



Vitamin E (α-tocopherol) was discovered about 100 years ago because it is required by pregnant rats to bring their fetuses to term(Reference Evans and Bishop1); it is still unknown as to whether vitamin E is needed for a specific function by the mother, the placenta or the developing embryo. It is well accepted that vitamin E functions as an antioxidant by scavenging lipid peroxyl radicals and preventing the propagation of lipid peroxidation (Fig. 1)(Reference Niki2), but it is unclear how the antioxidant function relates to the deficiency symptoms. Vitamin E deficiency in human subjects is well known to cause ataxia, which is a result of the dying back of the sensory neurons of the peripheral nervous system(Reference Sokol, Bove and Heubi3,Reference Traber4) . Further, long-term (decades) α-tocopherol supplementation can prevent progression of the degenerating nervous system caused by vitamin E deficiency(Reference Kohlschutter, Finckh and Nickel5).
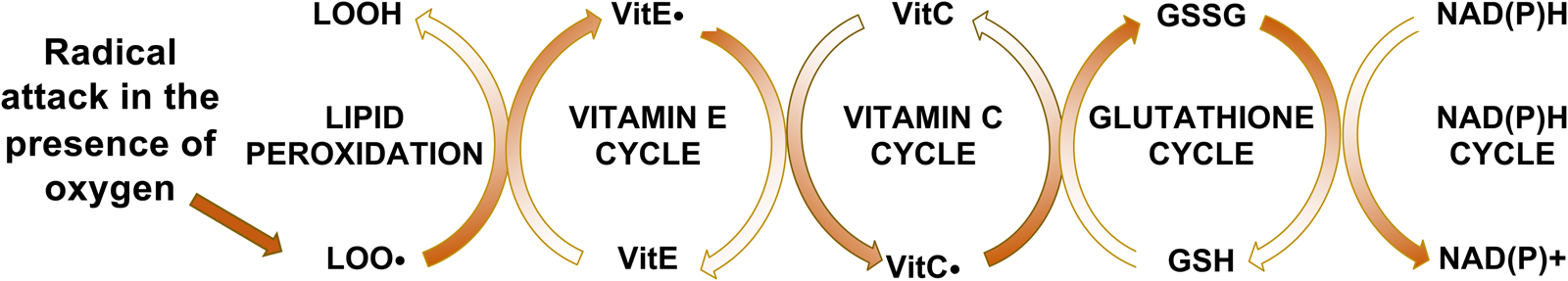
Fig. 1. Vitamin E interactions with lipid peroxidation and antioxidants. Vitamin E intercepts peroxyl radicals (LOO•), but becomes a radical itself (vitamin E•), which is reduced by VitC•, oxidizing it. Glutathione reduces the VitC. and becomes oxidised itself. The GSSG is then enzymatically reduced by glutathione reductase. Thus, the reversal of the entire oxidation process is energy (NADPH) dependent.
Perhaps the most important vitamin E-related discovery in the past century has been the existence of the α-tocopherol transfer protein (α-TTP)(Reference Ben Hamida, Doerflinger and Belal6–Reference Ouahchi, Arita and Kayden8). α-TTP facilitates the hepatic secretion of α-tocopherol, but not other forms of vitamin E (β-, γ-, δ-tocopherols, or α-, β-, γ-, δ-tocotrienols, or synthetic-α-tocopherol (2S-α-tocopherols)) into the circulation(Reference Traber, Burton and Ingold9,Reference Traber, Sokol and Burton10) . Thus, the liver via α-TTP maintains not only the plasma but the entire body α-tocopherol supply(Reference Leonard, Terasawa and Farese11). In addition to the α-TTP gene initially being identified in human subjects, the National Center for Biotechnology Information lists 322 jawed vertebrates, including bony fishes, amphibians, birds and mammals, which have been reported to have the α-TTP gene (https://www.ncbi.nlm.nih.gov/gene/7274/ortholog/?scope=7776&term=TTPA). Apparently, the gene product, α-TTP, is critical for vertebrates.
Vitamin E background information
The vitamin E dietary reference intake for adult men and women (and individuals 14–18 years) was set in 2000 in the USA at a daily estimated average requirement of 12 mg α-tocopherol and an RDA of 15 mg(12). Plants synthesise eight different forms of vitamin E(Reference Mene-Saffrane and DellaPenna13), which all have antioxidant activity. The vitamin E forms are not interconvertible by animals or human subjects, only plants have the appropriate enzymes(Reference Mene-Saffrane and DellaPenna13). However, the human body preferentially retains α-tocopherol; α-tocopherol is the only form that has been shown to reverse clinical vitamin E deficiency symptoms. Therefore, only α-tocopherol meets human vitamin E requirements(12).
Human vitamin E deficiency is based on circulating α-tocopherol concentrations (<12 μmol/L serum or plasma). An increase in the prevalence of human vitamin E deficiency has been reported, based on low circulating α-tocopherol concentrations(Reference Peter, Friedel and Roos14), which may be a result of increased consumption of vegetable oil that has become rancid through multiple frying uses(Reference Fišnar, Doležal and Réblová15) or other causes of rancidity (lipid peroxidation). Vitamin E food sources in addition to vegetable oils include nuts and seeds, and green/leafy vegetables(12).
α-Tocopherol absorption and transport have been studied using stable isotopes over the past 30 years. Recent studies show that intestinal absorption is about 55 %, α-tocopherol is then transported in chylomicrons from the intestine to the liver where it is preferentially secreted from the liver in newly synthesised lipoproteins into the plasma(Reference Traber, Leonard and Ebenuwa16). The α-tocopherol secretion from the liver mediated by α-TTP is the mechanism by which the plasma becomes α-tocopherol-enriched relative to the other forms of vitamin E(Reference Traber, Burton and Ingold9,Reference Traber, Sokol and Kohlschutter17) . Notably, the intestine does not require α-tocopherol absorption and secretion in chylomicrons(Reference Traber, Sokol and Burton10). Thus, the hepatic α-TTP is critical for plasma α-tocopherol enrichment.
Vitamin E and the nervous system
The clearest example of vitamin E deficiency in human subjects is caused by defective α-TTP and results in the disorder, ataxia with vitamin E deficiency (OMIM #277460). Ataxia with vitamin E deficiency is characterised by degeneration of sensory neurons, a progressive dying back of peripheral nerves, which causes a spinocerebellar ataxia with Purkinje cell death(Reference Di Donato, Bianchi and Federico18,Reference Schuelke19) . The defective α-TTP in ataxia with vitamin E deficiency causes low circulating α-tocopherol (<1 μm/l plasma) and low peripheral nerve and adipose tissue α-tocopherol concentrations(Reference Traber, Sokol and Ringel20). In addition, the ataxia may also be a result of impaired α-tocopherol trafficking in the brain because α-TTP is located in the brain in Bergmann glial cells surrounding Purkinje cells(Reference Ulatowski, Parker and Warrier21,Reference Marcos, Gonzalez-Fuentes and Castro-Vazquez22) , suggesting α-TTP in glial cells traffics α-tocopherol to neurons in the brain.
Although it is clear the human body needs α-tocopherol and that other forms of vitamin E do not fulfil this vitamin function because α-TTP does not maintain them, it remains unclear as to why the embryo specifically needs α-tocopherol and what is its molecular function. Critically, human subjects with deficient plasma α-tocopherol concentrations (<12 μm/l plasma) experience greater rates of miscarriage during early pregnancy(Reference Shamim, Schulze and Merrill23), suggesting embryonic defects due to α-tocopherol deficiency. The Traber laboratory has been trying to address these questions by using the premier model of vertebrate embryogenesis, the zebrafish embryo.
Zebrafish embryo model system
Zebrafish embryos are widely used for investigating the molecular mechanisms of vertebrate development because the transcriptional networks, molecular responses and physiology are evolutionary conserved and similar to those in human subjects(Reference Howe, Clark and Torroja24–Reference Garcia, Noyes and Tanguay26). Zebrafish are also highly relevant for antioxidant research because they require the same dietary antioxidants as do human subjects, specifically both vitamins E and C(Reference Drouin, Godin and Page27,Reference Kirkwood, Lebold and Miranda28) . Thus, the vitamin E-deficient zebrafish embryo model allows evaluation of developmental dysregulation in a highly relevant model to establish the mechanisms for the embryonic vitamin E requirement.
The Traber laboratory group has pioneered the use of vitamin E deficient or sufficient (E– or E+) diets fed to adult zebrafish that are spawned to obtain E– and E+ embryos(Reference Lebold, Kirkwood and Taylor29,Reference Miller, Labut and Lebold30) . These E– and E+ fish lay and fertilise eggs in similar numbers(Reference Lebold, Jump and Miller31). Biological variables, such as sex, age, weight and underlying health conditions (with the exception of vitamin E deficiency), are similar between the groups. Vitamin E deficiency causes >70 % of the E– embryos to die or be malformed by 72 h post-fertilisation (hpf)(Reference McDougall, Choi and Kim32) with significant histologic abnormalities as early as 12 hpf(Reference Head, La Du and Tanguay33), although the E+ and E– embryos appear phenotypically normal at 24 hpf(Reference McDougall, Choi and Kim32). Each embryo is a self-contained unit that does not require food until after 120 hpf. Thus, embryonic vitamin E deficiency can be studied as a progression of deleterious outcomes as the embryo progresses through the various developmental stages impacted by increasing lipid peroxidation.
Metabolic consequences of vitamin E deficiency in zebrafish embryos
Potentially, one reason cells in E– embryos die is due to increased lipid peroxidation because it is a self-propagating cycle that generates toxic compounds causing cell death(Reference Yang and Stockwell34,Reference Dixon, Lemberg and Lamprecht35) , while vitamin E prevents the propagation of lipid peroxidation(Reference Carlson, Tobe and Yefremova36–Reference Angeli, Shah and Pratt38). Thus, inadequate vitamin E in lipid peroxidation-susceptible cells, such as neural crest cells(Reference Eason, Williams and Chawla39–Reference Han, Schomacher and Schule42), could be a cause for cell death.
Both targeted and untargeted MS approaches (metabolomics and lipidomics) were used to determine why the E– embryo dies. McDougall et al.(Reference McDougall, Choi and Kim32) discovered that vitamin E deficiency causes a fatal depletion of energy-producing nutrients (e.g. glucose for NADPH production via the pentose phosphate pathway) and that glucose repletion of the embryo by injection at an early stage could be used for rescue. Additionally, the E– embryos at 24 hpf were hypermetabolic, based on their oxygen consumption(Reference McDougall, Choi and Kim32,Reference McDougall, Choi and Kim43) . Thus, metabolic adaptation and compensation occur in the developing E– embryo to alleviate molecular, morphologic and biochemical phenotypes caused by the inadequate vitamin E supply. In support of this statement, E– embryos demonstrated a dysregulation of a complex, interwoven set of metabolic networks (Fig. 2)(Reference McDougall, Choi and Kim32,Reference McDougall, Choi and Kim43,Reference McDougall, Choi and Magnusson44) . Further, quantitative measurements of glutathione, other thiols and methyl (one-carbon) donating molecules demonstrated that vitamin E deficiency leads to metabolic dysregulation, likely caused initially by lipid peroxidation of phosphatidyl choline-DHA (DHA-PC)(Reference McDougall, Choi and Kim43,Reference McDougall, Choi and Truong45,Reference Zhang, Head and Leonard46) , resulting in choline depletion and increased betaine production(Reference Zhang, Head and Leonard46). Vitamin E deficiency also dysregulates the methionine cycle(Reference Zhang, Head and Leonard46). These pathways are interconnected with the folate cycle, and it is well-appreciated that inadequate folate causes neural tube defects, in human subjects and in zebrafish(Reference Lee, Bonner and Bernard47,Reference Kao, Chu and Lee48) . During vitamin E deficiency, the depleted molecule is likely a thiol, probably glutathione, which then appears to dysregulate the balance of cysteine homostasis, both through generation from cystathionine and through the Xc– antiporter(Reference McBean49). Additionally, methyl group donors, such as S-adenosyl methionine, are dysregulated, possibly causing epigenetic dysregulation(Reference McDougall, Choi and Truong45).

Fig. 2. Vitamin E interactions with lipid peroxidation and dysregulation of metabolism. In the absence of vitamin E, lipid peroxidation becomes a chain reaction and depletes critical phospholipids, such as phosphatidyl choline (oxidised phosphatidyl choline shown as a lipid hydroperoxide-phosphatidyl choline, LOOH-phosphatidyl choline). To replace these molecules, choline is needed, but choline is also needed via betaine for maintenance of the methionine cycle. Critically, lipid hydroperoxides (LOOH) also consume thiols, such as glutathione, which must be synthesised from the limited amino acid, cysteine. To maintain cysteine, the cell depends both on the methionine cycle as well as the Xc– antiporter. Thus, with inadequate vitamin E, multiple overlapping pathways become depleted and dysregulated.
The critical role of vitamin E as an antioxidant and the relationship between lipid peroxidation, glutathione and other thiols, taken together, suggest that the abnormalities and lethality observed in the E– embryos are a result of lipid peroxidation-dependent death mechanisms, such as ferroptosis, as has been described for liver(Reference Carlson, Tobe and Yefremova36). Vitamin E deficiency impairs the zebrafish embryo at a time prior to when a woman knows she is pregnant, very similar to the actions of folic acid deficiency on neural tube development. Although overt vitamin E deficiency is rare, the prevalence of vitamin E deficiency (serum α-tocopherol concentrations 12 μmol/L) in Bangladesh is estimated at 70 % of women(Reference Shamim, Schulze and Merrill23). Bangladeshi women with low α-tocopherol concentrations were about1⋅8 times more likely to miscarry(Reference Shamim, Schulze and Merrill23). Additionally, Balogun et al. in a Cochrane Database Systematic Review(Reference Balogun, da Silva Lopes and Ota50) reported that ‘There was evidence of a decrease in the risk for stillbirth among women receiving multivitamins plus iron and folic acid compared to iron and folate only groups (RR 0⋅92, 95% CI 0⋅85 to 0⋅99, 10 trials, 79,851 women; high-quality evidence).’ The beneficial results from supplementation with both multivitamins plus folate and iron support the idea that the stillbirth in human subjects induced by vitamin E inadequacy can be reversed by multivitamins containing vitamin E, but further research is needed. Additionally, vitamin E deficiency in zebrafish embryos induces secondary deficiencies of DHA, choline and glucose. Choline depletion may be the most important for human subjects because people do not consume sufficient choline(Reference Wallace and Fulgoni25). Choline is a methyl donor that works in concert with folic acid and other B-vitamins and there is cross-talk between methylation status and energy homeostasis(Reference Zeisel51).
Vitamin E and neurogenesis
In early studies characterizing vitamin E deficiency in rodents, abnormalities were described to include exencephaly(Reference Cheng, Chang and Bairnson52,Reference Verma and Wei King53) , dorsal root ganglia degeneration and defective blood–brain barrier formation(Reference Verma and Wei King53). Importantly, neural tube defects were also described in vitamin E-deficient mice(Reference Jishage, Arita and Igarashi54–Reference Raabe, Flynn and Zlot58). Vitamin E protects zebrafish and rodent embryos at embryological states in which neural tube defects occur in human embryos 18–19 hpf in zebrafish(Reference Kimmel, Ballard and Kimmel59), 9–12 d in rats(60) and 22–30 d in human subjects(Reference O'Rahilly61–Reference Gilbert63) (Table 1).
Table 1. Comparisons of timing of developmental stages between zebrafish, rats and human subjects

Embryological stages in zebrafish(Reference Kimmel, Ballard and Kimmel59), 9–12 d in rats(60) and 22–30 d in human subjects(Reference O'Rahilly61–Reference Gilbert63).
Neural crest cells are also important for the evaluation of the impact of vitamin E deficiency during embryonic development. Neural crest cells are stem cells that differentiate into precursors of the peripheral nervous system, as well as the cardiovascular system, craniofacial skeleton and pigment epithelia(Reference Tang, Martik and Li64). They migrate and differentiate into distinct populations along the embryo body axis during embryogenesis. Neural crest cells have a limited supply of nutrients for their migration through the embryo and are, thus, especially susceptible to oxidative damage(Reference Eason, Williams and Chawla39). Studies in E– embryos indicated these cells need more vitamin E antioxidant protection(Reference Head, La Du and Tanguay33). Specifically, neural crest cell formation is the result of a well-orchestrated gene regulatory network(Reference Weider and Wegner65). SRY-related HMG-box 10 protein (Sox10), a transcription factor, has a direct role in sensory neuron specification(Reference Carney, Dutton and Greenhill66) and most peripheral nervous system neurons and glia are neural crest cell-derived and express Sox10(Reference Weider and Wegner65). These peripheral nervous system sites are also the most susceptible to damage in vitamin E-deficient human subjects(Reference El Euch-Fayache, Bouhlal and Amouri67). Importantly, E– zebrafish embryos demonstrated fewer cells expressing Sox10 (Reference Head, La Du and Tanguay33). Collectively these data suggest that impaired neurodevelopment and degeneration are associated with neural crest cell abnormalities and cells derived from neural crest cells. Apparently, metabolic adaptation of the neural crest cells to vitamin E deficiency limits their migration, proliferation, differentiation and survival.
Requirement for α-TTP and α-tocopherol in embryonic development
α-TTP was reported in the developing human embryo at 5–12 weeks of gestation, specifically, α-TTP is expressed in the yolk sac(Reference Jauniaux, Cindrova-Davies and Johns68). Thus, zebrafish embryos with their yolk sac and ease of visualisation make a good model for these studies. The zebrafish embryo Ttpa mRNA increases 7-fold by 12 hpf and remains elevated at 24–36 hpf, while its knockdown causes 100 % embryonic mortality by 24 hpf(Reference Miller, Ulatowski and Labut69). Further, α-TTP is essential for normal neural plate and neural tube formation(Reference Miller, Ulatowski and Labut69). Ttpa is also found in developing brain, eyes and tail bud(Reference Miller, Ulatowski and Labut69). Thus, vitamin E uptake and trafficking occurs in the nervous system prior to the development of the liver or circulatory system, suggesting it is critical in the nervous system for delivery of vitamin E to specific regions. Ttpa is also highly expressed at the leading edges of the brain cavities during brain ventricle formation(Reference Head, La Du and Tanguay33). Importantly, in E– embryos, the development of the brain, the migration of neural stem cells and the formation of the spinal cord were impaired(Reference Head, La Du and Tanguay33). Taken together, these data show that both α-TTP and vitamin E are critical molecules during embryonic development, especially during neurulation (neural plate and tube formation)(Reference Miller, Ulatowski and Labut69) and neural crest cell migration(Reference Head, La Du and Tanguay33).
Neurodegeneration and cognition
Recent developments in neuroscience have shown that the human hippocampus, the site of memory and learning, undergoes neurogenesis in adults, but declines with ageing, which may be linked to cognitive impairments(Reference Boldrini, Fulmore and Tartt70). In 2015, 46⋅8 million people worldwide were living with dementia and this number will reach 131⋅5 million in 2050(Reference Prince, Comas-Herrera and Knapp71) Brain neurodegenerative disorders (e.g. Alzheimer's disease and -related diseases, and Down syndrome) are associated with (1) cognitive decline(Reference Barone, Arena and Head72,Reference Butterfield and Boyd-Kimball73) , (2) increased lipid peroxidation(Reference Buijs, Doan and van Rooden74), (3) changes in metabolic function(Reference Fu, Shi and Westaway75), and (4) mitochondrial dysfunctions and metabolic reprogramming(Reference Grimm, Friedland and Eckert76,Reference Demetrius, Magistretti and Pellerin77) . The research community has focused on damaged proteins, but lipid peroxidation may be more dangerous in the brain because it is a self-propagating cycle that generates radicals and toxic lipid oxidation end-products (e.g. reactive aldehydes) that can damage proteins, DNA, etc. Our discoveries in adult zebrafish also show that low brain α-tocopherol is associated with a nearly 60 % depletion of 19 brain lysophosphatidyl cholines (lysoPL, combined P = 0⋅0003), especially 3 lysoPL containing DHA: lysoP-choline, -ethanolamine, -serine(Reference Choi, Leonard and Kasper78). The wide variety of lysoPL that are depleted suggests that the entire lysoPL substrate population is affected. LysoPL are needed for phospholipid remodelling during membrane synthesis, repair and replacement(Reference Shindou, Hishikawa and Harayama79). The brain acquires DHA as lysoPL-DHA(Reference Lagarde, Bernoud and Brossard80). A transporter from the major facilitator superfamily, MFSD2A, which is critical to maintain the blood–brain barrier(Reference Ben-Zvi, Lacoste and Kur81), facilitates brain lysoPL-DHA uptake(Reference Nguyen, Ma and Shui82). The MFSD2A transporter is a critical mechanism for lysoPL-DHA(Reference Lagarde, Bernoud and Brossard80) delivery to the brain(Reference Nguyen, Ma and Shui82), resolving a long-standing mystery of how the brain acquires DHA(Reference Pelerin, Jouin and Lallemand83). Importantly, lysoPL-DHA depletion is linked to Alzheimer's disease(Reference Semba84).
Protection from lipid peroxidation is provided by a network of antioxidants, including vitamin E and glutathione(Reference Cadenas, Packer and Traber85), and is dependent on energy production (NADPH)(Reference McDougall, Choi and Kim32). The brain is particularly susceptible to lipid peroxidation due to its high concentration of polyunsaturated lipids (e.g. DHA-PC (18:0/22:6))(Reference Cobley, Fiorello and Bailey86,Reference Wu, Song and Wang87) . DHA-PC is a significant membrane component in the brain(Reference Farkas, Kitajka and Fodor88) and a human serum biomarker of Alzheimer's disease(Reference Yuki, Sugiura and Zaima89). To replace peroxidised DHA-PC requires (1) GSH to detoxify the oxidised lipids(Reference Cadenas, Packer and Traber85) and (2) choline(Reference Bernhard, Bockmann and Maas90), a one-carbon donor for DHA-PC synthesis(Reference Bernhard, Bockmann and Maas90,Reference Yan, Ginsberg and Powers91) . The metabolic connection linking choline and one-carbon donors is through homocysteine.
Since homocysteine is an oxidation product and vitamin E is an antioxidant, oxidative damage has long been a focus in Alzheimer's disease research. Homocysteine elevation has been long recognised as a risk factor for dementia(Reference Smith and Refsum92), is used as a biomarker to predict Alzheimer's disease pathology in human subjects(Reference Dayon, Guiraud and Corthesy93), and homocysteinemia is decreased by increased B-vitamin intakes(Reference Cordaro, Siracusa and Fusco94). Nonetheless, clinical trials using B-vitamin supplements have shown no benefit for improving cognitive impairment or dementia(Reference Aisen, Schneider and Sano95), despite the slowing of brain shrinkage(Reference Douaud, Refsum and de Jager96). Could hyperhomocysteinemia be a result of inadequate brain vitamin E? Human clinical trials have shown that vitamin E supplements slowed the onset of dementia in patients with Alzheimer's disease(Reference Dysken, Sano and Asthana97,Reference Sano, Ernesto and Thomas98) . However, meta-analyses of vitamin E supplements used in a number of Alzheimer's disease trials have shown no statistical benefit(Reference Farina, Isaac and Clark99,Reference Farina, Llewellyn and Isaac100) . By contrast, low blood vitamin E levels are associated with high AD incidence(Reference de Wilde, Vellas and Girault101). Importantly, improved cognition and less brain shrinkage were associated with long-term dietary patterns that increase blood levels of both B-vitamins and vitamin E(Reference de Wilde, Vellas and Girault101,Reference Bowman, Silbert and Howieson102) . Based on the molecular interrelationships between vitamin E and B-vitamins in neurodegeneration, it is clear that chronic poor dietary choices such as diets low in vitamins B and E can promote neurodegeneration and cognitive decline.
Conclusions
Significant progress has been made in understanding the role of vitamin E in embryogenesis. The Traber laboratory has taken on these investigations because: (1) vitamin E is critical during neuro-embryogenesis; (2) the mechanisms by which vitamin E prevents defects during neural differentiation are heretofore unstudied; (3) the pathophysiological mechanisms of embryonic neurodegeneration are investigated in a highly relevant paradigm since women are increasingly consuming inadequate amounts of vitamin E(Reference Peter, Friedel and Roos14,103) ; and (4) the role of lipid peroxidation, as a mediator of embryonic neurodegeneration has largely been overlooked, although the embryonic environment is recognised to be under oxidative stress(Reference Rodriguez-Rodriguez, Ramiro-Cortijo and Reyes-Hernandez104) and redox status is tightly regulated(Reference Jones and Sies105).
The Traber laboratory has focused on the unknown mechanism of an antioxidant vitamin in a vertebrate embryo to prevent nervous system abnormalities. Elucidation of the cellular and metabolic pathways involved in the molecular dysregulation caused by vitamin E deficiency will lead to important insights into abnormal neurogenesis and embryonic malformations. Identification of vitamin E-dependent pathways is necessary to provide critical knowledge necessary for effective progress in public policy concerning nutritional and therapeutic interventions to prevent malformations, such as neural tube defects during early embryonic development and potentially associated miscarriages(Reference Shamim, Schulze and Merrill23).
Importantly, for human disease pathophysiology, the brain is particularly susceptible to lipid peroxidation due to its high concentration of polyunsaturated lipids, especially DHA-PC(Reference Cobley, Fiorello and Bailey86,Reference Wu, Song and Wang87) . Based on the molecular interrelationships between vitamin E and B-vitamins in neurodegeneration, it is clear that chronic poor dietary choices may exacerbate deficiencies that can promote neurodegeneration and cognitive decline. What is less clear is whether any dietary changes can reverse damage or improve cognition.
Acknowledgements
M. G. T is supported in part by the Ava Helen Pauling Endowment to the Linus Pauling Institute.
Financial Support
The research reported in this publication was partially supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award P30ES030287. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
None.
Authorship
The author had sole responsibility for all aspects of preparation of this paper.



