



Chikungunya fever is caused by Chikungunya virus (CHIKV), an arbovirus that belongs to the genus Alphavirus and is transmitted by Aedes mosquitoes. The most common symptoms are fever, asthenia, arthralgia, myalgia, headache and rash [Reference Sharma and Jain1, Reference McFee2] and even though it is a self-limiting disease, it can last from days to years thus posing a significant impact in public health and serious economic burden [Reference Cunha3].
CHIKV outbreaks have occurred in Africa, Asia, Europe, Indian and Pacific regions and was introduced into the Caribbean in 2013, from where it spread to Central and South America [Reference Sharma and Jain1, Reference Nunes4]. In Brazil, the first autochthonous case was identified in 2014, in the northern state of Rondônia, caused by the Asian genotype. A week later, autochthonous East/Central/South/African (ECSA) genotype cases were confirmed in Bahia, northeast region of Brazil [Reference Nunes4].
More recently, our group had access to sera samples from a suspected CHIKV outbreak in Governador Valadares, state of Minas Gerais (Oswaldo Cruz Foundation IRB approval 1.920.256). Patients with history of arthralgia and fever were recruited during two visits (June and August 2017) to a local Arbovirus Healthcare Center. From a total of 80 patients presenting with compatible clinical manifestations, 25 reported symptoms for less than 14 days (median (IQR): 1 (1–1.75)). Those were classified as acute-phase patients and were further investigated for Chikungunya disease by ELISA (Euroimmun, Germany) and qRT-PCR (ZDC kit, Bio-Manguinhos, Brazil) according to manufacturers’ instructions.
Four patients were borderline for IgM antibodies and one of them was qRT-PCR positive. Four patients were IgM positive, all with negative qRT-PCR as IgM usually becomes detectable after the end of viraemia. Of the remaining 17 patients with negative serology, nine tested positive by qRT-PCR. None of the patients had detectable IgG and all were negative for Zika and Dengue by ZDC molecular multiplex test.
In order to determine which genotype was responsible for the outbreak, viral RNA from the 10 qRT-PCR positive samples was extracted with QIAmp RNA Viral Mini Kit (Qiagen, Germany) and 3 µl was used to amplify a fragment of 1014 bp of E1 sequence, with previously described primers [Reference Santhosh5], in a 25 µl reaction with SuperScript™ III Platinum™ One-Step RT-PCR Kit (Invitrogen, USA). Primer concentration was adjusted to 0.4 µM and cycling conditions were as follows: 55 °C – 30 m, 94 °C – 2 m, 35 cycles of 94 °C – 15 s, 57 °C – 15 s, 68 °C – 1 m and a final extension of 68 °C – 7 m. PCR products were purified with Wizard® SV Gel and PCR Clean-Up System (Promega, USA) and sequenced with BigDye 3.1 Cycle Sequencing Kit (Applied Biosystems, USA) using ABI 3500XL Genetic Analyzer (Life Technologies, USA) following manufacturer's recommendations. CHIKV_seq_10688–10707 F: CCATACTCTCAGGCACCATC and CHIKV_seq_10688–10707 R: GATGGTGCCTGAGAGTATGG were used as internal primers.
Final consensus sequences were obtained with SeqManII v5:05 (DNASTAR, USA) and were deposited in GenBank under accession numbers MH215572-MH215581. When compared to each other, sequences were not identical but overall identity was greater than 99%. Moreover, none of the patients had the A226 V mutation in E1 protein previously described to facilitate CHIKV transmission by Aedes albopictus mosquitoes [Reference Schuffenecker6].
Consensus sequences were then aligned with E1 partial sequence (genome position 10145–11158) edited from 41 reference genomes, including ECSA (n = 12), Asian (n = 15) and West African (n = 11) genotypes. Viral phylogenies were performed with MEGA 5.2 software [Reference Tamura7] using the neighbour-joining method [Reference Saitou and Nei8] and trees were constructed using the Tamura-Nei model [Reference Tamura and Nei9]; the reliability of the branches was validated by the generation of 1000 ‘bootstrap’ replicates.
A phylogenetic tree, including representative sequences for all known genotypes, revealed that all 10 cases from Minas Gerais belonged to monophyletic group associated with ECSA genotype (Fig. 1). They clustered with sequences isolated in previous years in Bahia, probably due to states’ geographic proximity (Fig. 2). Indeed, a recent study [Reference Cunha3] also showed that ECSA is the circulating genotype in the state of Sergipe, which borders Bahia to the north. As expected by Nunes and collaborators [Reference Nunes4], ECSA is spreading from Feira de Santana, Bahia to the northeast and southeast of Brazil and even though they have predicted that both Asian and ECSA genotypes would eventually spread to the whole country, we have not detected Asian samples in Governador Valadares, Minas Gerais.
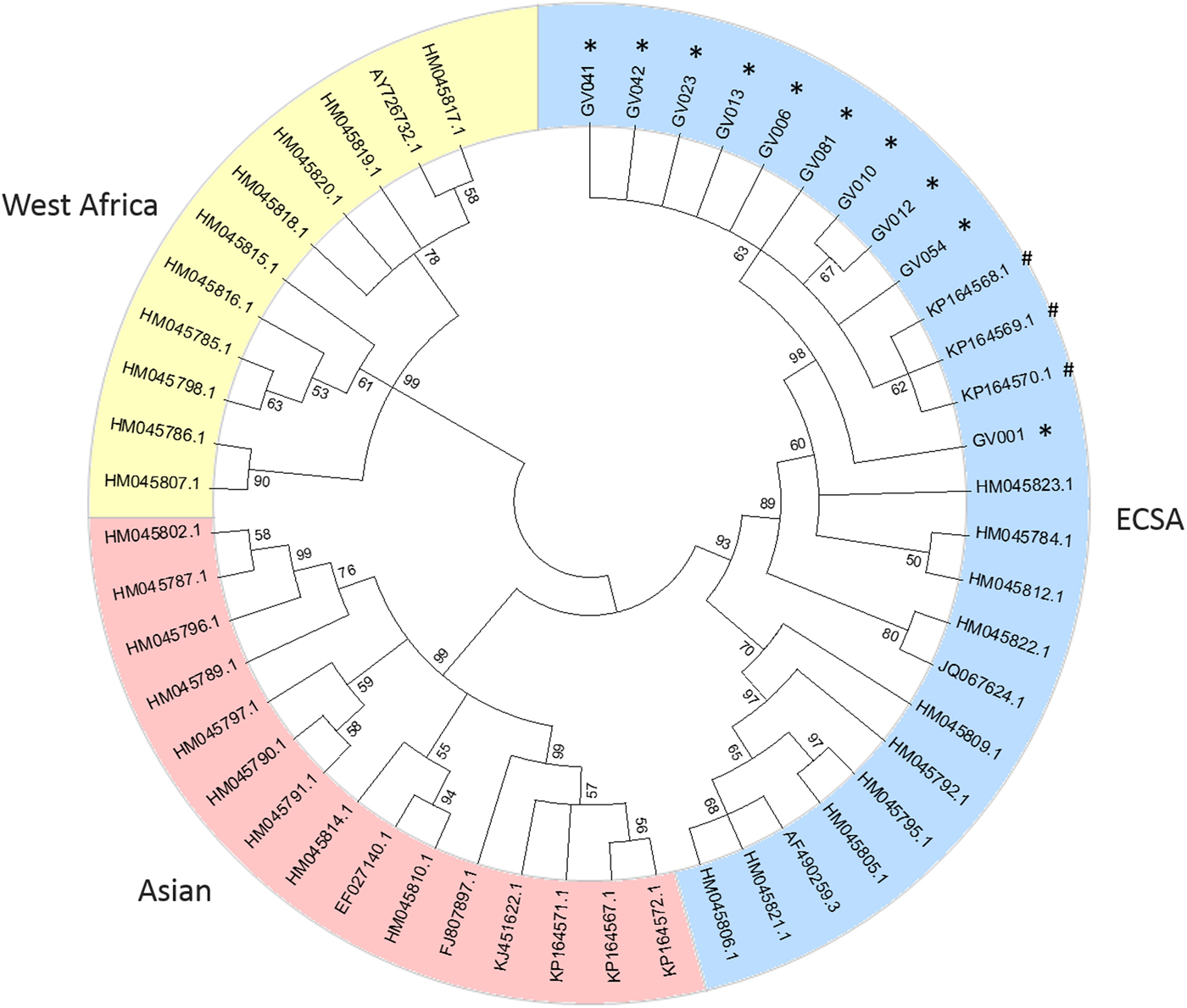
Fig. 1. Phylogenetic tree of 51 Chikungunya E1 partial sequences. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches [Reference Felsenstein10]. Sequences are identified by the corresponding GenBank accession number.
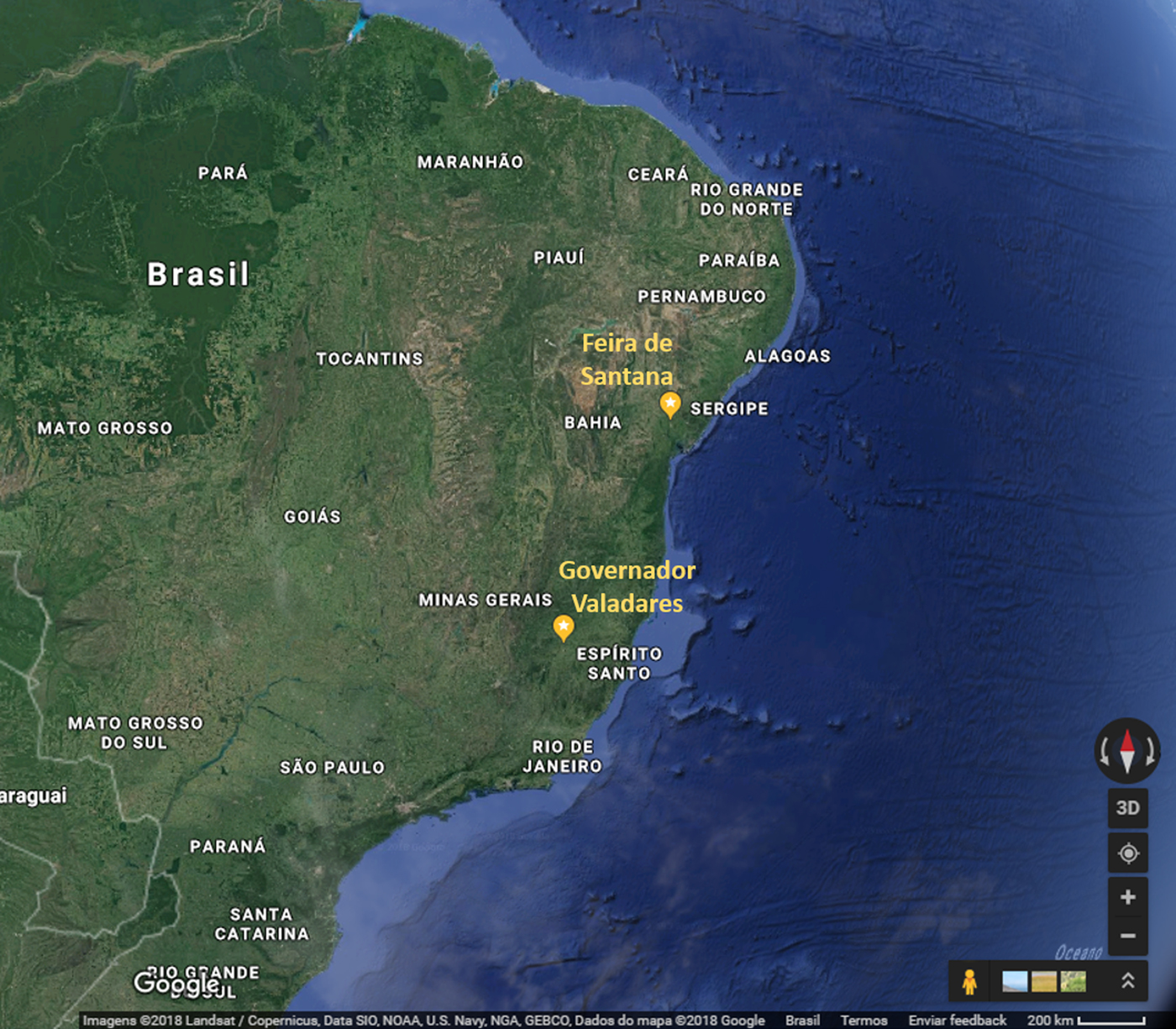
Fig. 2. Partial map of Brazil showing the geographic proximity of Feira de Santana, state of Bahia and Governador Valadares, state of Minas Gerais. Source: Google Maps. https://www.google.com.br/maps/@-17.0364381,-51.9291697,1706185m/data=!3m1!1e3 (Accessed 23 July 2018).
Acknowledgements
The authors would like to thank Tatiane Bettoni, Rômulo Gusmão and Raylaine Castro from Governador Valadares Epidemiology Surveillance and the staff of Rui Pimenta Arbovirus Unit for their support in Chikungunya infection studies. Special acknowledgment also goes to Bio-Manguinhos’ Development Department for providing the ZDC molecular kits and to all patients involved in this study.
Ethics statement
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.


