interval, 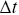 ), but this quantity is sensitive to the choice of
), but this quantity is sensitive to the choice of 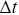 e.g. a smaller choice of
e.g. a smaller choice of 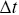 can correspond to higher values of velocity for sub-ballistic processes. Lots of values of motor protein velocities in the literature are mishandled due to such issues and when faster cameras are manufactured, the quoted motor protein velocities often also increase. More robust methods to quantify velocities are to consider velocity autocorrelation functions
can correspond to higher values of velocity for sub-ballistic processes. Lots of values of motor protein velocities in the literature are mishandled due to such issues and when faster cameras are manufactured, the quoted motor protein velocities often also increase. More robust methods to quantify velocities are to consider velocity autocorrelation functions 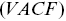 or velocities calculated via first passage probabilitiesReference Rogers, Flores-Rodriguez, Allan, Woodman and Waigh39 (see later).
or velocities calculated via first passage probabilitiesReference Rogers, Flores-Rodriguez, Allan, Woodman and Waigh39 (see later). 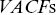 (Figure 1.7) can be defined as
(Figure 1.7) can be defined as
 (1.3)
(1.3) (1.4)
(1.4)
where 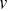 is the velocity at time
is the velocity at time  ,
, 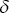 is the time interval,
is the time interval, 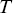 is the duration of the experiment and
is the duration of the experiment and  is the displacement.
is the displacement.

Figure 1.7 Velocity autocorrelation function (VACF) as a function of lag time for a particle moving inside a cell. The negative values of the VACF are due to anti-correlation.
The use of probability distributions of survival times has its origins in medicine (Figure 1.8a).Reference Aalen, Borgan and Gjessing40 Histograms of the number of patients in a medical trial can be plotted as a function of time. If the death rate occurs at a constant value per unit time, the survival distribution has an exponential decay (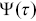 , a Poisson process). Decays due to more complex processes can be non-exponential, and hazard rates
, a Poisson process). Decays due to more complex processes can be non-exponential, and hazard rates 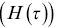 can be introduced to make it easier to visualise them i.e. a constant hazard rate as a function of time is equivalent to a single exponential decay for the survival time (Figure 1.8b). The hazard rate is the rate of death of a subject of age
can be introduced to make it easier to visualise them i.e. a constant hazard rate as a function of time is equivalent to a single exponential decay for the survival time (Figure 1.8b). The hazard rate is the rate of death of a subject of age  .
.

Figure 1.8 Plots of (a) the survival time distribution 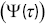 , (b) the hazard function
, (b) the hazard function 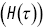 and the probability density function
and the probability density function 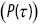 as a function of time
as a function of time 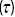 for the run times of a bacterium.
for the run times of a bacterium.
A practical problem for calculating survival times is when patients leave trials before they die, which biases the data due to a form of censuring. Kaplan–Meier estimators can be used to correct for these biases,
 (1.5)
(1.5)
where 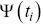 is the survival distribution at time
is the survival distribution at time 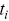 ,
, 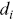 is the number of events that happen at
is the number of events that happen at 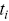 and
and 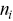 is the number of events that survive up to
is the number of events that survive up to 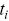 . Survival times can be used in the more general context of biological physics using such Kaplan–Meier corrections e.g. the run times of bacteria can be considered as a distribution of survival times. Biases introduced by the finite length of tracks in tracking experiments can be corrected using Equation (1.5). Survival times can also be used to understand the residence times of bacteria on surfaces.Reference Blee, Roberts and Waigh41 Survival times (e.g. for runs, Figure 1.8a) can be simpler than just considering histograms (or pdfs), since they are monotonic decreasing functions (in contrast, the run time pdfs,
. Survival times can be used in the more general context of biological physics using such Kaplan–Meier corrections e.g. the run times of bacteria can be considered as a distribution of survival times. Biases introduced by the finite length of tracks in tracking experiments can be corrected using Equation (1.5). Survival times can also be used to understand the residence times of bacteria on surfaces.Reference Blee, Roberts and Waigh41 Survival times (e.g. for runs, Figure 1.8a) can be simpler than just considering histograms (or pdfs), since they are monotonic decreasing functions (in contrast, the run time pdfs, 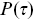 , will be peaked, Figure 1.8c).
, will be peaked, Figure 1.8c).
The first passage probability (FPP) for particle tracks is defined as the probability distribution for the times a particle takes to travel a specific distance for the first timeReference Redner43 (Figure 1.9a). The mean FPP 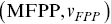 is the mean of the FPP distribution. In numerous biological situations, the FPP is the crucial statistical quantity of interest e.g. times for a chemical reaction to occur or for a particle to leave a maze. MFPPs for particles with multiple scaling regimes as a function of time imply that the reaction kinetics of the particle will also exhibit multiple regimes. Furthermore, FPPs can also provide more robust alternatives to instantaneous velocities to quantify motility and can help separate up the motility of particles using the average FPP velocity
is the mean of the FPP distribution. In numerous biological situations, the FPP is the crucial statistical quantity of interest e.g. times for a chemical reaction to occur or for a particle to leave a maze. MFPPs for particles with multiple scaling regimes as a function of time imply that the reaction kinetics of the particle will also exhibit multiple regimes. Furthermore, FPPs can also provide more robust alternatives to instantaneous velocities to quantify motility and can help separate up the motility of particles using the average FPP velocity 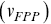 as a function of transit length (
as a function of transit length (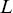 , Figure 1.9b) e.g. the question can be asked as to whether long-range transits happen at larger velocities, which is useful with endosomal transport.
, Figure 1.9b) e.g. the question can be asked as to whether long-range transits happen at larger velocities, which is useful with endosomal transport.

(a) First passage probability (FPP) of particles moving inside a cell as a function of time 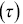 , red < blue < navy blue correspond to longer transit lengths.
, red < blue < navy blue correspond to longer transit lengths.

(b) Mean first passage velocity 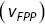 of the particles as a function of transit length
of the particles as a function of transit length 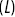 .Reference Rogers, Flores-Rodriguez, Allan, Woodman and Waigh39, Reference Flores-Rodriguez, Rogers, Kenwright, Waigh, Woodman and Allan42
.Reference Rogers, Flores-Rodriguez, Allan, Woodman and Waigh39, Reference Flores-Rodriguez, Rogers, Kenwright, Waigh, Woodman and Allan42
MSDs are insensitive to direction (so too are the survival times and the FPPs), and they just provide a measure of the amplitude of motion as a function of time. Angular correlations of particle displacements thus provide crucial information to understand particle motilityReference Harrison, Kenwright, Waigh, Woodman and Allan44 with respect to direction. Analogous to an MSD, the average direction cosine 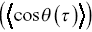 for segments along a track can be quantified as a function of time interval (averaged in an analogous manner to a TAMSD, Equation (1.1), although three points are required to define the consecutive displacements
for segments along a track can be quantified as a function of time interval (averaged in an analogous manner to a TAMSD, Equation (1.1), although three points are required to define the consecutive displacements 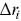 and
and 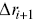 ), and a scalar product is used,
), and a scalar product is used,
 (1.6)
(1.6)
Cosines are bounded functions; 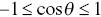 . Negative values of the angular correlation function correspond to anti-persistent motion i.e. the particle is constantly changing direction and tends to move back on itself.
. Negative values of the angular correlation function correspond to anti-persistent motion i.e. the particle is constantly changing direction and tends to move back on itself. 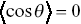 corresponds to no average directional bias and is expected for an unbiased random walk. Positive values of
corresponds to no average directional bias and is expected for an unbiased random walk. Positive values of 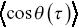 correspond to directional persistence. Such measures of directionality are useful for the development of models for bacteria, since bacteria act as stochastic swimmers, and for the motility of intracellular cargoes within bacterial cells. Some similar information is encoded in velocity correlation functions (Equation 1.2), but it is useful to have both measures.
correspond to directional persistence. Such measures of directionality are useful for the development of models for bacteria, since bacteria act as stochastic swimmers, and for the motility of intracellular cargoes within bacterial cells. Some similar information is encoded in velocity correlation functions (Equation 1.2), but it is useful to have both measures.
More sophisticated statistical measures are needed to describe the correlated motion of particles. Two-point correlation functions are one possibility, 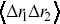 , where
, where 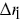 and
and 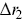 are the displacements for two different particles, which has been studied from the perspective of two-particle microrheology.Reference Levine and Lubensky45 Velocity cross-correlation functions
are the displacements for two different particles, which has been studied from the perspective of two-particle microrheology.Reference Levine and Lubensky45 Velocity cross-correlation functions 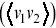 are also useful to study the mutual motion of cells e.g. in chemotactic fields, and provide similar information to
are also useful to study the mutual motion of cells e.g. in chemotactic fields, and provide similar information to 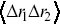 .
.
Flocking order parameters 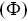 have also been introduced to describe phase transitions during coherent motion in motile particlesReference Vicsek, Czirok, Ben-Jacob, Cohen and Shochet46 (such as bacteria, starlings and ants) e.g.
have also been introduced to describe phase transitions during coherent motion in motile particlesReference Vicsek, Czirok, Ben-Jacob, Cohen and Shochet46 (such as bacteria, starlings and ants) e.g.
 (1.7)
(1.7)
where 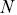 is the number of particles,
is the number of particles, 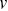 is the average velocity and
is the average velocity and 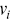 is the velocity of each particle. Care must be taken in calculating
is the velocity of each particle. Care must be taken in calculating 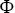 for particles that experience anomalous transport, since the values of
for particles that experience anomalous transport, since the values of 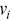 will depend on time for non-ballistic particle motion, and other order parameters have been suggested to make them more robust.Reference Cavagna, Giardina and Grigera47
will depend on time for non-ballistic particle motion, and other order parameters have been suggested to make them more robust.Reference Cavagna, Giardina and Grigera47
A sophisticated modern approach to the motility of both particles inside bacteria and cellular motility follows a framework of heterogeneous anomalous transport (HAT).Reference Itto and Beck48 This attempts to quantify the heterogeneity of the anomalous transport of particles in both space and time by considering generalised diffusion coefficients 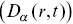 and scaling exponents
and scaling exponents 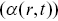 that vary in space and time, defined via the MSDs of the distributions using
that vary in space and time, defined via the MSDs of the distributions using
 (1.8)
(1.8)
where 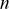 is the number of dimensions. Note that
is the number of dimensions. Note that 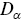 has fractional units, which provides some challenges e.g. it is not possible to plot
has fractional units, which provides some challenges e.g. it is not possible to plot 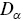 on a single axis. Rescaling
on a single axis. Rescaling 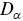 by characteristic length and time scales solves many of these problems. Thus, values of both
by characteristic length and time scales solves many of these problems. Thus, values of both 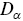 and
and 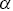 are allowed to vary with time and space during the analysis, which corresponds to a multi-fractal model.Reference Waigh and Korabel23 There is good evidence that HAT occurs for the majority of cellular motility and intracellular motility of large molecules and aggregates.
are allowed to vary with time and space during the analysis, which corresponds to a multi-fractal model.Reference Waigh and Korabel23 There is good evidence that HAT occurs for the majority of cellular motility and intracellular motility of large molecules and aggregates.
Experiments with extended linear objects, such as single molecules, aggregates of cells, organelles or individual cells, lend themselves to Fourier analysis of data segmented using snakes algorithmsReference Gittes, Mickey, Nettleton and Howard49 (Figure 1.4). The equipartition theorem can be used to calculate the energy of each Fourier mode assuming the fibres are in thermal equilibrium and simple continuum models are used for the energy of the snakes e.g. all the energy is stored in Hookean bending modes.Reference Gittes, Mickey, Nettleton and Howard49 Similar analysis is also possible with cell membranes in two dimensions.Reference Monzel and Sengupta50 Challenges occur to describe systems in which quenched disorder or active transport affect the conformations of the extended objects.Reference Perkins, Allan and Waigh3, Reference Cox, Xu, Waigh and Lu17
Other less direct methods of calculating stochastic processes occur in the literature. For example, the square of the Fourier transform of the particle displacement as a function of the correlation time is called the power spectral density 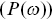 and is often measured in optical tweezer experiments. The information content is similar to a MSD as a function of time interval, but MSDs are often simpler to work with.
and is often measured in optical tweezer experiments. The information content is similar to a MSD as a function of time interval, but MSDs are often simpler to work with.
1.5 Scattering Alternatives
Instead of working directly with images, scattering experiments function in reciprocal space. Historically, scattering techniques were used in situations where imaging was not possible e.g. in experiments with hard X-rays or thermal neutrons where it is hard to construct an imaging lens, such as the Braggs’ initial work to study the structure of simple crystals, such as sodium chloride. Images tend to be preferred in modern-day biological physics experiments, since they are easier to interpret and tend to be less ambiguous, but scattering data can also be useful. Inelastic scattering experiments (e.g. dynamic light scattering, X-ray photon correlation spectroscopy or quasi-elastic neutron scattering) detect small energy changes in scattered radiation and often provide much faster dynamic information than is currently possible with imaging experiments e.g. point detectors can stream data much faster than pixel arrays. In bacterial biophysics, fluorescence correlation spectroscopy (FCS) and dynamic differential microscopy (DDM) are commonly used scattering techniques and can be microscope based to improve their spatial sensitivity.
In DDM, a movie of a biological system is made with a microscope and versions are possible using both coherent (e.g. bright-field contrast) and incoherent (e.g. fluorescence) scattering. Software correlators can then be used with stacks of the images to calculate correlation functions that describe the image dynamics (Figure 1.10). The correlation functions allow access to identical information as inelastic scattering experiments, although they are at relatively slow time scales due to the update times of pixel arrays used on standard digital cameras in optical microscopes.

Figure 1.10 (a) A schematic diagram of the algorithm used to analyse DDMReference Germain, Leocmach and Gibaud51 experiments. (b) An example of the difference of two images 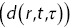 . (c) The square of the Fourier transform of the difference of two images. (d)
. (c) The square of the Fourier transform of the difference of two images. (d) 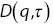 (the square of the Fourier transform of
(the square of the Fourier transform of 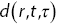 averaged over
averaged over  ) as a function of momentum transfer,
) as a function of momentum transfer, 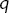 . (e)
. (e) 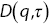 as a function of time interval
as a function of time interval 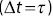 .
.
Intermediate scattering functions 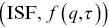 are a key statistical tool used in inelastic scattering experiments to quantify the dynamics e.g. with light, neutrons and X-ray.Reference Berne and Percora52 DDM can be used to extract the ISF from stacks of images. There needs to be a source of speckle on the images (which is non-necessarily due to coherent scattering) and the images do not need to be particularly well resolved. DDM works well on images from both bright-field (with both laser and light-emitting diode [LED] illumination) and fluorescence microscopy (the calculations are slightly different in each case). Taking differences between images suppresses the noise due to stationary particles and detector heterogeneitiesReference Cerbino and Cicuta53 (hence the name DDM), which gives
are a key statistical tool used in inelastic scattering experiments to quantify the dynamics e.g. with light, neutrons and X-ray.Reference Berne and Percora52 DDM can be used to extract the ISF from stacks of images. There needs to be a source of speckle on the images (which is non-necessarily due to coherent scattering) and the images do not need to be particularly well resolved. DDM works well on images from both bright-field (with both laser and light-emitting diode [LED] illumination) and fluorescence microscopy (the calculations are slightly different in each case). Taking differences between images suppresses the noise due to stationary particles and detector heterogeneitiesReference Cerbino and Cicuta53 (hence the name DDM), which gives
 (1.9)
(1.9)
Next, these image differences are Fourier transformed in space (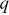 is the momentum transfer) and squared,
is the momentum transfer) and squared,
 (1.10)
(1.10)
The ISF 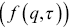 is constructed by fitting
is constructed by fitting 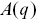 and
and 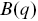 using
using
 (1.11)
(1.11)
where 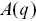 and
and 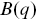 are assumed arbitrary smooth functions.
are assumed arbitrary smooth functions.
In DDM, a major advantage compared with tracks from direct imaging is that the dynamics can be averaged and quantified without segmentation of the images. The neglect of segmentation can be an advantage for complex hierarchical structures e.g. the endoplasmic reticulum in eukaryotic cells, where it can be hard to unambiguously locate the objects’ boundaries.Reference Perkins, Allan and Waigh3 Challenges with DDM are that some spatial information is lost in the averaging procedures (e.g. during the calculation of the Fourier transforms), analytic calculations are slightly harder in reciprocal space and it can be more challenging to determine which specific structures are being analysed.
Fluorescence correlation spectroscopy considers the fluctuations in fluorescent emission from a small volume that is illuminated in a sampleReference Rigler and Elson54 (Figure 1.11). The fluctuations in fluorescent emission can be related to the motion of the fluorophores through the calculation of correlation functions (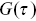 , where
, where 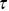 is the correlation time) and can be performed relatively quickly (using fast point photodetectors) and thus experiments can be performed with quickly photobleaching fluorophores. If only a single detection volume is used in the sample, it is harder to explore the length dependence of the dynamic processes using FCS (via
is the correlation time) and can be performed relatively quickly (using fast point photodetectors) and thus experiments can be performed with quickly photobleaching fluorophores. If only a single detection volume is used in the sample, it is harder to explore the length dependence of the dynamic processes using FCS (via 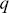 , the momentum transfer). This can make the exploration of anomalous transport more challenging (the spatial dependence of the motility is ambiguous) and a partial solution is to use a range of pin hole sizes, so different volumes are illuminated in the sample.Reference Stolle and Fradin55 In this case, the fluorophores need to be long-lived and the statistical processes must be stationary (they should not evolve with time).
, the momentum transfer). This can make the exploration of anomalous transport more challenging (the spatial dependence of the motility is ambiguous) and a partial solution is to use a range of pin hole sizes, so different volumes are illuminated in the sample.Reference Stolle and Fradin55 In this case, the fluorophores need to be long-lived and the statistical processes must be stationary (they should not evolve with time).

Figure 1.11 (a) Apparatus for FCS is based on a fluorescence microscope, which uses a pinhole to define the detection volume. (b) The intensity of light 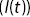 emitted by fluorophores in the detection volume as a function of time. (c) A correlation function
emitted by fluorophores in the detection volume as a function of time. (c) A correlation function 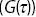 of the intensity fluctuations as a function of time interval
of the intensity fluctuations as a function of time interval 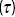 from (b). The correlation function can be used to quantify the motion of the fluorescence particles within the detection volume.
from (b). The correlation function can be used to quantify the motion of the fluorescence particles within the detection volume.

















