



Introduction
Over the past four decades, molecular population genetics has proven to be a fruitful field in addressing fundamental questions regarding the population biology of helminth parasites. Although its implementation by parasitologists initially lagged behind those in other fields (Criscione, Reference Criscione, Janovy and Esch2016), molecular population genetic methodologies have since been used to help elucidate the ecology, epidemiology/epizoology and evolution for a number of parasitic helminths (Nadler, Reference Nadler1995; Criscione et al., Reference Criscione, Poulin and Blouin2005; de Meeûs et al., Reference De Meeûs, McCoy, Prugnolle, Chevillon, Durand, Hurtrez-Bousses and Renaud2007; Cole & Viney, Reference Cole and Viney2018). Moreover, the field has been integral in overcoming inherent challenges (e.g. laboratory maintenance of complex life cycles, need to dissect hosts, etc.) by allowing indirect inferences on population biology (de Meeûs et al., Reference De Meeûs, McCoy, Prugnolle, Chevillon, Durand, Hurtrez-Bousses and Renaud2007). By sequencing one or a few targeted loci (e.g. a region of the mitochondria or ribosomal DNA) or genotyping a panel of five to 20 codominant genetic markers (e.g. allozymes or microsatellites), parasitologists could begin to elucidate previously unknown or inaccessible helminth population dynamics. For example, molecular markers have proven essential in parasite studies on cryptic species (Nadler & de Leon, Reference Nadler and De Leon2011), hybridization (Detwiler & Criscione, Reference Detwiler and Criscione2010), modes of reproduction (Tibayrenc & Ayala, Reference Tibayrenc and Ayala2013), mating systems (Detwiler et al., Reference Detwiler, Caballero and Criscione2017), contemporary or historical effective population sizes (Ne) (Archie & Ezenwa, Reference Archie and Ezenwa2011; Strobel et al., Reference Strobel, Hays, Moody, Blum and Heins2019), phylogeography (Nieberding et al., Reference Nieberding, Morand, Libois and Michaux2006), local scale transmission (Criscione et al., Reference Criscione, Anderson, Sudimack, Subedi, Upadhayay, Jha, Williams, Williams-Blangero and Anderson2010) and anthelmintic drug resistance (Gilleard & Beech, Reference Gilleard and Beech2007).
Molecular population genetics has helped advance our knowledge about parasites; nonetheless, this ‘first generation’ of parasite population genetics often required large time investments to collect data and/or monetary constraints that largely restricted studies to the use of one or a few targeted loci. Many molecular ecological questions can be addressed with a few genetic markers, but inferences in some areas remain limited or intractable. For instance, drug resistance studies may have missed novel loci due to a focus on candidate gene approaches (Gilleard & Beech, Reference Gilleard and Beech2007; Doyle & Cotton, Reference Doyle and Cotton2019), historical demographic estimates may have been less accurate (Putman & Carbone, Reference Putman and Carbone2014), evolutionary history conclusions based on a single marker could reflect the history of the locus rather than the species (Casillas & Barbadilla, Reference Casillas and Barbadilla2017), or mito-nuclear discordance patterns might be inconclusive in disentangling incomplete lineage sorting from hybridization (Joly et al., Reference Joly, McLenachan and Lockhart2009; Perea et al., Reference Perea, Vukić, Šanda and Doadrio2016).
In 2005, the first next generation sequencing (NGS) technologies were developed (Margulies et al., Reference Margulies, Egholm and Altman2005; Pareek et al., Reference Pareek, Smoczynski and Tretyn2011). Numerous genetic markers across the genome, for example, thousands of single nucleotide polymorphisms (SNPs), could now be obtained in a short time frame and at a reduced cost per locus (Leshchiner et al., Reference Leshchiner, Alexa and Kelsey2012). Thus, emerged the field of population genomics, which as stated by Charlesworth (Reference Charlesworth2010) ‘is a new term for a field of study that is as old as the field of genetics itself, assuming that it means the study of the amount and causes of genome-wide variability in natural populations.’ While the primary benefits of population genomics may not necessarily be the generation of novel questions (Charlesworth, Reference Charlesworth2010), Luikart et al. (Reference Luikart, Kardos, Hand, Rajora, Aitken, Hohenlohe and Rajora2018) discussed how population genomics has led to ‘conceptually novel approaches to address questions intractable by traditional genetic methods by using high-density genome-wide markers (e.g. DNA, RNA, epigenetic marks),’ especially questions centred on adaptive evolution. Thus, genomic approaches have increased the accessibility of the population genetics field to further our understanding of parasite ecology and evolution.
The advantages in population genomics data lie in two non-mutually exclusive areas. First, NGS enables researchers to assemble the genomes of target organisms with no existing or limited genomic resources (Luikart et al., Reference Luikart, Kardos, Hand, Rajora, Aitken, Hohenlohe and Rajora2018). In turn, an assembly, especially a highly contiguous assembly, allows annotation to characterize functional and structural variants in the genome (Thomma et al., Reference Thomma, Seidl, Shi-Kunne, Cook, Bolton, van Kan and Faino2016). Second, the availability of additional genetic markers improves the ability to accurately predict population genetic parameters (e.g. demography), provides a backdrop to identify loci under selection (i.e. outlier-based tests) and allows tracking genetic changes along genomic regions (i.e. inferences are along haplotypes vs. individual SNPs) (Allendorf et al., Reference Allendorf, Hohenlohe and Luikart2010; Luikart et al., Reference Luikart, Kardos, Hand, Rajora, Aitken, Hohenlohe and Rajora2018). For example, reductions in nucleotide diversity along chromosomal segments help identify strong selective sweeps (Stephan, Reference Stephan2019; Garud et al., Reference Garud, Messer and Petrov2021); the frequency and length of stretches of homozygosity (runs of homozygosity (ROH)) estimate inbreeding and demographic history more accurately (Ceballos et al., Reference Ceballos, Joshi, Clark, Ramsay and Wilson2018); and the length of haplotypes with derived alleles (extended haplotype homozygosity (EHH)) have greater ability to detect soft selective sweeps (Garud et al., Reference Garud, Messer and Petrov2021). Such data enable greater access to complex evolutionary questions related to the genetic architecture of adaptive traits or differentiation, loci affecting phenotypic variation or fitness, hybridization and adaptive introgression and associations of landscape/environmental features to genomic variation (Charlesworth, Reference Charlesworth2010; Luikart et al., Reference Luikart, Kardos, Hand, Rajora, Aitken, Hohenlohe and Rajora2018; Rochus et al., Reference Rochus, Tortereau, Plisson-Petit, Restoux, Moreno-Romieux, Tosser-Klopp and Servin2018; Orteu & Jiggins, Reference Orteu and Jiggins2020). Taken together, the rapidly expanding field of population genomics presents new opportunities to elucidate parasite population biology by giving a more complete picture of the genome.
Here, we review population genomics studies on parasitic helminths of animals. Although interesting, between-species comparative genomics (e.g. synteny comparisons) is beyond the scope of our review (see Coghlan et al., Reference Coghlan, Tyagi and Cotton2019). We also do not review various statistical methodologies for population genomics analyses (we refer readers to reviews by Hahn, Reference Hahn2018; Luikart et al., Reference Luikart, Kardos, Hand, Rajora, Aitken, Hohenlohe and Rajora2018; Bourgeois & Warren, Reference Bourgeois and Warren2021). Rather, a primary goal is to highlight how current studies are applying NGS and population genomics to study helminth population biology and microevolution. We cover many classical topics in molecular ecology outlined in earlier reviews (Nadler, Reference Nadler1995; Criscione et al., Reference Criscione, Poulin and Blouin2005; Detwiler & Criscione, Reference Detwiler and Criscione2010; Gorton et al., Reference Gorton, Kasl, Detwiler and Criscione2012) including species identification, phylogeography, demography, hybridization and loci under selection. As an additional goal, we discuss these topics with renewed interest in the expanded inferences that can be drawn from population genomics data. We also cover the fields of parabiome research and linkage mapping given that these areas often have interrelated objectives and/or methods with population genomics. Finally, we provide quantitative summaries of the taxa studied in the recent literature to not only emphasize the current application of population genomics, but to also identify where we believe major strides can be made using population genomics data to address the breadth of parasitic helminth life cycles, life histories and ecological and evolutionary diversity.
Parabiome
Helminth species do not exist in isolation within a host, rather, a host is a microcosm of an ecosystem for which there may be a community of microbes (bacteria and viruses), protozoans, or helminths concurrently infecting a host (Pedersen & Fenton, Reference Pedersen and Fenton2007; Graham, Reference Graham2008). It is recognized that the makeup of a microbial community can impact both the host and the ‘ecosystem’ which the host represents (see Ha et al., Reference Ha, Lam and Holmes2014 for a review). Likewise, infection by multiple helminths and/or protozoan parasites is a major concern in livestock and wildlife given that co-infections have the potential to exacerbate morbidity or mortality of the host (Graham, Reference Graham2008; Ezenwa & Jolles, Reference Ezenwa and Jolles2011; Griffiths et al., Reference Griffiths, Pedersen, Fenton and Petchey2011; Gorsich et al., Reference Gorsich, Ezenwa and Jolles2014; Lee et al., Reference Lee, Ngui, Tan, Muhammad Aidil and Lim2014; Ezenwa, Reference Ezenwa2016). Thus, knowing the helminth and protozoan parasite community composition in a host (what we describe as the parabiome) is critical given the potential for the community composition to influence the host immune response, other organisms residing in the host (e.g. dysbiosis), as well as the variable pathogenicity of parasites (Supali et al., Reference Supali, Verweij and Wiria2010). Moreover, characterization of a parabiome is useful in understanding broader community ecology questions concerning species interactions, diversity and abundance (see Poulin, Reference Poulin2001, Reference Poulin2014; Ezenwa & Jolles, Reference Ezenwa and Jolles2011; Titcomb et al., Reference Titcomb, Jerde and Young2019). We note that the term parabiome represents a subset (i.e. helminths and protozoans of a host) of the term holobiont, which is defined as a metazoan organism with all associated microorganisms living on or in it (Hodžić et al., Reference Hodžić, Dheilly, Cabezas-Cruz and Berry2023).
With regard to helminths, parabiome work has largely centred on nematodes (also known as the nemabiome; Avramenko et al., Reference Avramenko, Redman, Lewis, Yazwinski, Wasmuth and Gilleard2015). Prior to the use of molecular markers, morphological analysis of third larval stage (L3) larvae was used for livestock nematode identification, which was largely limited to the genus level (Roeber et al., Reference Roeber, Larsen, Anderson, Campbell, Anderson, Gasser and Jex2012; Roeber & Kahn, Reference Roeber and Kahn2014; Avramenko et al., Reference Avramenko, Redman, Lewis, Yazwinski, Wasmuth and Gilleard2015). Taking advantage of the ability of NGS to generate large amounts of sequence data, Avramenko et al. (Reference Avramenko, Redman, Lewis, Yazwinski, Wasmuth and Gilleard2015) conducted a proof-of-concept study to determine if deep amplicon sequencing, that is, metabarcoding, of nematode ITS-2 (internal transcribed spacer unit 2 of the rDNA complex) could be used to identify L3 larvae to species as well as ascertain species composition in a host. Using eight laboratory-reared species of nematodes, they found that ITS-2 reliably differentiated species. However, when pooling L3 larvae of different species, sequencing produced species-specific amplification biases. Applying a correction factor to account for the biased amplification, their method was able to detect species at low frequencies within samples and access species proportions. In addition, their method identified species that were difficult to identify morphologically. For example, the sequence data revealed L3 Haemonchus contortus, whereas morphological identifications only reported Haemonchus placei.
Subsequently, several studies have used the nemabiome approach for various metabarcoding applications (table 1). A primary focus is survey work of domestic farm animals to determine species composition and identify pathogenic nematodes. For example, Avramenko et al. (Reference Avramenko, Redman, Lewis, Bichuette, Palmeira, Yazwinski and Gilleard2017) found lower nematode species diversity in Canadian cattle herds, which were dominated by Ostertagia ostertagi and Cooperia oncophora, compared to herds in the central/south-eastern United States and São Paulo, Brazil, where Cooperia punctata and H. placei were more common. In addition, the proportion of Cooperia spp. increased and O. ostertagi decreased following macrocyclic lactone treatment in Canadian herds. Unexpectedly, C. punctata, a highly pathogenic nematode thought to be better adapted to warmer climates, was found in Ontario (central Canada). In a subsequent study across western Canada, De Seram et al. (Reference De Seram, Redman and Wills2022) also found a high proportion of C. punctata in Manitoba cattle herds and hypothesized a range expansion due to a combination of animal movement, changes in climate and anthelmintic treatment. In a survey of sheep and neighbouring roe deer populations, Beaumelle et al. (Reference Beaumelle, Redman and Verheyden2022) examined if livestock farming was modifying the roe deer parasite community composition. They found higher infection intensities and prevalence of generalist nematode species (e.g. H. contortus) compared to wild-deer specialist species (e.g. Ostertagia leptospicularis) in roe deer. Their findings were in contrast to a previous roe deer nemabiome study wherein nematodes commonly associated with cervids were mostly found in two isolated roe deer populations (Beaumelle et al., Reference Beaumelle, Redman and de Rijke2021). Beaumelle et al. (Reference Beaumelle, Redman and Verheyden2022) hypothesized that the nematode community of roe deer near sheep farms had been displaced by generalist livestock parasites after several decades of sheep farming. Additional parabiome approaches have been extended to horses (Poissant et al., Reference Poissant, Gavriliuc and Bellaw2021), and have included applications such as comparison of community composition of faecal pats on pastures before and after winter (Wang et al., Reference Wang, Avramenko, Redman, Wit, Gilleard and Colwell2020) and comparison among cattle, commercial bison and wild bison populations (Avramenko et al., Reference Avramenko, Bras, Redman, Woodbury, Wagner, Shury, Liccioli, Windeyer and Gilleard2018) (table 1).
Table 1. Parabiome.

[1] Avramenko et al. (Reference Avramenko, Redman, Lewis, Yazwinski, Wasmuth and Gilleard2015); [2] Avramenko et al. (Reference Avramenko, Redman, Lewis, Bichuette, Palmeira, Yazwinski and Gilleard2017); [3] Avramenko et al. (Reference Avramenko, Bras, Redman, Woodbury, Wagner, Shury, Liccioli, Windeyer and Gilleard2018); [4] De Seram et al. (Reference De Seram, Redman and Wills2022); [5] Wang et al. (Reference Wang, Avramenko, Redman, Wit, Gilleard and Colwell2020); [6] Redman et al. (Reference Redman, Queiroz, Bartley, Levy, Avramenko and Gilleard2019); [7] Beaumelle et al. (Reference Beaumelle, Redman and de Rijke2021); [8] Beaumelle et al. (Reference Beaumelle, Redman and Verheyden2022); [9] Poissant et al. (Reference Poissant, Gavriliuc and Bellaw2021); [10] Titcomb et al. (Reference Titcomb, Pansu, Hutchinson, Tombak, Hansen, Baker, Kartzinel, Young and Pringle2022); [11] Gogarten et al. (Reference Gogarten, Calvignac-Spencer and Nunn2020).
Other parabiome studies have focused on foundational parasite biodiversity and community ecology questions. For example, Gogarten et al. (Reference Gogarten, Calvignac-Spencer and Nunn2020) used a 18s rDNA region to provide family-level identification of helminth, fungi and protozoan parasites across 11 non-human primate species. They found previously unreported families from some primates and that closely related primate species had greater parabiome similarity. Titcomb et al. (Reference Titcomb, Pansu, Hutchinson, Tombak, Hansen, Baker, Kartzinel, Young and Pringle2022) tested for associations between the nemabiome and host traits or phylogenetic relatedness across 17 species of sympatric mammalian herbivores in Kenya as well as assessed parasite-sharing networks among hosts. Key findings included 53% of the nemabiome dissimilarity among faecal samples explained by host species, significant congruence between host and parasite phylogenies, and that host gut morphology predicted nematode community composition. The parasite-sharing analyses indicated that most nematode species were host specific, but a few did have broad host ranges suggesting potential for exchange between wildlife and livestock. Additionally, they suggested that central host species (i.e. hosts that shared parasite species with many other hosts) could be targeted for management strategies where deworming these hosts might limit the spread of parasites to threatened wildlife (Titcomb et al., Reference Titcomb, Pansu, Hutchinson, Tombak, Hansen, Baker, Kartzinel, Young and Pringle2022).
A primary utility of a parabiome approach is in estimating parasite richness, especially regarding the detection of low frequency and morphologically indistinguishable species. However, both Avramenko et al. (Reference Avramenko, Redman, Lewis, Yazwinski, Wasmuth and Gilleard2015) and Titcomb et al. (Reference Titcomb, Pansu, Hutchinson, Tombak, Hansen, Baker, Kartzinel, Young and Pringle2022) have noted important caveats about quantifying parasite abundance. In particular, cell number, rDNA copy number, variation in primer binding efficiency across species and amplification efficiency of sequence variants are factors that can skew true abundance. Controlled experiments can be used to correct for some of these latter factors (see Avramenko et al., Reference Avramenko, Redman, Lewis, Yazwinski, Wasmuth and Gilleard2015), but many natural systems may have undescribed species or species not yet represented with sequence data (e.g. Titcomb et al., Reference Titcomb, Pansu, Hutchinson, Tombak, Hansen, Baker, Kartzinel, Young and Pringle2022). Also, as source material are parasite offspring from faecal samples, abundance inferences need to be restricted to what will be seeding the next generation of infections. For example, the presence of a highly fecund species may lead to a higher proportion in faecal samples relative to the adult population in the host itself. Lastly, the need to collect L3 larvae is also a logistical issue if rapid species detection is critical (e.g. agricultural producers). Using eggs, Francis & Šlapeta (Reference Francis and Šlapeta2022) recently developed a quick and simple nemabiome protocol. Such a protocol may facilitate the parabiome approach to a wider range of host–parasite systems.
Hybridization
Hybridization between genetically diverged groups of populations can lead to the formation of stable hybrid zones, reinforcement, speciation, or homogenization of the groups (Runemark et al., Reference Runemark, Vallejo-Marin and Meier2019; Moran et al., Reference Moran, Payne, Langdon, Powell, Brandvain and Schumer2021). Moreover, hybridization is a potential source of genetic variation via introgression into the diverged genetic backgrounds of the interbreeding groups (Barton, Reference Barton2001). For parasites in particular, introgression events could influence parasite pathogenicity, virulence, fitness, drug resistance and host specificity (Ziętara & Lumme, Reference Ziętara and Lumme2002; Detwiler & Criscione, Reference Detwiler and Criscione2010; King et al., Reference King, Stelkens, Webster, Smith and Brockhurst2015). For example, hybrid offspring may be able to colonize novel hosts or be able to infect both host species for which the parental parasite lines were host specific (Henrich et al., Reference Henrich, Benesh and Kalbe2013). As such, detection of helminth hybrids is critical from both an evolutionary viewpoint and an epidemiological perspective.
The use of molecular markers to study parasite hybridization dates to the late 1970s (e.g. Bullini et al., Reference Bullini, Nascetti, Carrè, Rumore and Biocca1978; Vrijenhoek, Reference Vrijenhoek1978). Many pre-NGS population genetic studies inferred hybridization via nuclear-mitochondrial discordance (primarily comparing a rDNA region such as ITS-1 to a mtDNA region such as cytochrome c oxidase subunit 1). The caveat with these data is that one cannot tease apart incomplete lineage sorting, historical introgression, or contemporary hybridization (Detwiler & Criscione, Reference Detwiler and Criscione2010). Also, only a couple of studies have inferred natural contemporary helminth hybridization via a small panel of microsatellite loci (Criscione et al., Reference Criscione, Anderson, Sudimack, Peng, Jha, Williams-Blangero and Anderson2007; Steinauer et al., Reference Steinauer, Hanelt and Mwangi2008). Population genomics data and analyses greatly improve inferences of hybridization by enabling detection of cryptic species/species complexes, timing of hybridization events, as well as introgressed genomic regions that are likely to confer a selective advantage in their new ‘species background’ (Payseur & Rieseberg, Reference Payseur and Rieseberg2016; Moran et al., Reference Moran, Payne, Langdon, Powell, Brandvain and Schumer2021). Moreover, there are genomic methods that enable teasing apart lineage sorting from historical introgression (Bourgeois & Warren, Reference Bourgeois and Warren2021).
The use of population genomics to study helminth hybridization has largely been conducted among schistosome species. Indeed, interest in schistosome hybridization dates to the 1950s (reviewed in Southgate et al., Reference Southgate, Jourdane and Tchuenté1998; Morgan et al., Reference Morgan, DeJong, Lwambo, Mungai, Mkoji and Loker2003; Detwiler & Criscione, Reference Detwiler and Criscione2010; Steinauer et al., Reference Steinauer, Blouin and Criscione2010; Leger & Webster, Reference Leger and Webster2017). Hybridization between Schistosoma haematobium and Schistosoma bovis has received the most recent attention. Oey et al. (Reference Oey, Zakrzewski and Gravermann2019) compared a de novo genome assembly of S. bovis to the assembly of an Egyptian isolate of a S. haematobium. The genomes were highly similar with 97% sequence identity. However, there were also a few distinct genome regions with >99% sequence identity. Oey et al. (Reference Oey, Zakrzewski and Gravermann2019) concluded that the Egyptian isolate of S. haematobium most likely contained S. bovis DNA via hybridization.
Subsequently, Platt et al. (Reference Platt, McDew-White and Le Clec'h2019) compared the S. bovis genome to multiple S. haematobium samples from Niger and Zanzibar. Analyses assessing ancestry proportions did not reveal any admixture in the Zanzibar samples, but 0.1 to 2.7% of S. bovis and Schistosoma curassoni (the latter two are closely related) ancestry was detected in several Nigerien samples. Using lengths of introgressed haplotype blocks, admixture was estimated to occur between 108 and 612 years ago. There was no evidence of filial 1 (F1) or early generation hybrids. Moreover, these admixed regions in the Nigerien S. haematobium samples showed fixed or nearly fixed S. bovis alleles. Analyses of allelic differentiation and EHH provided strong evidence of selection in these regions. In particular, one region on chromosome 4, annotated to contain an invadolysin gene, showed a large reduction of nucleotide diversity indicating a selective sweep. Interestingly, this region is also one of the introgressed regions identified in the Egyptian isolate of S. haematobium by Oey et al. (Reference Oey, Zakrzewski and Gravermann2019). Overall, Platt et al. (Reference Platt, McDew-White and Le Clec'h2019) provided evidence of ancient, regional hybridization along with selection for an introgressed invadolysin gene, which they speculated may be involved in tissue penetration or immune evasion of the mammalian host. Additional sampling by Rey et al. (Reference Rey, Toulza and Chaparro2021a) supports an ancient introgression where the same S. bovis invadolysin-containing tract was found in isolates of S. haematobium from Egypt, Corsica and Mali; Madagascar samples did not show evidence of introgression. Interestingly, the Corsican sample had 77% S. haematobium and 23% S. bovis genomic content (Kincaid-Smith et al., Reference Kincaid-Smith, Tracey and de Carvalho Augusto2021), which is an admixture pattern consistent with more recent hybridization such as a mating between a F1 hybrid and S. haematobium parent (but see Rey et al., Reference Rey, Toulza and Chaparro2021a for other explanations).
The above work on S. curassoni/S. bovis x S. haematobium hybrids currently suggests ancient introgression, but it is important to note that contemporary hybridization can easily go undetected (especially if hybrids are selected against) by sparse sampling typical of genomic studies (e.g. one to ten samples per country or broad geographical region). Targeted local scale sampling in areas of documented sympatry of potentially hybridizing species may yield new insights. Indeed, based on prior survey and nuclear-mtDNA discordance data (Webster et al., Reference Webster, Diaw, Seye, Webster and Rollinson2013), Berger et al. (Reference Berger, Léger and Sankaranarayanan2022) used genomic data to test if there was contemporary hybridization between S. bovis and S. curassoni from naturally infected cattle in Northern Senegal. Using miracidia and adult parasites from four hosts, they identified bidirectional hybridization with admixture results consistent with F1 hybrids and backcrosses (between F1 hybrids and S. curassoni). Ongoing hybridization was inferred in this system as hybrid-identified samples came from multiple hosts and stages and showed no evidence of relatedness.
In additional studies, RADSeq data suggested another possible contemporary hybridization between S. haematobium and Schistosoma guineensis in samples from Cameroon (Landeryou et al., Reference Landeryou, Rabone, Allan, Maddren, Rollinson, Webster, Tchuem-Tchuenté, Anderson and Emery2022). Recent genome assemblies of other S. haematobium isolates from Africa have shown evidence of introgressed regions matching other species in the S. haematobium-group (Stroehlein et al., Reference Stroehlein, Korhonen and Lee2022). As noted by Stroehlein et al. (Reference Stroehlein, Korhonen and Lee2022), the S. haematobium-group may be a complex genetic landscape resulting from a history of genomic admixture. If such reticulate evolution characterizes this group, the concept of a ‘pure’ S. haematobium isolate becomes obscure.
Population genomics of hybridization has received little attention in other helminths with just a few studies on human-associated and pig-associated roundworms (Ascaris) and monogeneans in the genus Gyrodactylus. For the former, species status and the interbreeding capabilities of ascarid worms infecting humans (Ascaris lumbricoides) and pigs (Ascaris suum) has been the subject of debate for some time (Peng & Criscione, Reference Peng and Criscione2012). It is clear that in sympatry human-roundworm and pig-roundworm samples show genetic differentiation (reviewed in Peng & Criscione, Reference Peng and Criscione2012; Wang, Reference Wang2021). Nonetheless, it has also been shown that sympatric samples from both hosts can be genetically more similar than they are to their host-associated counterparts from a distant location (Criscione et al., Reference Criscione, Anderson, Sudimack, Peng, Jha, Williams-Blangero and Anderson2007). Based on results from pre-NGS population genetic studies, Peng & Criscione (Reference Peng and Criscione2012) proposed the hypothesis that geographical isolation along with multiple host colonization (i.e. host jump followed by differentiation) events may characterize the evolutionary history of human and pig Ascaris.
Zhou et al. (Reference Zhou, Chen, Niu, Ouyang and Wu2020a, Reference Zhou, Guo, Deng, He, Ouyang and Wub) conducted population genomics analyses of sympatric Ascaris samples from China based on autosomal genome-wide SNPs and mito-genomes, respectively. Zhou et al. (Reference Zhou, Chen, Niu, Ouyang and Wu2020a) found clear genetic differentiation between host-associated samples, but a small sample size (six nematodes per host species) may have precluded detection of hybrids. Moreover, while the mito-genome study of Zhou et al. (Reference Zhou, Guo, Deng, He, Ouyang and Wu2020b) included pre-identified hybrid samples (based on prior microsatellite and ITS genotypes), Zhou et al. (Reference Zhou, Chen, Niu, Ouyang and Wu2020a) only included pre-identified non-hybrid samples (see their methods). Unfortunately, mtDNA alone is not sufficient to address hybridization. As demonstrated by Easton et al. (Reference Easton, Gao and Lawton2020), the inclusion of both mtDNA and nuclear DNA provides additional insight into possible hybridization between human and pig Ascaris. They sampled 68 roundworms from human hosts in Kenyan villages, where pig husbandry is rare. In a comprehensive analysis of existing mtDNA data from around the world, there was no consistent pattern of mtDNA clade with host or geographical association (e.g. their Kenyan samples from human roundworms had haplotypes falling in the clade predominantly comprising pig-sampled roundworms). Moreover, the genomic analysis of autosomal SNPs among the Kenyan samples in comparison to an A. suum reference genome showed a mosaic of A. suum-like or A. lumbricoides-like inheritance patterns. Overall, these patterns are consistent with an interbred Ascaris species complex and are consistent with a history of multiple host colonization events. Nevertheless, to test the latter hypothesis, global sampling along with concurrent sympatric sampling remains necessary.
Population genomics has also provided an interesting perspective on the mode of reproduction and hybridization in monogeneans of the genus Gyrodactylus. Gyrodactylids have a unique ‘Russian nesting doll’ mode of reproduction wherein they give birth to a fully developed offspring that already contains a developing embryo. First-born are produced asexually, second-born through parthenogenesis and possible sexual reproduction or parthenogenesis thereafter (Cable & Harris, Reference Cable and Harris2002; Bakke et al., Reference Bakke, Cable and Harris2007). As such, there is the potential for the propagation of clonal lines. In the context of asexual reproduction, ‘hybridization’ occurs between two diverged clonal lines as observed in some protozoan parasites (Detwiler & Criscione, Reference Detwiler and Criscione2010). The extent to which Gyrodactylus spp. have clonal lines that persist and hybridize in nature had limited support from a study on Gyrodactylus salaris showing fixed heterozygosity (hypothesized to have resulted from a hybridization event) at a single nuclear marker (Kuusela et al., Reference Kuusela, Ziętara and Lumme2008). Two recent population genomics studies on Gyrodactylus spp. provide evidence of hybridization between diverged lineages (Konczal et al., Reference Konczal, Przesmycka and Mohammed2020, Reference Konczal, Przesmycka, Mohammed, Hahn, Cable and Radwan2021).
Konczal et al. (Reference Konczal, Przesmycka, Mohammed, Hahn, Cable and Radwan2021) sampled 30 Gyrodactylus turnbulli across multiple rivers from Trinidad and Tobago. Relative to the samples from Tobago, the samples from Trinidad (N = 14) had low levels of genome-wide heterozygosity and were more similar to one another and a reference genome generated from parasites of commercial guppies. In contrast, only one sample from Tobago had comparable low heterozygosity with most samples (N = 13) from Tobago having much higher heterozygosity. Consistent with hybridization of diverged lineages, the highly heterozygous Tobagonian samples consisted of two divergent haplotypes: one similar to the low heterozygosity Tobagonian sample; and the other similar to the reference assembly. There were also two additional samples from Tobago that had mosaic patterns of heterozygous and homozygous blocks (of each ‘parental’ haplotype), indicating recombination after the initial hybridization event.
In Konczal et al. (Reference Konczal, Przesmycka and Mohammed2020), a reference genome of Gyrodactylus bullatarudis originating from Tobago was compared to 10 samples from Trinidad. Inference of hybridization was inferred based on bi-modal distribution of divergence across the genome from the Trinidad samples compared to the reference. In Konczal et al. (Reference Konczal, Przesmycka and Mohammed2020, Reference Konczal, Przesmycka, Mohammed, Hahn, Cable and Radwan2021), the authors invoke greater hybrid fitness to the ‘hybrid’ genotypes in their sampling. We advise caution in these adaptive inferences for the following reasons. First, genetic drift alone could explain the greater frequency of hybrid individuals. A single hybrid individual could colonize a new location (i.e. a founder event) where clonal reproduction subsequently maintains highly heterozygous genotypes. Indeed, Konczal et al. (Reference Konczal, Przesmycka, Mohammed, Hahn, Cable and Radwan2021) discuss how the short generation time of G. turnbulli likely leads to mostly first and second births, both of which produce clonal offspring. Second, in Konczal et al. (Reference Konczal, Przesmycka and Mohammed2020), the G. bullatarudis reference genome could in itself be the ‘hybrid’ genome rather than all of the samples from Trinidad.
Population structure across multiple scales
A central aim in population genetics is to determine the factors that shape the amount and distribution of genetic variation within and among units at different scales (e.g. individuals, subpopulations and vicariant geological features). As such, topics may range from how hermaphroditic mating systems increase individual homozygosity to estimating current rates of gene flow among subpopulations to determining if past geological/environmental events altered population growth or influenced the presence of lineages across a landscape (i.e. what shaped the phylogeography of a species; Avise, Reference Avise2004). As sexually mature adults of many helminth parasites are further subdivided among individual hosts or host species, parasitologists have also utilized population genetics to elucidate transmission foci or the presence of host races, respectively (Criscione et al., Reference Criscione, Poulin and Blouin2005; Huyse et al., Reference Huyse, Poulin and Theron2005; Gorton et al., Reference Gorton, Kasl, Detwiler and Criscione2012). More recently, population genetics has been applied to monitor the effectiveness of helminth control programmes via changes in genetic diversity (N e) (Criscione, Reference Criscione and Holland2013; see review on schistosomes in Rey et al., Reference Rey, Webster, Huyse, Rollinson, Van den Broeck, Kincaid-Smith, Onyekwere and Boissier2021b). The nature of population genetic questions has largely remained the same among population genomic studies (table 2). Nevertheless, the larger amounts of data afforded by NGS along with various genomic-based analyses enable finer resolution of parameter estimates such as relatedness as well as more accurate historical inference (e.g. Wang et al., Reference Wang, Santiago and Caballero2016; Terhorst et al., Reference Terhorst, Kamm and Song2017).
Table 2. Population structure.

[1] Han et al. (Reference Han, Lan and Li2022); [2] Durrant et al. (Reference Durrant, Thiele and Holroyd2020); [3] Sallé et al. (Reference Sallé, Doyle, Cortet, Cabaret, Berriman, Holroyd and Cotton2019); [4] Doyle et al. (Reference Doyle, Tracey and Laing2020); [5] Choi et al. (Reference Choi, Tyagi and McNulty2016); [6] Doyle et al. (Reference Doyle, Bourguinat and Nana-Djeunga2017); [7] Shortt et al. (Reference Shortt, Card, Schield, Liu, Zhong, Castoe, Carlton and Pollock2017); [8] Shortt et al. (Reference Shortt, Timm and Hales2021); [9] Nikolakis et al. (Reference Nikolakis, Hales and Perry2021); [10] Berger et al. (Reference Berger, Crellen and Lamberton2021); [11] Vianney et al. (Reference Vianney, Berger and Doyle2022); [12] Crellen et al. (Reference Crellen, Walker, Lamberton, Kabatereine, Tukahebwa, Cotton and Webster2016); [13] Platt et al. (Reference Platt, McDew-White and Le Clec'h2022); [14] Doyle et al. (Reference Doyle, Søe and Nejsum2022a); [15] Small et al. (Reference Small, Reimer, Tisch, King, Christensen, Siba, Kazura, Serre and Zimmerman2016); [16] Small et al. (Reference Small, Labbé, Coulibaly, Nutman, King, Serre and Zimmerman2019).
Various local scale applications of helminth population genomics have included studying the potential for transmission foci, the potential impacts of chemotherapeutic control measures and assessment of reservoir hosts (table 2). An example addressing transmission dynamics is provided by Shortt et al. (Reference Shortt, Timm and Hales2021) where miracidia of Schistosoma japonicum were collected from 12 villages (maximum distance ~25 km) in Sichuan, China, in 2007, 2008, 2010 and 2016. Using model-based and non-model-based clustering analyses, individuals from the same village largely belonged to the same cluster, regardless of the time-period sampled. Also, the proportion of rare alleles shared among villages declined as distance increased. These patterns, which indicate that transmission is primarily restricted to within villages, held in a reduced data set correcting for the possibility that miracidia from a host may be siblings (see Steinauer et al., Reference Steinauer, Christie and Blouin2013). Indeed, several instances of full or half-sibling parasites were found among miracidia from the same host and a few cases between hosts in the same village. The latter finding is suggestive of clonemate adults (resulting from the asexual stage in snails) in different hosts and thus, denotes the same source of infection for these hosts. Sibling miracidia were also found in pre-praziquantel and post-praziquantel treatment of the same host, indicating that individuals likely retained infections of adult flukes. Collectively, the results of Shortt et al. (Reference Shortt, Timm and Hales2021) parallel findings from a microsatellite-based landscape genetics study on the roundworm A. lumbricoides (Criscione et al., Reference Criscione, Anderson, Sudimack, Subedi, Upadhayay, Jha, Williams, Williams-Blangero and Anderson2010) in that there are local parasite transmission foci and that these foci are stable over time and after drug treatment.
In contrast to the focal transmission of S. japonicum, S. mansoni in shoreline and island villages of Lake Victoria (~100 km apart) show markedly less population structure (Berger et al., Reference Berger, Crellen and Lamberton2021; Vianney et al., Reference Vianney, Berger and Doyle2022). Although, a nearby inland district (~40 km from the shoreline villages) was genetically distinct. At the individual host level, Berger et al. (Reference Berger, Crellen and Lamberton2021) did not find evidence of highly related S. mansoni miracidia from individuals in shoreline villages of Lake Victoria. Vianney et al. (Reference Vianney, Berger and Doyle2022), however, did find some related S. mansoni miracidia from island villages of Lake Victoria using the relatedness measure of Shortt et al. (Reference Shortt, Timm and Hales2021). The relatedness measure of Shortt et al. (Reference Shortt, Timm and Hales2021) is a within-study relative measure as the proportion of shared alleles cut-off to determine relatedness differs in Vianney et al. (Reference Vianney, Berger and Doyle2022); thus, a direct comparison between the S. japonicum and S. mansoni studies is not possible. Nonetheless, a face value comparison suggests that human hosts of S. mansoni around Lake Victoria harbour more breeding adults compared to human hosts of S. japonicum in the villages of Sichuan.
Berger et al. (Reference Berger, Crellen and Lamberton2021) and Vianney et al. (Reference Vianney, Berger and Doyle2022) also examined whether praziquantel treatment impacted the local population structure of S. mansoni. Berger et al. (Reference Berger, Crellen and Lamberton2021) found no difference in nucleotide diversity and no evidence of genetic differentiation between one round of pre-praziquantel and post-praziquantel treatment samples of S. mansoni from shoreline villages of Lake Victoria. Moreover, despite nine rounds of mass drug administration in these villages, N e has remained stable and large (~33,000) in recent history. In mild contrast, S. mansoni from the island villages of Lake Victoria did show a slight reduction in nucleotide diversity, 0.00325 to 0.0032, in one round of pre-treatment vs. post-treatment, respectively (Vianney et al., Reference Vianney, Berger and Doyle2022). In addition, island villages with drug treatment four times per year had slightly lower nucleotide diversity compared to villages treated one time per year; N e was estimated to be 100,000 (Vianney et al., Reference Vianney, Berger and Doyle2022). Collectively, mass drug administration with praziquantel is having a mild impact, at best, on reducing the N es of S. mansoni around Lake Victoria, supporting conclusions from pre-NGS population genetic studies (reviewed in Rey et al., Reference Rey, Webster, Huyse, Rollinson, Van den Broeck, Kincaid-Smith, Onyekwere and Boissier2021b).
The use of reservoir hosts by human parasites is an important epidemiological consideration as reservoirs could maintain a local parasite population despite the reduction in human infections. Indeed, extensive eradication efforts since 1986 have drastically reduced Guinea worm, Dracunculus medinensis, infections in humans in Chad, but there has been a concurrent rise in reports of dog infections (Durrant et al., Reference Durrant, Thiele and Holroyd2020). Both mitochondrial and nuclear genome data show that human and dog samples of Guinea worms are part of a single population. These authors acknowledge that their historical (>2000 thousand years ago) N e estimate of ~31,000 in Chad may not capture a population bottleneck in the past few decades. Nonetheless, the high level of nucleotide diversity (0.0252) still indicated a currently large N e despite the near absence of human infections in the past two decades. Hence, these authors advise vigilance by surveying both dogs and humans as control programmes continue.
On broader geographical scales, topics of assessing ecotypes and historical migration/colonization patterns have been addressed in helminth population genomic studies. For example, the genomics data of Choi et al. (Reference Choi, Tyagi and McNulty2016) showed genetic distinction between the savanna and forest ecotypes (possibly associated with blackfly host specificity) of the filarial nematode Onchocerca volvulus in West Africa. Several helminths show a history of dispersal that reflect human movement (table 2). Crellen et al. (Reference Crellen, Walker, Lamberton, Kabatereine, Tukahebwa, Cotton and Webster2016) found that S. mansoni has an East African origin and diverged from Schistosoma rodhaini approximately 107.5–147.6 thousand years ago, which is in line with the time frame for the origins of human fishing in this region. Additionally, the data indicated that S. mansoni spread to the Americas between the 16th and 19th centuries, which coincides with the transatlantic slave trade. Additional sampling by Platt et al. (Reference Platt, McDew-White and Le Clec'h2022) also found an East African origin for S. mansoni with an expansion, and reduction in genetic diversity, into West Africa and then to the Americas. Similar levels of nucleotide diversity in West Africa and Brazil may suggest repeated colonization events that occurred during the transatlantic slave trade. Population genomic evidence of helminths transported to the Americas via the slave trade also exists for H. contortus (Sallé et al., Reference Sallé, Doyle, Cortet, Cabaret, Berriman, Holroyd and Cotton2019), O. volvulus (Choi et al., Reference Choi, Tyagi and McNulty2016) and Wuchereria bancrofti (Small et al., Reference Small, Labbé, Coulibaly, Nutman, King, Serre and Zimmerman2019). Global sampling of H. contortus also revealed reduced genetic diversity among isolates outside of Africa, an additional link reflecting British colonization of Australia and some phylogenetic clustering that likely reflects mixed-origin sheep breeding (Sallé et al., Reference Sallé, Doyle, Cortet, Cabaret, Berriman, Holroyd and Cotton2019). In a non-human example where the parasite parallels host history, Han et al. (Reference Han, Lan and Li2022) found that changes in the N e of the roundworm Baylisascaris schroederi slightly lagged behind that of its giant panda host. In particular, the authors suggest the sharp N e decline of roundworms in the last 10,000 years could reflect the human-induced N e decline in giant pandas in this time frame.
Surveys of candidate drug-resistant loci and scans for loci under selection
In parasitology, the identification of loci under selection has largely centred on the origin and genetic basis for drug resistance (see Gilleard, Reference Gilleard2006; Gilleard & Beech, Reference Gilleard and Beech2007; Doyle & Cotton, Reference Doyle and Cotton2019 for reviews). Prior to NGS, population genetic methods were used to determine if parasite candidate genes were under selection where candidate genes were sometimes identified in non-parasitic species such as Caenorhabditis elegans or suspected based on possible drug mode of action. As described in Gilleard (Reference Gilleard2006), the candidate gene approach ‘involves making an “educated guess” as to which genes might be involved in resistance and then conducting experimental work to test the hypothesis.’ Doyle & Cotton (Reference Doyle and Cotton2019) note, however, that few candidate genes have shown associations with resistance phenotypes in the field. The high genetic diversity commonly found in many nematodes along with high among-species life history diversity has likely led to the evolution of multiple independent mechanisms for drug resistance in helminth parasites (Doyle & Cotton, Reference Doyle and Cotton2019). The main exception to where a candidate locus has shown association to resistance in pre-NGS studies is with the β-tubulin isotype 1 gene among livestock nematodes. In particular, three non-synonymous substitutions at three codons can be found to accompany benzimidazole treatment failure (reviewed in Doyle & Cotton, Reference Doyle and Cotton2019).
Deep amplicon NGS approaches (similar to the parabiome approach) have been applied to survey the frequencies of the possible β-tubulin isotype 1, resistant alleles among several nematode species (table 3). Compared to traditional polymerase chain reaction, the deep amplicon methods enable high sample throughput and in certain instances, can be applied to multiple nematode species simultaneously (Avramenko et al., Reference Avramenko, Redman, Melville, Bartley, Wit, Queiroz, Bartley and Gilleard2019; Chihi et al., Reference Chihi, Andersen, Aoun, Bouratbine and Stensvold2022). Studies on human and horse roundworms (A. lumbroides and Parascaris equorum, respectively) did not find any resistant alleles at the three candidate codons of β-tubulin despite use of benzimidazoles (Tydén et al., Reference Tydén, Dahlberg, Karlberg and Höglund2014; Roose et al., Reference Roose, Avramenko and Pollo2021). In contrast, several studies reported the resistant-associated SNP F200Y in the β-tubulin gene of various trichostrongylid nematodes collected from sheep or cattle (Ali et al., Reference Ali, Rashid, Shabbir, Shahzad, Ashraf, Sargison and Chaudhry2019; Avramenko et al., Reference Avramenko, Redman, Melville, Bartley, Wit, Queiroz, Bartley and Gilleard2019; Avramenko et al., Reference Avramenko, Redman, Windeyer and Gilleard2020; Melville et al., Reference Melville, Redman and Morrison2020). Interestingly, phylogenetic analysis of β-tubulin haplotype by Ali et al. (Reference Ali, Rashid, Shabbir, Shahzad, Ashraf, Sargison and Chaudhry2019) revealed that the F200Y allele arose independently multiple times in samples of H. contortus from Pakistan, but only once among samples of H. placei. Although the deep amplicon surveys of β-tubulin isotype 1 have enabled detection of resistant alleles at low frequency and have identified other possible resistant alleles, no formal tests of selection or drug-resistant association have been conducted in these studies.
Table 3. Targeted drug-resistant loci surveys.

[1] Roose et al. (Reference Roose, Avramenko and Pollo2021); [2] Jimenez Castro et al. (Reference Jimenez Castro, Howell, Schaefer, Avramenko, Gilleard and Kaplan2019); [3] Ali et al. (Reference Ali, Rashid, Shabbir, Shahzad, Ashraf, Sargison and Chaudhry2019); [4] Melville et al. (Reference Melville, Redman and Morrison2020); [5] Tydén et al. (Reference Tydén, Dahlberg, Karlberg and Höglund2014); [6] Chevalier et al. (Reference Chevalier, Le Clec'h and McDew-White2019); [7] Le Clec'h et al. (Reference Le Clec'h, Chevalier and Mattos2021b); [8] Avramenko et al. (Reference Avramenko, Redman, Melville, Bartley, Wit, Queiroz, Bartley and Gilleard2019); [9] Avramenko et al. (Reference Avramenko, Redman, Windeyer and Gilleard2020).
Two studies on S. mansoni (table 3) also conducted targeted drug-resistant loci surveys, but in Chevalier et al. (Reference Chevalier, Le Clec'h and McDew-White2019) and Le Clec'h et al. (Reference Le Clec'h, Chevalier and Mattos2021b), the candidate loci were identified a priori via linkage mapping (Valentim et al., Reference Valentim, Cioli and Chevalier2013) or a laboratory-based association analysis (Le Clec'h et al., Reference Le Clec'h, Chevalier, McDew-White, Menon, Arya and Anderson2021a). In addition, these two studies used exome capture rather than a targeted amplicon approach to examine the loci of interest. Exome capture provides extended haplotype information beyond the small genetic regions obtained from deep amplicon sequencing. In Chevalier et al. (Reference Chevalier, Le Clec'h and McDew-White2019), samples of S. mansoni from the Middle East, Africa, South America and the Caribbean were surveyed for variants of the sulphotransferase gene SmSULT-OR, the locus that was identified in mapping studies of oxamniquine resistance (Valentim et al., Reference Valentim, Cioli and Chevalier2013; Chevalier et al., Reference Chevalier, Valentim, LoVerde and Anderson2014). The mapped variant associated with oxamniquine resistance, p.E142del, along with six other likely resistance variants were found in African samples. Moreover, they identified an identical haplotype block between a Caribbean and West African sample, suggesting a common origin of p.E142del that predates the use of oxamniquine. These results support the origin of drug-resistance stemming from standing genetic variation rather than a de novo origin (Chevalier et al., Reference Chevalier, Valentim, LoVerde and Anderson2014). In Le Clec'h et al. (Reference Le Clec'h, Chevalier and Mattos2021b) a transient receptor potential channel (TRPM) on chromosome 3 was associated with praziquantel resistance in a laboratory-based assay. Interestingly, this TRPM was shown to be activated by nano-molar quantities of praziquantel, suggesting it is a likely target for praziquantel (Park et al., Reference Park, Friedrich, Yahya, Rohr, Chulkov, Maillard, Rippmann, Spangenberg and Marchant2021). A subsequent field survey of the TRPM alleles by Le Clec'h et al. (Reference Le Clec'h, Chevalier and Mattos2021b), though, only found a single possible resistance allele in a heterozygous state across 122 individuals from Africa, South America and the Middle East.
While targeted approaches of candidate loci have their role, a primary limitation is the ability to identify novel genes involved in resistance (Doyle & Cotton, Reference Doyle and Cotton2019). In contrast, various methods now exist to scan genome data for signatures of selection (Ahrens et al., Reference Ahrens, Rymer, Stow, Bragg, Dillon, Umbers and Dudaniec2018; Luikart et al., Reference Luikart, Kardos, Hand, Rajora, Aitken, Hohenlohe and Rajora2018; Bourgeois & Warren, Reference Bourgeois and Warren2021). Such methods may rely on within population samples (e.g. scanning for regions of reduced nucleotide diversity or various EHH statistics) or outlier analyses of population differentiation (e.g. absolute differentiation (F ST)). The latter are often conducted between two or more samples that differ in phenotype (e.g. drug resistance). Inference of selection is reinforced with association-based analyses that aim to detect relationships between environmental variables and genetic variants. Although such genome scans are useful to identify regions under selection, there are caveats regarding both genome scans and association tests (see Barrett & Hoekstra, Reference Barrett and Hoekstra2011; Cruickshank & Hahn, Reference Cruickshank and Hahn2014; Luikart et al., Reference Luikart, Kardos, Hand, Rajora, Aitken, Hohenlohe and Rajora2018; Doyle & Cotton, Reference Doyle and Cotton2019). Importantly, not accounting for population structure among samples can lead to false associations. In addition, differentiation scans between two phenotypically characterized groups (e.g. drug-resistant and susceptible) assume that the cause of differentiation is related to the driver of the phenotype (e.g. drug selection pressure). However, and especially if the two samples are confounded with different geographical origins, additional selection pressures may be driving differentiation (Barrett & Hoekstra, Reference Barrett and Hoekstra2011; Doyle & Cotton, Reference Doyle and Cotton2019). Also, differentiation tests based on F ST can be affected by linked selection reducing diversity in areas of low recombination (Cruickshank & Hahn, Reference Cruickshank and Hahn2014).
Several helminth studies have used genome-wide approaches to scan for loci under selection with an emphasis on detecting selection from drug pressure (table 4). For example, Berger et al. (Reference Berger, Crellen and Lamberton2021) conducted within population tests for S. mansoni from Ugandan districts Mayuge and Tororo that had long and short histories of praziquantel treatment, respectively. While multiple genome regions in Mayuge showed evidence of selection consistent with higher drug pressure, only a few possible impactful variants could be functionally annotated and these were not found directly under the peak signals of selection. The TRPM locus identified by Le Clec'h et al. (Reference Le Clec'h, Chevalier and Mattos2021b) also did not show evidence of selection. One region on chromosome 3, though, showed evidence of selection from multiple tests and annotation indicated ion exchange proteins, which have been implicated in blockage of praziquantel uptake (Kohn et al., Reference Kohn, Anderson, Roberts-Misterly and Greenberg2001; Valle et al., Reference Valle, Troiani, Festucci, Pica-Mattoccia, Liberti, Wolstenholme, Francklow, Doenhoff and Cioli2003). Global studies using genome scans in H. contortus have revealed both within-sample and between-sample signatures of selection around the β-tubulin isotype 1 locus, consistent with extensive use of benzimidazoles (Sallé et al., Reference Sallé, Doyle, Cortet, Cabaret, Berriman, Holroyd and Cotton2019; Doyle et al., Reference Doyle, Tracey and Laing2020). In addition, differentiation analyses between isolates that differ in ivermectin resistance revealed elevated F ST along a segment of chromosome 5 (Sallé et al., Reference Sallé, Doyle, Cortet, Cabaret, Berriman, Holroyd and Cotton2019; Doyle et al., Reference Doyle, Tracey and Laing2020), a region identified in mapping studies (Doyle et al., Reference Doyle, Illingworth and Laing2019a, Reference Doyle, Laing and Bartley2022b). On a Swedish sheep farm with suspected ivermectin treatment failure, a pre-treatment and post-treatment comparison of nucleotide diversity also implicated this same region of chromosome 5 to be involved in ivermectin resistance (Baltrušis et al., Reference Baltrušis, Doyle, Halvarsson and Höglund2022).
Table 4. Genome-wide scans for loci under selection.
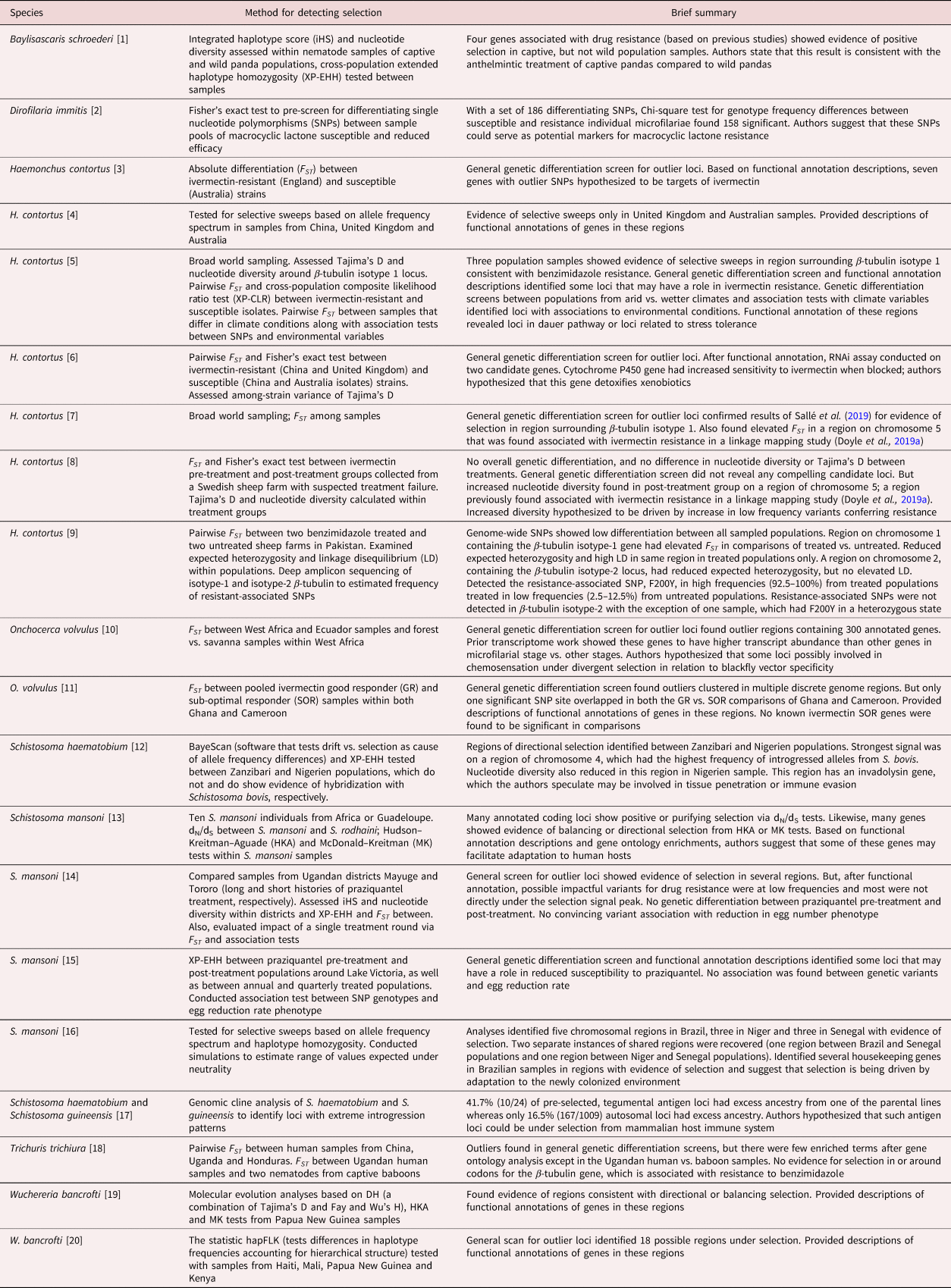
[1] Han et al. (Reference Han, Lan and Li2022); [2] Bourguinat et al. (Reference Bourguinat, Lee and Lizundia2015); [3] Luo et al. (Reference Luo, Shi, Yuan, Ai, Ge, Hu, Feng and Yang2017); [4] Khan et al. (Reference Khan, Zhao, Hou, Yuan, Li, Luo, Liu and Feng2019); [5] Sallé et al. (Reference Sallé, Doyle, Cortet, Cabaret, Berriman, Holroyd and Cotton2019); [6] Khan et al. (Reference Khan, Nisar and Yuan2020); [7] Doyle et al. (Reference Doyle, Tracey and Laing2020); [8] Baltrušis et al. (Reference Baltrušis, Doyle, Halvarsson and Höglund2022); [9] Wit et al. (Reference Wit, Workentine and Redman2022); [10] Choi et al. (Reference Choi, Tyagi and McNulty2016); [11] Doyle et al. (Reference Doyle, Bourguinat and Nana-Djeunga2017); [12] Platt et al. (Reference Platt, McDew-White and Le Clec'h2019); [13] Crellen et al. (Reference Crellen, Walker, Lamberton, Kabatereine, Tukahebwa, Cotton and Webster2016); [14] Berger et al. (Reference Berger, Crellen and Lamberton2021); [15] Vianney et al. (Reference Vianney, Berger and Doyle2022); [16] Platt et al. (Reference Platt, McDew-White and Le Clec'h2022); [17] Landeryou et al. (Reference Landeryou, Rabone, Allan, Maddren, Rollinson, Webster, Tchuem-Tchuenté, Anderson and Emery2022); [18] Doyle et al. (Reference Doyle, Søe and Nejsum2022a); [19] Small et al. (Reference Small, Reimer, Tisch, King, Christensen, Siba, Kazura, Serre and Zimmerman2016); [20] Small et al. (Reference Small, Labbé, Coulibaly, Nutman, King, Serre and Zimmerman2019).
Even with most studies focusing on selection from drug pressures, a few studies have found signatures of selection for genes that are likely not under anthelmintic pressure (table 4). For example, analyses from schistosome hybrids have identified loci that may be under selection from host immune systems (Platt et al., Reference Platt, McDew-White and Le Clec'h2019; Landeryou et al., Reference Landeryou, Rabone, Allan, Maddren, Rollinson, Webster, Tchuem-Tchuenté, Anderson and Emery2022). Choi et al. (Reference Choi, Tyagi and McNulty2016) propose that some of the highly differentiated loci between forest and savanna populations of O. volvulus may be involved in blackfly vector specificity. In Sallé et al. (Reference Sallé, Doyle, Cortet, Cabaret, Berriman, Holroyd and Cotton2019) outlier differentiation tests between populations with arid vs. wetter climates and/or association tests with climate variables identified potential loci involved in drought stress or the dauer (developmental arrest) pathway in H. contortus. The authors call for a better understanding of genetic links between traits such as hypobiosis (arrested larval development in the host) and unfavourable environmental conditions. In a study on the filarial nematode W. bancrofti, Small et al. (Reference Small, Labbé, Coulibaly, Nutman, King, Serre and Zimmerman2019) found evidence of selection at a locus annotated as providing resistance to acetylcholinesterase inhibitors. Such inhibitors are a common mode of action in pesticides used to control vectors. Thus, the use of pesticides on the mosquito host of W. bancrofti may be resulting in inadvertent selection on the parasite.
Linkage mapping
Linkage mapping uses experimental crosses along with genotypical and phenotypical characterization of parents and their offspring to identify co-segregation between genetic markers and quantifiable phenotypes (quantitative trait loci (QTL)) (Broman et al., Reference Broman, Wu, Sen and Churchill2003; Falconer & Mackay, Reference Falconer and Mackay2009). Genetic cross data have been used to determine the mode of inheritance of genetic markers or phenotypes in parasitic helminths (Le Jambre & Royal, Reference Le Jambre and Royal1977; Le Jambre et al., Reference Le Jambre, Royal and Martin1979; Habe et al., Reference Habe, Agatsumam and Hirai1985; Cioli et al., Reference Cioli, Picamattoccia and Moroni1992; Hunt et al., Reference Hunt, Kotze, Knox, Anderson, McNally and Le Jambre2010; Detwiler & Criscione, Reference Detwiler and Criscione2011; Redman et al., Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). However, parasitic helminth linkage maps are relatively recent, with the first maps generated by targeted genotyping of microsatellites or other nuclear markers (Criscione et al., Reference Criscione, Valentim, Hirai, LoVerde and Anderson2009; Nemetschke et al., Reference Nemetschke, Eberhardt, Viney and Streit2010). The advent of NGS has enabled rapid, cost-effective, and genome-wide linkage and QTL mapping (Bailey-Wilson & Wilson, Reference Bailey-Wilson and Wilson2011; Jaganathan et al., Reference Jaganathan, Bohra, Thudi and Varshney2020). Since 2013, NGS has been utilized in linkage and QTL mapping studies in the trematode S. mansoni and the nematodes H. contortus and Teladorsagia circumcincta (table 5).
Table 5. Linkage mapping.

[1] Doyle et al. (Reference Doyle, Laing and Bartley2018); [2] Doyle et al. (2019); [3] Niciura et al. (Reference Niciura, Tizioto and Moraes2019); [4] Doyle et al. (Reference Doyle, Laing and Bartley2022b); [5] Valentim et al. (Reference Valentim, Cioli and Chevalier2013); [6] Chevalier et al. (Reference Chevalier, Valentim, LoVerde and Anderson2014); [7] Le Clec'h et al. (Reference Le Clec'h, Chevalier, McDew-White, Menon, Arya and Anderson2021a); [8] Choi et al. (Reference Choi, Bisset, Doyle, Hallsworth-Pepin, Martin, Grant and Mitreva2017).
Anderson et al. (Reference Anderson, LoVerde, Le Clec'h and Chevalier2018) provided a detailed review of linkage and QTL mapping of oxamniquine resistance in S. mansoni based on the studies of Valentim et al. (Reference Valentim, Cioli and Chevalier2013) and Chevalier et al. (Reference Chevalier, Valentim, LoVerde and Anderson2014). Here, we just highlight that S. mansoni is amenable to standard F2 design because crosses between parental worms are facilitated by asexual reproduction in snails. Hence, many individuals of the same clone (i.e. a single female or male parent genotype) can be crossed to many individuals of another clone. Downstream phenotyping is possible on individuals but pooling large numbers of individuals to conduct bulk segregant analysis (BSA) is also feasible. The latter approach genotypes two pooled groups either with different phenotypes or where one pool is subjected to a selective pressure to identify regions enriched for alleles from one of the parents. Variations on BSA have been termed extreme QTL (X-QTL) or linkage group selection (Michelmore et al., Reference Michelmore, Paran and Kesseli1991; Culleton et al., Reference Culleton, Martinelli, Hunt and Carter2005; Ehrenreich et al., Reference Ehrenreich, Gerke and Kruglyak2009). In S. mansoni, both individual phenotyping (Valentim et al., Reference Valentim, Cioli and Chevalier2013) and X-QTL (Chevalier et al., Reference Chevalier, Valentim, LoVerde and Anderson2014) approaches combined with whole genome or exome data from NGS enabled fine mapping of a sulphotransferase encoding gene involved in oxamniquine resistance.
Anderson et al. (Reference Anderson, LoVerde, Le Clec'h and Chevalier2018) also discussed other epidemiologically relevant traits, such as host specificity, virulence and cercarial shedding that could be mapped in S. mansoni. Recently, Le Clec'h et al. (Reference Le Clec'h, Chevalier, McDew-White, Menon, Arya and Anderson2021a) performed a QTL analysis of cercarial shedding, an important transmission trait, and snail-host virulence (using laccase-like activity and haemoglobin rate in the haemolymph as a proxy) by performing reciprocal crosses of high and low cercarial shedding individuals. Whole genome data from the F0 parents and exome data from the F1 and F2 generations revealed five QTLs that explained 28.56% of the variance in cercarial production. While there was good support for the polygenic inheritance of cercarial production, no significant QTLs were found for the snail haemolymph phenotypes. To our knowledge, this study is the only non-drug-resistant phenotype to be mapped in a helminth.
In contrast to linkage mapping in S. mansoni, mapping studies in livestock nematodes have been challenging due to high levels of within strain diversity that make creating inbred lines difficult, the need for surgical transfer to stage crosses and the need for assays to phenotype individuals for drug resistance (Gilleard, Reference Gilleard2006; Gilleard & Redman, Reference Gilleard and Redman2016). For example, pairing a single male and single female in host may not yield a successful mating (contrast with the many individuals of a clone in S. mansoni). Consequently, various other crossing strategies have been employed to create inbred lines (Sargison et al., Reference Sargison, Redman and Morrison2018), estimate recombination rates, or to map traits (table 5). For example, Doyle et al. (Reference Doyle, Laing and Bartley2018) generated a recombination map in H. contortus with a pseudo-testcross, which enables recombination estimates among loci that are heterozygous in a mother and that segregate 1:1 in her F1 offspring. SNP variants were called from Illumina reads mapped to a reference genome and a map of recombination rates across chromosomes was generated by overlaying the physical and linkage maps.
Choi et al. (Reference Choi, Bisset, Doyle, Hallsworth-Pepin, Martin, Grant and Mitreva2017) and Doyle et al. (Reference Doyle, Illingworth and Laing2019a) mapped drug-resistant loci in T. circumcincta and H. contortus, respectively, using introgression-mapping approaches (table 5). There are variations on the method, but in brief, introgression mapping for drug resistance entails crossing parental lines that are drug-resistant and susceptible. The F1 is backcrossed to the susceptible line and offspring are subjected to drug exposure within hosts. Backcrossing to the susceptible line as well as subsequent drug exposure is repeated to create a largely ‘susceptible’ genetic background that is enriched for the drug-resistant allele(s) at one or more loci. Enriched regions are identified by testing for regions of differentiation (often inferred by FST) between pools of individuals from the susceptible line compared to the introgressed–backcrossed line or other downstream comparisons based on BSA. Using a multi-drug-resistant line of T. circumcincta, Choi et al. (Reference Choi, Bisset, Doyle, Hallsworth-Pepin, Martin, Grant and Mitreva2017) found putative resistance genes for the drugs oxfendazole, levamisole and ivermectin. However, Choi et al. (Reference Choi, Bisset, Doyle, Hallsworth-Pepin, Martin, Grant and Mitreva2017) stated that the fragmented genome of assembly of T. circumcincta meant the number of differentiated regions could not be accurately estimated and precluded assessment of the number of independent loci involved in drug resistance. Indeed, Doyle et al. (2019) demonstrated the benefits of a contiguous-assembled genome in mapping. With the chromosome-level assembly of H. contortus, they found a single QTL for ivermectin drug resistance on chromosome 5. In contrast, when using older fragmented assemblies, signals of selection were dispersed across multiple scaffolds. The latter would have led to an incorrect conclusion of multiple loci being involved in resistance (Doyle et al., Reference Doyle, Illingworth and Laing2019a). Recently, X-QTL has been used to map drug resistance in H. contortus (Niciura et al., Reference Niciura, Tizioto and Moraes2019; Doyle et al., Reference Doyle, Laing and Bartley2022b). When compared to introgression mapping, X-QTL is faster, less expensive and requires fewer hosts and crossing cycles; these advantages make X-QTL a promising method for future QTL mapping studies among helminths.
Concluding remarks
Genomic approaches have enabled parasitologists to delve deeper into helminth population biology and evolution. For example, NGS facilitated efficient methodologies to classify parasite community richness and changes in community composition (table 1, Online supplementary table S1). Population genomic methods enabled improved inferences on the timing of hybridization/introgression events among schistosomes (e.g. Platt et al., Reference Platt, McDew-White and Le Clec'h2019; Berger et al., Reference Berger, Léger and Sankaranarayanan2022), as well as provided a means to monitor the genetic impacts of helminth control measures including surveillance of drug-resistant alleles (tables 2 and 3), or the use of reservoir hosts (Durrant et al., Reference Durrant, Thiele and Holroyd2020). Genomic tests of selection have not only verified known loci but have also led to the discovery of novel candidate loci conferring adaptation to drug resistance (table 4). In addition, selection has been detected in novel regions that may influence parasite host specificity, development, or immune evasion traits (Choi et al., Reference Choi, Tyagi and McNulty2016; Platt et al., Reference Platt, McDew-White and Le Clec'h2019; Sallé et al., Reference Sallé, Doyle, Cortet, Cabaret, Berriman, Holroyd and Cotton2019).
Although there have been advancements, there remain hurdles in the application of and inferences drawn from NGS and consequently, population genomics of parasites. A main limitation is the limited amount of quality DNA template per individual due to the inherent small size of many helminth life cycle stages. One common approach has been to utilize pooled samples (e.g. Bourguinat et al., Reference Bourguinat, Lee and Lizundia2015; Doyle et al., Reference Doyle, Bourguinat and Nana-Djeunga2017; Khan et al., Reference Khan, Zhao, Hou, Yuan, Li, Luo, Liu and Feng2019), but this limits many of the advantages of using single specimen-based data. For example, analysis of individuals enables use of ROH to estimate inbreeding (Ceballos et al., Reference Ceballos, Joshi, Clark, Ramsay and Wilson2018), easier assessment of sequencing error vs. a true rare variant (Anand et al., Reference Anand, Mangano and Barizzone2016) and the use of relatedness measures to examine local scale transmission (e.g. Shortt et al., Reference Shortt, Timm and Hales2021). Moreover, unequal contributions of genetic material per individual in a pooled sample can create allele frequency bias and thus, downstream summary statistic (e.g. F ST) bias if sample sizes are small (Schlötterer et al., Reference Schlötterer, Tobler, Kofler and Nolte2014; Hivert et al., Reference Hivert, Leblois, Petit, Gautier and Vitalis2018). Haplotype construction and inferences using linkage disequilibrium are also hindered with short-read pooled data (Schlötterer et al., Reference Schlötterer, Tobler, Kofler and Nolte2014). Doyle et al. (Reference Doyle, Sankaranarayanan and Allan2019b) explored different extraction methods on individual helminth eggs/larvae, but DNA quantification was ‘inconsistent and largely unsuccessful’. Other studies have used whole genome amplification to increase template in helminth eggs/larvae (Shortt et al., Reference Shortt, Card, Schield, Liu, Zhong, Castoe, Carlton and Pollock2017; Le Clec'h et al., Reference Le Clec'h, Chevalier and McDew-White2018; Platt et al., Reference Platt, McDew-White and Le Clec'h2019); however, the method leads to fragmented DNA. Low yield or fragmented DNA precludes use of third generation sequencing (TGS), that is, long-read data, of individuals. An advantage of TGS is the creation of highly contiguous sequences that can span long repetitive regions (Amarasinghe et al., Reference Amarasinghe, Su, Dong, Zappia, Ritchie and Gouil2020). As such, structural variation (e.g. inversions, large insertions or deletions) can be assessed and phased haplotypes can be constructed, enabling analyses with ROH and linkage disequilibrium patterns (Wit & Gilleard, Reference Wit and Gilleard2017; Ceballos et al., Reference Ceballos, Joshi, Clark, Ramsay and Wilson2018).
Inferences from helminth population genomics have also been limited in scope. Although genome scans have identified regions under selection, the context of what is driving the selection is often ambiguous. Studies often rely on post hoc annotations to explain why there might be selection in that genomic region. Even when there are strong signals of selection in regions with assumed relation to drug resistance, only a handful of studies followed up these findings with functional validation (Khan et al., Reference Khan, Nisar and Yuan2020; Le Clec'h et al., Reference Le Clec'h, Chevalier and Mattos2021b). In general, with few exceptions (e.g. Sallé et al., Reference Sallé, Doyle, Cortet, Cabaret, Berriman, Holroyd and Cotton2019), there has been little attempt to correlate environmental variables or historical geological features with helminth genomic variation. The latter type of studies often requires large sample sizes, which understandably might be difficult to achieve in many helminth systems due to technical constraints in collecting specimens.
In general, the application of NGS and population genomics has yet to reach its realized potential in parasitology. Figure 1 illustrates a heuristic comparison (see fig. 1 legend for methods, Online supplementary table S1) of the frequency of ‘population genomic’ studies per year between a common vertebrate group (‘fish’) and helminth parasites. Upon the introduction of NGS in 2005, the number of studies among fish has shown a continued gradual increase since 2008 (a total of 402 studies in all years analysed). However, the number of studies among helminths is largely non-existent prior to 2014 with the number of studies per year remaining rather stagnant since 2014 (a total of 33 studies in all years analyzed). The current trend of population genomics in parasitology mirrors historical trends in the application of allozyme and microsatellite markers to parasite population genetics where there is an approximate ten-year lag compared to studies on ‘fish’ (compare fig. 1 to fig. 1 in Criscione, Reference Criscione, Janovy and Esch2016, which provides a parallel heuristic analysis based on the use of microsatellite markers).

Fig. 1. Comparison of the number of population genomic studies per year between helminths and fish. We conducted a Web of Science search where the search term [(‘population genomics’ or ‘population genomic’)] was searched with [fish] and then searched with [(helminth* or trematod* or nematod* or cestod* or monogene* or fluke* or tapeworm* or roundworm*)]. In both, the date range was from 1 January 2005 to 31 December 2022 and type of study was restricted to ‘article, review article, and book chapter’. In the parasite search, we excluded studies including Caenorhabditis. Extreme caution is advised in strictly interpreting results as we recognize there are caveats in the search (e.g. plant nematodes may be included or studies in this review did not end up in the search). As such, we regard the analysis as a simple heuristic means to reflect overall trends in the field. See Online supplementary table S1.
In addition to the slow integration of NGS and population genomics in parasitology, the taxonomic breadth of the studies available for our review is very limited and is biased to parasites of humans and/or domestic animals (fig. 2, Online supplementary table S2). Furthermore, the vast majority of the studies we reviewed are centred on drug resistance, despite the many other aspects of parasite biology that could potentially be addressed using genomic data. Helminth parasites are incredibly species rich (estimates range from 100,000–350,000 species, though 85%–95% are unknown; Carlson et al., Reference Carlson, Dallas, Alexander, Phelan and Phillips2020), inhabit diverse ecosystems, and display a myriad of life histories, life cycles and host use. As such, there are numerous opportunities to use population genomics to elucidate unknown helminth biology and genetic diversity. For example, there are many gaps in our knowledge about parasite life cycles where described species are known from only a single stage (Blasco-Costa & Poulin, Reference Blasco-Costa and Poulin2017). The expansion of parabiome-like approaches could prove very useful in both elucidating host use and cryptic parasite species. In addition, the diversity of helminths themselves can be used to address the consequential challenge of linking comparative population genomic patterns to species’ life history and ecology (Glémin et al., Reference Glémin, François, Galtier and Anisimova2019). For instance, there are also various life cycle patterns, mating systems (e.g. selfing vs. outcrossing), or modes of reproduction (asexual vs. sexual) among helminths (e.g. Detwiler et al., Reference Detwiler, Caballero and Criscione2017; Kasl et al., Reference Kasl, Font and Criscione2018; Criscione et al., Reference Criscione, Hulke and Goater2022) that enable among-species population genomic comparisons. Unfortunately, such comparisons cannot be drawn from the existing studies we reviewed. For example, most of the parasites reviewed here, barring schistosomes and filarial nematodes, have direct life cycles. Certainly, future fruitful avenues will be to use population genomics to explore how life cycle complexity may shape population structure or to ascertain if loci under selection might be influencing complex life cycle evolution itself. In conclusion, we emphasize that there is great potential for population genomics to elucidate helminth population biology and evolution as well as the potential for helminths to contribute to our understanding of broader ecological and evolutionary concepts. In doing so, we reiterate the arguments made by Carlson et al. (Reference Carlson, Dallas, Alexander, Phelan and Phillips2020) to increase resources (e.g. funding, taxonomic and classical parasitology training) for expanding our knowledge base about helminth biodiversity. As such, we hope that future trends in helminth population genomics reflect the taxonomic and life history breadth displayed by these parasites.
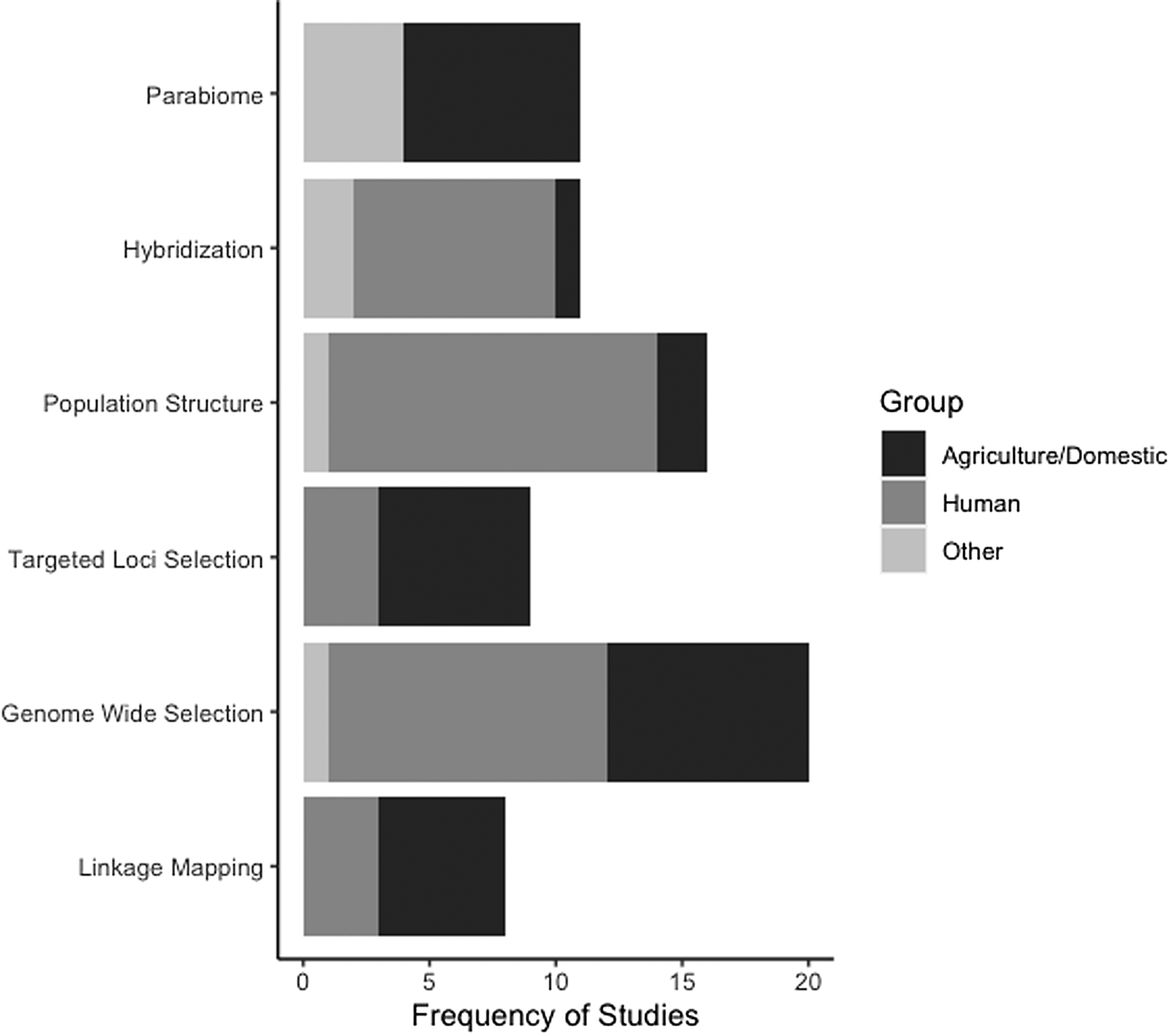
Fig. 2. Frequencies of studies examining helminth parasites in either domesticated animals, humans, or other hosts from each topical section of the review (S2). Host groups were determined based on the focal host group in the study. A study could be included under more than one topical section. See Online supplementary table S2.
Supplementary material
To view supplementary material for this article, please visit 10.1017/S0022149X23000123.
Acknowledgement
We thank P. Pilling for insightful discussions on topics in this review.
Financial support
K.E.D.'s studies on speciation, hybridization and genomics are supported by the United States National Science Foundation (NSF) Grant IOS-2143004 and C.D.C.'s studies on the population genetics and evolution of parasitic helminths are supported by the NSF Grant DEB-1655147.
Conflicts of interest
None.
Ethical standards
Not applicable.










 Open access
Open access