


The classical risk factors for cardiovascular and metabolic disease include smoking, obesity, sedentary lifestyle, high blood pressure and a obesogenic diet(Reference Forouhi and Sattar1). More recently, epidemiological evidence and animal models have demonstrated that the in utero environment may also be an important risk factor for the development of adult disease. Epidemiological and experimental evidence highlights a relationship between the periconceptional, fetal and early infant phases of life and the subsequent development of adult disease. This concept, the ‘developmental origins of health and disease’ model, proposes that the fetus adapts to adverse environmental cues in utero with permanent readjustments in homeostatic systems to aid survival(Reference Osmond, Barker, Winter, Fall and Simmonds2, Reference Kajantie, Osmond, Barker, Forsen, Phillips and Eriksson3). However, these adaptations, known as predictive adaptive responses, may ultimately be disadvantageous in postnatal life and may lead to an increased risk of chronic disease in adulthood(Reference Gluckman and Hanson4). More recent evidence has indicated that the greater risk is borne in those who were small at birth but underwent rapid postnatal growth(Reference Eriksson, Forsen, Tuomilehto, Osmond and Barker5).
It has been proposed that the greater the mismatch between the in utero and extra-uterine environment, the greater the risk of adult disease(Reference Gluckman and Hanson4). These insights have been strengthened by the development of several animal models of unbalanced nutrition in pregnancy which leads to cardiovascular and metabolic dysfunction in the offspring(Reference McMillen and Robinson6). In the rat, restriction of maternal protein or total dietary intake throughout gestation leads to raised blood pressure and endothelial dysfunction in the offspring(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson7–Reference Torrens, Brawley, Anthony, Dance, Dunn, Jackson, Poston and Hanson9). To better describe the interaction between the pre- and postnatal environment we have developed a rat model which gives rise to offspring exhibiting raised blood pressure, obesity, reduced activity, insulin insensitivity, hyperleptinaemia and hyperphagia, particularly in the presence of a postnatal high-fat diet(Reference Vickers, Breier, Cutfield, Hofman and Gluckman10–Reference Vickers, Gluckman, Coveny, Hofman, Cutfield, Gertler, Breier and Harris12). The cluster of conditions in this model resembles the human metabolic syndrome, a cluster of metabolic and vascular disorders associated with insulin resistance, hypertension, dyslipidaemia, obesity as well as hyperleptinaemia, microalbuminuria and a range of vascular injury markers(Reference Bonora13).
The vascular endothelium plays an important role in the control of vascular tone, through the release of both vasodilatory factors such as NO, prostacyclin (PGI2) and endothelial-derived hyperpolarising factor (EDHF)(Reference Busse and Fleming14), as well as vasoconstrictors such as endothelin (ET)(Reference Yanagisawa, Kurihara, Kimura, Tomobe, Kobayashi, Mitsui, Yazaki, Goto and Masaki15). Endothelial dysfunction is associated with atherosclerosis(Reference Landmesser, Hornig and Drexler16), hypertension(Reference Panza, Casino, Kilcoyne and Quyyumi17) but is also a common feature of the metabolic syndrome(Reference Fornoni and Raij18). The aim of the present study was to determine whether vascular dysfunction was also present in offspring following developmental programming, and, if so, whether it was exacerbated by a postnatal high-fat dietary challenge on vascular function.
Methods
A previously developed maternal undernutrition model of developmental programming was utilised in the present study(Reference Vickers, Breier, Cutfield, Hofman and Gluckman10). In brief, virgin Wistar rats (age 100 ± 5 d) were time-mated using a rat oestrous cycle monitor to assess the stage of oestrous of the animals before introducing the male. After confirmation of mating, rats were housed individually in standard rat cages with free access to water. Rats were kept in the same room with a constant temperature maintained at 25°C and a 12 h light–12 h darkness cycle. Animals were assigned to either a control diet of ad libitum intake throughout gestation (AD; n 16) or an undernutrition diet (UN, 30 % of ad libitum; n 16). Food intake and maternal weights were recorded daily until birth. After birth, pups were weighed, and litter size was recorded. Pups from undernourished mothers were cross-fostered onto dams that received ad libitum feeding throughout pregnancy. Litter size was adjusted to eight pups per litter to ensure adequate and standardised nutrition until weaning. At weaning, male AD and UN offspring were placed on a standard rat chow (Harlan Teklad Diet 2018; Harlan Teklad, Bicester, Oxon, UK) fed ad libitum (ADC, UNC). At postnatal day 240, a cohort from each group was challenged with a high-fat diet (D12451, 45 % energy from fat; Research Diets, Inc., New Brunswick, NJ, USA) for the remainder of the study (ADHF, UNHF). The fat source in the high-fat diet was derived from lard (39·5 % of total energy) and soyabean oil (5·5 % of total energy) with fat content by weight 5 and 24 % for the chow and high-fat diets respectively. The energy density of the chow diet was 14·2 kJ/g compared with 19·8 kJ/g for the high-fat diet. The mineral and vitamin content in the two diets was identical and in accordance with the requirements for standard rat diets. At age 365 d, rats were euthanised with pentobarbitone (60 mg/kg) followed by decapitation. All animal procedures were approved by The Animal Ethics Committee of the University of Auckland.
Vascular protocol – adult offspring mesenteric arteries
Mesenteric artery segments (internal diameter about 300 μm) were dissected, cleaned of connective tissue and mounted on a wire myograph (Danish Myo Technology A/S, Aarhus, Denmark). Segments were bathed in physiological salt solution of the following composition: NaCl, 119; KCl, 4·7; CaCl2, 2·5; MgSO4, 1·17; NaHCO3, 25; KH2PO4, 1·18; EDTA, 0·026; d-glucose, 5·5 mm, heated to 37°C and continuously gassed with 95 % O2 and 5 % CO2. The passive tension–internal circumference relationship was determined by incremental increases in tension to achieve an internal circumference equivalent to a transmural pressure of 100 mmHg (IC100) using the Laplace relationship. Arteries were set to a diameter equivalent to 0·9 × IC100 as previously described(Reference Mulvany and Halpern19). Functional integrity of the smooth muscle was assessed with four 2 min washes with 125 mm-KPSS solution (physiological salt solution with an equimolar substitution of KCl for NaCl). Vessels failing to produce an active tension equivalent to 13·3 kPa were discarded from the study.
Following normalisation and tests of functional integrity, cumulative concentration–response curves were constructed for the α1-adrenoceptor agonist phenylephrine (PE; 10 nm to 100 μm) and ET (1 pm to 10 nm). Then, following pre-constriction with PE (-log effective concentration equal to 80 % of the maximal response; pEC80), cumulative concentration–response curves were constructed to the endothelium-dependent vasodilator acetylcholine (ACh; 0·1 nm to 10 μm), the hormone leptin (0·01 to 10 ng/ml) and the NO donor sodium nitroprusside (0·1 nm to 10 μm). To investigate the factors involved in ACh-mediated vasodilatation, responses to ACh were repeated in vessels incubated with the non-selective NO synthase (NOS) inhibitor N ω-nitro-l-arginine methyl ester (l-NAME; 100 μm) and the cyclo-oxygenase (COX) inhibitor indomethacin (10 μm) 30 min before commencing the ACh response. These inhibitors were given only in combination and not independently, based on our previous experience that the PGI2 and therefore COX-sensitive component of the ACh response is negligible in this particular vascular bed (C Torrens, unpublished results). Leptin was investigated due to our previous observation that early acute leptin treatment could reverse the effects of the diets(Reference Vickers, Gluckman, Coveny, Hofman, Cutfield, Gertler, Breier and Harris12). Leptin has previously been shown to be an endothelial-dependent vasodilator in isolated arteries(Reference Lembo, Vecchione, Fratta, Marino, Trimarco, d'Amati and Trimarco20), and we also investigated responses to leptin in arterial segments before and following pre-incubation with l-NAME (100 μm) for 30 min.
All drugs and chemicals were obtained from Sigma-Aldrich (Auckland, New Zealand) with the exception of recombinant rat leptin (Arieh Gertler, University of Rehovot, Israel).
Calculations and statistical analysis
Data are expressed as mean values with their standard errors. Constrictor responses were calculated as percentage of maximum contraction induced by 125 mm-KPSS and relaxant responses as percentage inhibition of PE-induced contraction. Cumulative concentration–response curves to agonists were analysed by fitting to a four-parameter logistic equation using non-linear regression to obtain the -log effective concentration equal to 50 % of the maximal response (pEC50) and maximum response. Differences were assessed by one-way ANOVA with Bonferroni post hoc correction. When the curve produced by non-linear regression was dissimilar to the unfitted data, curve-fitted data were not used. Where curves were not sigmoidal, calculation of the pEC50 was deemed inappropriate and concentration–response curves were compared using two-way ANOVA. Significance was accepted at the P < 0·05 level.
Results
Maternal undernutrition
Maternal undernutrition resulted in fetal growth retardation reflected by significantly decreased birth weight (AD, 6·05 (sem 0·01) g; UN, 4·34 (sem 0·02) g; P < 0·0001) and nose–anus length (AD, 48·6 (sem 0·01) mm; UN, 41·3 (sem 0·01) mm; P < 0·0001) in the offspring from UN dams. Litter size was not different between the two groups (AD, 12·3 (sem 1·8); UN, 11·9 (sem 2·0)). UN animals remained significantly lighter and shorter than AD animals for the duration of the trial.
Effect of postnatal high-fat dietary challenge
At postnatal day 240, animals were weight matched within group and placed on either chow or a high-fat diet (45 % energy as fat, Research Diets D12451; Research Diets, Inc., New Brunswick, NJ, USA) fed ad libitum for the remainder of the trial (day 365). Total body-weight gain was significantly increased in all high-fat-fed animals (ADC, 799 (sem 10) g; ADHF, 977 (sem 39) g; UNC, 706 (sem 24) g; UNHF, 864 (sem 26) g) and there were no differences in the susceptibility to diet-induced weight gain in UN animals on the high-fat diet compared with AD animals (Fig. 1). There was a small but significant increase (P < 0·05) in energy intake in UN animals compared with AD animals and a significant increase (P < 0·001) in total energy intake in high-fat-fed animals compared with chow-fed offspring (kJ/g body weight; ADC, 0·540 (sem 0·008); ADHF, 0·623 (sem 0·013); UNC, 0·586 (sem 0·013); UNHF, 0·653 (sem 0·008)).

Fig. 1 Diet-induced body-weight gain (%) in ad libitum-fed (AD) and undernourished (UN) animals fed either chow (C) or challenged with a high-fat (HF) diet from day 240 until the end of the trial. Data are means for twelve animals per group, with standard errors represented by vertical bars. The effect of the HF diet was significant (P < 0·0001).
Mesenteric artery reactivity
In all arteries the depolarising KPSS wash produced a vasoconstriction that did not differ between the four groups (data not shown). The α1-adrenoceptor agonist PE produced a concentration-dependent vasoconstriction in all arteries. In both groups of animals fed the high-fat diet, sensitivity to PE was significantly reduced compared with controls; this was independent of the earlier challenge and was not different to the UN chow response (pEC50; ADC, 5·90 (sem 0·04) (n 7); ADHF, 5·67 (sem 0·04) (n 8); UNC, 5·81 (sem 0·04) (n 8); UNHF, 5·70 (sem 0·05) (n 8); P < 0·05; Fig. 2 (A)).

Fig. 2 Cumulative additions of (A) the α1-adrenoceptor agonist phenylephrine (PE) and (B) endothelin (ET) to mesenteric arteries of 365-d-old male rats from ad libitum chow-fed (ADC) (○, n 7–9), ad libitum high-fat-fed (ADHF) (□, n 8–9), undernourished chow-fed (UNC) (●, n 8) and undernourished high-fat-fed (UNHF) (■, n 7–9) groups. Data are means, with standard errors represented by vertical bars. * Mean value of -log effective concentration equal to 50 % of the maximal response (pEC50) for the AD rats was significantly different from that for the ADHF and UNHF rats (P < 0·05). ** Mean value of pEC50 for the ADC rats was significantly different from that for the ADHF, UNC and UNHF rats (P < 0·01).
Similarly, ET1 produced a potent vasoconstriction in all arteries, but which was significantly more potent in each of the three treatment groups compared with the controls (pEC50; ADC, 9·30 (sem 0·07) (n 7); ADHF, 9·79 (sem 0·11) (n 8); UNC, 10·01 (sem 0·11) (n 8); UNHF, 10·02 (sem 0·15) (n 8); P < 0·01; Fig. 2 (B)).
Endothelial-dependent vasodilatation
In all groups the endothelial-dependent vasodilator ACh produced a concentration-dependent vasodilatation. In all groups vasodilatation to ACh was significantly impaired compared with the AD group (% maximum response; ADC, 72·4 (sem 3·3) (n 7); ADHF, 47·5 (sem 4·4) (n 8); UNC, 44·4 (sem 4·4) (n 8); UNHF, 45·5 (sem 3·3) (n 8); P < 0·01; Fig. 3 (A)).

Fig. 3 (A) Cumulative additions of the endothelial-dependent vasodilator acetylcholine (ACh) to mesenteric arteries of 365-d-old rats from ad libitum chow-fed (ADC) (○, n 7), ad libitum high-fat-fed (ADHF) (□, n 8), undernourished chow-fed (UNC) (●, n 8) and undernourished high-fat-fed (UNHF) (■, n 8) groups. Data are means, with standard errors represented by vertical bars. ** Mean value of percentage maximum response for the ADC rats was significantly different from that for the ADHF, UNC and UNHF rats (P < 0·01). (B) Maximal response to ACh in small mesenteric arteries in the absence (□, n 7–9) and presence of N ω-nitro-l-arginine methyl ester (100 μm) and indomethacin (10 μm) (■, n 6–8). Data are means, with standard errors represented by vertical bars. *** Mean value was significantly different from that of the naive preparation (P < 0·001). a,b Mean values with unlike letters were significantly different (P < 0·01).
ACh responses in the presence of the NOS inhibitor l-NAME (100 μm) and the COX inhibitor indomethacin (10 μm) were significantly impaired compared with naive preparations in all groups (P < 0·001; Fig. 3 (B)). In the presence of the inhibitors, the previously observed differences in the ACh response between the groups were abolished. Moreover, the size of response to ACh following NOS and COX blockade would indicate a sizeable NO/PGI2 component with a relatively poor EDHF component.
Leptin-induced vasodilatation
In all groups, leptin produced a weak inhibition of PE-induced tone that did not differ between the groups (Fig. 4 (A)). What dilatation there was observed to leptin was unaffected in a subgroup of arteries (n 3), when repeated in the presence of the NOS inhibitor l-NAME (100 μm) in all groups (data not shown).
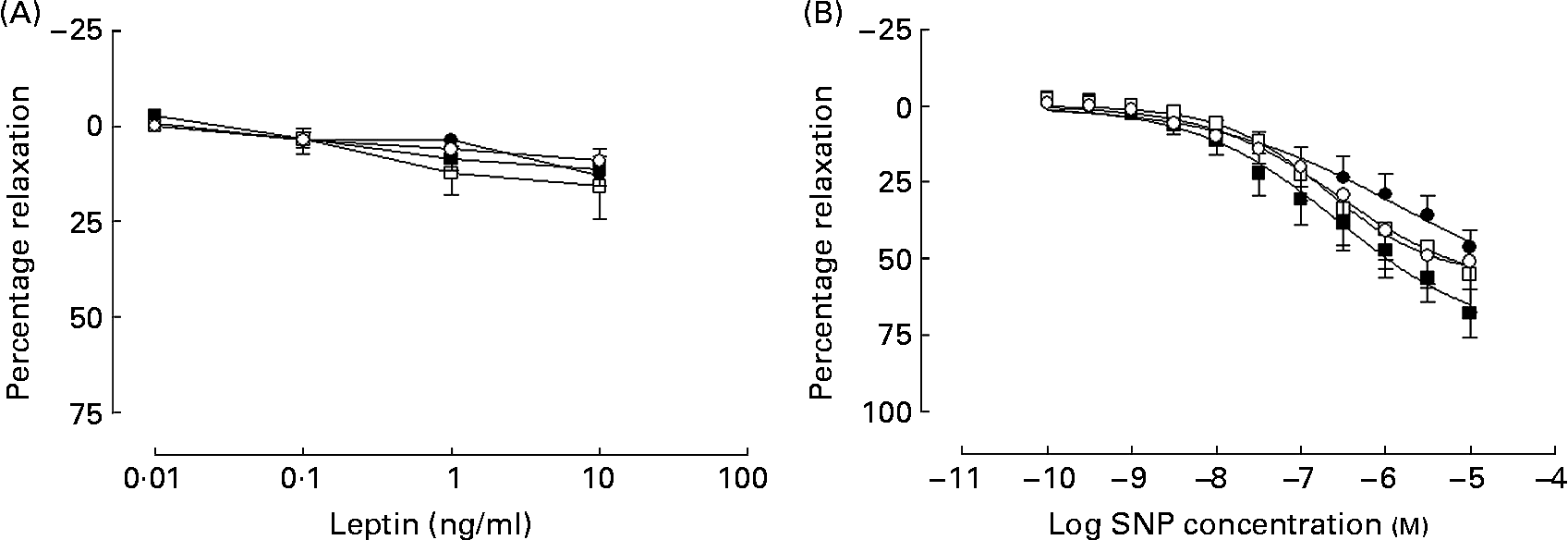
Fig. 4 Cumulative additions of (A) the appetite hormone leptin and (B) the NO donor sodium nitroprusside (SNP) to mesenteric arteries from 365-d-old rats from ad libitum chow-fed (○, n 5–6), ad libitum high-fat-fed (□, n 7), undernourished chow-fed (●, n 5–8) and undernourished high-fat-fed (■, n 6–8) rats. Data are means, with standard errors represented by vertical bars.
Endothelium-independent vasodilatation
In all groups, the NO donor sodium nitroprusside produced a concentration-dependent vasodilatation in all four experimental groups (Fig. 4 (B)).
Discussion
An adverse intra-uterine environment is associated with long-term metabolic consequences, in particular obesity and CVD. Data from epidemiological as well as animal studies have given rise to the concept of developmental programming, whereby an unfavourable prenatal environment is believed to trigger adaptive responses that improve fetal survival or prepare the fetus in expectation of a particular range of environments postnatally. However, if the pre- and postnatal environments are widely discrepant, these adaptive responses may prove to be the origins of later disease(Reference Gluckman and Hanson4). Our model of maternal undernutrition followed by postnatal high fat(Reference Vickers, Breier, Cutfield, Hofman and Gluckman10–Reference Vickers, Gluckman, Coveny, Hofman, Cutfield, Gertler, Breier and Harris12) supports epidemiological evidence that the greatest risk is seen in those who are born small but have rapid postnatal weight gain(Reference Eriksson, Forsen, Tuomilehto, Osmond and Barker5). The present study shows that endothelial dysfunction is manifest in offspring of undernourished mothers, independently of postnatal dietary environment and is a permanent consequence of adverse prenatal conditions.
The vascular endothelium is important in the control of vascular tone, and endothelial dysfunction is a common feature of atherosclerosis(Reference Landmesser, Hornig and Drexler16) and hypertension(Reference Panza, Casino, Kilcoyne and Quyyumi17) as well as type 2 diabetes and the metabolic syndrome(Reference Fornoni and Raij18). Impaired endothelial function following maternal dietary manipulations have been previously reported in the offspring of fat-fed dams(Reference Khan, Taylor, Dekou, Seed, Lakasing, Graham, Dominiczak, Hanson and Poston21–Reference Taylor, Khan, Hanson and Poston23) as well as offspring from protein-restricted dams(Reference Brawley, Itoh, Torrens, Barker, Bertram, Poston and Hanson7, Reference Torrens, Brawley, Anthony, Dance, Dunn, Jackson, Poston and Hanson9). Similarly to the present study, endothelial dysfunction has also been seen in the offspring of dams following global nutrient restriction. However, these have not always been consistent and may depend on the severity and duration of the challenge as to whether effects on endothelial function are seen(Reference Franco, Fortes and Akamine8, Reference Holemans, Gerber, Meurrens, De Clerck, Poston and Van Assche24), or not(Reference Williams, Hemmings, Mitchell, McMillen and Davidge25). While evidence for altered endothelial function exists from the offspring of nutrient-restricted dams, the present study is the first to examine the possible effects of nutritional mismatches on postnatal endothelial function.
A diet high in fat has long been considered a risk factor for such CVD, and the impact of high-fat diets on blood pressure and vascular function is well documented(Reference Gerber, Holemans, O'Brien-Coker, Mallet, van Bree, Van Assche and Poston26–Reference Song, Gao, Di, Pan, Zhou and Ye29). The present study confirms these observations, noting that endothelial function is impaired following a high-fat diet. In addition, the relative hyperphagia and predisposition to sedentary activity in this model may have contributed to the endothelial dysfunction observed in offspring of undernourished mothers(Reference Vickers, Breier, Cutfield, Hofman and Gluckman10, Reference Vickers, Breier, McCarthy and Gluckman11). We have previously described increased systolic blood pressure in a similar animal model(Reference Vickers, Breier, Cutfield, Hofman and Gluckman10–Reference Vickers, Gluckman, Coveny, Hofman, Cutfield, Gertler, Breier and Harris12) and, taken together, our finding of endothelial dysfunction may not be surprising. What is interesting about these findings, however, is whilst the effects on systolic blood pressure showed a synergistic interaction between the maternal undernutrition and postnatal diet(Reference Vickers, Breier, Cutfield, Hofman and Gluckman10–Reference Vickers, Gluckman, Coveny, Hofman, Cutfield, Gertler, Breier and Harris12), no such interaction was observed in reference to endothelial function. The lack of such an interaction in the present study may suggest that the timing of the postnatal challenge is important, as the postnatal high-fat feeding began later in the present study compared with previous studies. However, despite the shorter duration of the high-fat challenge in the present study it is clear that the energy intake and weight gain was still significantly greater in the high-fat groups, suggesting that the pre-pubertal timing in previous reports may be of importance to the synergistic effects of pre- and postnatal dietary interactions(Reference Vickers, Gluckman, Coveny, Hofman, Cutfield, Gertler, Breier and Harris12). Alternatively, it is possible that postnatal high-fat diets and maternal undernutrition have comparable effects on the vascular endothelium and the mediators of vasodilatation and that whilst postnatal diet and lifestyle have an undoubted effect on cardiovascular health, the prenatal environment may be of equal importance.
Our evidence of endothelial dysfunction is inferred from our observation of an attenuated dilatation to ACh in all three experimental groups at 1 year of age. In rat small mesenteric arteries, ACh-mediated vasodilatation involves the release of NO, PGI2 and EDHF from the endothelium(Reference Busse and Fleming14), with EDHF providing the major contribution, a smaller, yet sizeable NO component and only a small PGI2 component(Reference Taylor, Khan, Hanson and Poston23). As such, the impaired dilatation reported in the present study is likely to be due to alteration in one or more of these pathways, and experiments were conducted in the presence of NOS and COX blockade to identify the component parts of the ACh response. There are two interesting findings from the more in-depth analysis of the ACh response. First, it is clear that following NOS and COX blockade, the response to ACh was almost abolished, indicating a relatively small EDHF component in comparison with younger animals. Second, the differences previously noted in the ACh response are no longer present. Taken together, these data indicate that the EDHF component is equivalent in all groups and, given the minor role for PGI2 in these arteries, the impairment in the ACh response is likely to be due to decrease in the NO pathway.
A decrease in the bioavailability NO could arise from a decrease in the endothelial NOS expression or activity as well as an increase in oxidative stress. Decreased levels of the enzyme endothelial NOS have been reported following maternal undernutrition(Reference Torrens, Brawley, Anthony, Dance, Dunn, Jackson, Poston and Hanson9, Reference Franco, Arruda, Dantas, Kawamoto, Fortes, Scavone, Carvalho, Tostes and Nigro30) and postnatal high-fat diets(Reference Roberts, Barnard, Sindhu, Jurczak, Ehdaie and Vaziri28, Reference Song, Gao, Di, Pan, Zhou and Ye29), while increased oxidative stress has been proposed as a mechanism for the vascular dysfunction following maternal undernutrition(Reference Franco, Fortes and Akamine8). Previous studies suggest that decreased NO bioavailability enhances the response to sodium nitroprusside, both in the thoracic aorta of endothelial NOS-null mice(Reference Waldron, Ding, Lovren, Kubes and Triggle31), and also alongside endothelial dysfunction in the offspring of nutrient-restricted dams(Reference Holemans, Gerber, Meurrens, De Clerck, Poston and Van Assche24). The present study did not note any change in response to sodium nitroprusside indicating that whilst endothelial-dependent vasodilatation is attenuated, the smooth muscle soluble guanylate cyclase pathway is unaltered by the dietary challenges. In addition, the adipokine leptin has also been suggested to induce vasodilatation through an endothelial-dependent mechanism in isolated arteries(Reference Lembo, Vecchione, Fratta, Marino, Trimarco, d'Amati and Trimarco20). In the present study, leptin-induced vasodilatation was poor in comparison with the other vasodilators tested, although previous reports do not indicate leptin to be a particularly potent dilator(Reference Lembo, Vecchione, Fratta, Marino, Trimarco, d'Amati and Trimarco20, Reference Jaffar, Myers, Hainsworth, Hainsworth and Drinkhill32). Whilst some have reported leptin acting through an NO-dependent pathway(Reference Jaffar, Myers, Hainsworth, Hainsworth and Drinkhill32), in the mesenteric arteries akin to those investigated in the present study this appears to be an NO-independent, EDHF pathway(Reference Lembo, Vecchione, Fratta, Marino, Trimarco, d'Amati and Trimarco20). Owing to the poor response to leptin, only a small number of leptin responses were repeated in the presence of l-NAME (n 3 from all groups). Where performed, NOS blockade had no effect on the leptin response and would seem to suggest that leptin response is NO-independent; the lack of difference between the groups would therefore not contradict a decrease in NO bioavailability.
A novel finding from the present study is the enhanced vasoconstriction to ET1. ET1 is a potent vasoconstrictor peptide released from the endothelium(Reference Yanagisawa, Kurihara, Kimura, Tomobe, Kobayashi, Mitsui, Yazaki, Goto and Masaki15) which acts on two receptor subtypes: ETA and ETB. Whilst the ETA mediates constriction, the ETB receptor mediates vasodilatation through NO(Reference Fujitani, Ueda, Okada, Urade and Karaki33) and can modulate the ETA-mediated constrictor response(Reference Goddard, Johnston, Hand, Cumming, Rabelink, Rankin and Webb34). As such, the enhanced constriction could be explained by loss of this balance. Alternatively, the endothelium also releases a number of constricting factors including ET1 and thromboxane in addition to the endothelial-derived relaxing factors(Reference Busse and Fleming14), the enhanced release of which may also be responsible. Both hyperinsulinaemia(Reference Piatti, Monti, Conti, Baruffaldi, Galli, Phan, Guazzini, Pontiroli and Pozza35) and hypercholesterolaemia(Reference Song, Gao, Di, Pan, Zhou and Ye29, Reference Lerman, Edwards, Hallett, Heublein, Sandberg and Burnett36) have been shown to increase release of ET1. Such enhanced sensitivity to vasoconstrictors following a high-fat diet would also fit with our observation of enhanced PE constriction in both the ADHF and UNHF groups, although not the UNC group. However, while enhanced ET1 release has been implicated in altered ACh responses observed in aortas of dyslipidaemic rabbits, in contrast to the present study responses to exogenous ET1 were similar(Reference Maeso, Aragoncillo, Navarro-Cid, Ruilope, Diaz, Hernandez, Lahera and Cachofeiro37).
In summary, our data demonstrate that undernutrition in utero and/or a high-fat diet post-weaning leads to endothelial dysfunction in the offspring at 1 year of age. Interestingly, the present data suggest that while each challenge in isolation is detrimental to endothelial function, the effects are not cumulative when the offspring are subjected to both challenges. Importantly, while these data support the traditional high-fat risk factor for endothelial dysfunction they also suggest that poor maternal nutrition could be as detrimental to endothelial function and an equivalent risk factor.
Acknowledgements
C. T. and M. H. V. performed the experimental work. C. T., M. A. H., P. D. G. and M. H. V. wrote the paper. P. D. G. and M. H. V. designed the experiment. Financial support for the study was provided by the National Research Centre for Growth and Development (M. H. V. and P. D. G.). C. T. and M. A. H. received salary support from the British Heart Foundation. There are no conflicts of interest.




