


Multiple sclerosis (MS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) are both immune-mediated demyelinating disorders of the nervous system. However, in MS, autoimmune demyelination comprises the central nervous system (CNS), whereas CIDP is restricted to the peripheral nervous system (PNS). The literature has a limited number of reports concentrated on the occurrence of these demyelinating diseases in the same patient.Reference Quan, Pelak, Tanabe, Durairaj and Kleinschmidt-Demasters 1 - Reference Zephir, Stojkovic and Latour 12 There is still controversy whether this cooccurrence is a specific new entity or there is heterogeneity among this group as well. Previous reports identified some commonalities among these patients including absence of oligoclonal bands (OCBs) and conduction block. In a recent study, antibodies against neurofascin antigen; which is found in the Ranvier nodes of both the CNS and PNS, were detected in 86% of such patients.Reference Kawamura, Yamasaki and Yonekawa 3 These reports claimed that these patients may have a distinct disease with features different from both typical MS and CIDP.
Up until now, there has been no well-documented description of cooccurrence of central and peripheral demyelination at the same relapse in the early period of disease. It may be because of rarity and underrecognition of this phenomenon or lack of detailed radiological workup. In this study, we present clinical, laboratory, electrophysiological, and radiological findings of five patients diagnosed with both MS and CIDP. Cases 1, 2, and 3 need special attention because of diffuse demyelination of the nervous system and hypertrophic peripheral nerves during the first severe relapse in their lives.
Methods
We retrospectively evaluated our database of patients with CNS demyelinating disease that includes 1890 patients between January 2005 and December 2011. Five patients who were eventually diagnosed with both MS and CIDP were included in this study. Cases 1, 2, and 3 (group I) were diagnosed with peripheral demyelinating disease during a severe disease episode that extensively involved both the CNS and PNS. Cases 4 and 5 (group II) were diagnosed with CIDP during a separate and less severe relapse of peripheral demyelinating disease. For diagnosis of MS, the study applied the revised 2010 McDonald criteria.
Laboratory tests for connective tissue disorders, cerebrospinal fluid (CSF) analysis; immunoelectrophoresis, enzyme-linked immunosorbent assay for Lyme and syphilis; and serology for brucellosis were studied in all of the patients. OCBs were studied with isoelectric focusing. Anti-neurofascin antibodies (NF155 and NF186) were tested in sera of patients 1-4 using the methods described in Ng et al.Reference Ng, Malotka and Kawakami 13
Imaging studies were performed on a 1.5 T MR scanner (Magnetom, Symphony TIM system, Siemens, Erlangen, Germany). Brain magnetic resonance imaging (MRI) included routine contrast-enhanced studies. Spinal MRI included sagittal and axial T1-weighted (W) and T2W in addition to fat-suppressed coronal T2W images. Gadolinium-based contrast material was administered intravenously to all patients both for brain and spinal studies. Retrospective evaluation of cranial and spinal MRI studies was performed by two experienced neuroradiologists (R.G., K.K.O.) in consensus. Cranial imaging findings were assessed using Barkhof criteria. For those patients with longitudinal MRI studies, a visual evaluation was performed to assess changes in lesion load.
Conventional nerve conduction studies (NCS) were performed at the upper and lower extremities. Both the 1991 Ad Hoc Subcommittee criteria and the 2010 European Federation of Neurological Societies/Peripheral Nerve Society criteria were used for CIDP diagnosis.
Results
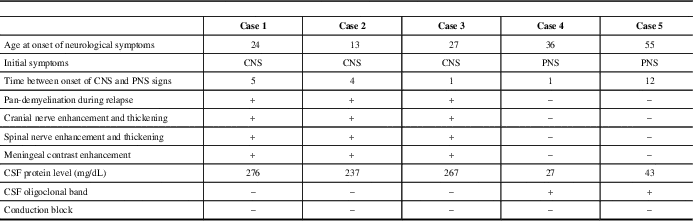
Distinctive clinical, radiological, electrophysiological, and laboratory findings of the patients are summarized in Table 1.
Table 1 Summary of the distinctive findings of patients
Clinical Findings
Patients in group I (one female and two males) presented with CNS symptoms and were diagnosed with MS before CIDP. All of the patients were suspected to have a peripheral demyelinating disease for the first time during a severe relapse that involved both the CNS and PNS. None of the patients had a positive family history or history of infection, vaccination, or toxic exposure. All of the patients had multiple cranial nerve involvement, quadriparesis, hypoesthesia, and loss of position sensation which was more severe in distal lower extremities. Deep tendon reflexes were absent and all patients became bedridden before treatment. Clinical features of patients are shown in supplementary Table 1. Patient 1 had one and patient 2 had three more relapses during follow-up, some of which were purely CNS demyelinating, whereas others were involving both the CNS and PNS. None of the subsequent relapses were as severe as the initial diffusely demyelinating episode. Expanded Disability Status Scale score and MRI follow-up of cases 1 and 2 are shown as timeline graphs in Figure 1A and 1B, respectively.

Figure 1 Clinical and radiological follow-up of patients 1 (A), and 2 (B). EDSS: Expanded Disability Status Scale.
Group II (one female, one male) presented with PNS symptoms of mild paraparesis, glove-and-stocking type paresthesia, and gait problems. Patient 4 fulfilled the McDonald’s criteria at the age of 39, 2 years after the diagnosis of CIDP. The fifth patient’s PNS symptoms appeared up to 12 years before CNS signs and he had both diagnoses at the age of 71. Neither of them had an episode involving both the CNS and the PNS. Peripheral demyelination was insidious, chronic, and more debilitating in comparison to CNS in both of these patients.
Laboratory Findings
Laboratory tests including routine biochemistry, complete blood count, thyroid function tests, vitamin B12, folate, C-reactive protein, sedimentation rate, lipid profile, antinuclear antibodies, anti-dsDNA, rheumatoid factor, antibodies for antiphospholipid syndrome, p-ANCA, c-ANCA, extractable nuclear antigens, and serum immunoelectrophoresis; enzyme-linked immunosorbent assay from serum and CSF for Lyme, syphilis, HSV1, HSV2, HIV, and hepatitis B and C; serology for brucellosis and toxoplasmosis, other (syphilis, varicella, mumps, parvovirus and HIV), rubella cytomegalovirus, and herpes simplex were within normal limits in all five patients. CSF was acellular, protein levels were very high, OCB were negative in group I, and protein levels were within normal limits and OCB were positive in group II (Table 1). Serum anti-neurofascin antibodies were negative.
Radiological Findings
All of the patients had cranial and cervical spinal MRI. Chronic demyelinating lesions in supra- and infratentorial white matter and cervical spinal cord meeting Barkhof criteria were present in all patients except patient 4, who fulfilled two of the criteria. Patients in group I had additional MRI of thoracic and lumbosacral regions. Multiple active, contrast-enhancing cerebral parenchymal lesions, thickening and enhancement of cranial nerves III and V-VIII were present in all patients of group I. Cases 1 and 2 had also thickening and enhancement of cranial nerves IX-X. Widespread thickening and contrast enhancement of spinal nerve roots, brachial (cases 1, 2, and 3) and lumbosacral (cases 2 and 3) plexuses, trunks, cords, and divisions were noted. Enhancement and thickening of cranial nerves and spinal nerve roots persisted during follow-up in cases 1 and 2 (Figure 1A, B). Meningeal contrast enhancement was also present in these patients. Extensive involvement of the cerebral parenchyma, cranial nerves, spinal nerves, and plexuses during the same relapse is shown in Figure 2. Neither patient 4 nor 5 had any contrast enhancement in either brain or spinal roots. Spinal cord lesions had a round or triangular shape, involved short segment (fewer than two vertebral body segments), and were located peripherally. Imaging findings of the patients at the time of CIDP diagnosis and anatomical distribution of disease are shown in Table 2.

Figure 2 Imaging findings of central and peripheral demyelination in all three patients in group I. Transverse contrast-enhanced T1-W images (A-F) show multiple contrast-enhanced acute demyelinating plaques in brain (A-C) as well as symmetrically thickened and contrast-enhanced cisternal segment of both trigeminal (D) (arrows), glossopharyngeal-vagal (E) (arrows), and oculomotor nerves (F) (arrows). (D) Diffuse abnormal meningeal enhancement. Reconstructed by maximum intensity projection (MIP) coronal contrast-enhanced T1W (G) and fat-suppressed T2W (H-J) images of the cervical spine reveal symmetric marked thickened and slightly contrast-enhanced extradural nerve root complexes, ganglia, and brachial plexuses (divisions, segments, and cords) bilaterally. Reconstructed by MIP coronal fat-suppressed T2W images of the lumbar spine (K, L) demonstrate thickening and high signal intensity of lumbar plexuses. Note that the thickening of lumbar nerves of case 3 is milder than others (L).
Table 2 Imaging findings at the time of CIDP diagnosis

B/L, bilateral; N/A, not applicable; ND, not done; R, right.
Electrophysiological Findings
NCS of patients are presented in supplementary Table 2. All of our patients fulfilled the European Federation of Neurological Societies/Peripheral Nerve Society electrodiagnostic criteria and four fulfilled the more strict 1991 Ad Hoc Subcommittee criteria for CIDP. Four of the patients had follow-up NCS. In the first group of patients, improvement in demyelinating features was detected after treatment. This was especially prominent in the second and third patients, which was parallel to the clinical improvement. Follow-up NCS of patient 4, which was performed 2 years later, showed mild worsening of peripheral nerve involvement. In four patients, there was no conduction block; in case 5, there was possible conduction block only in the peroneal nerve.
Treatment and Follow-up
During the acute flair of the disease, all patients in group I continued to deteriorate under 5-day intravenous methylprednisolone (IVMP) therapy, but had a moderate response to plasma exchange. Both the CNS and PNS symptoms of the patients responded well to treatment. As maintenance therapy, the first patient received intravenous immunoglobulin (IVIG) monthly and methotrexate. This patient had a severe and secondarily progressive course during 8-year follow-up. In the second case, we used Interferon-β-1b plus plasma exchange as initial maintenance therapy. After three relapses with moderate recovery, treatment was switched to fingolimod. During 2 years of follow-up, not only the central but also the PNS-related symptoms and MRI activity diminished gradually. Despite the presence of hypoactive deep tendon reflexes, his distal muscle bulk, distal muscle strength, and vibration and position senses were nearly normal and he regained independence. There has been no relapse or progression since that time. Repeat NCS 15 months after fingolimod showed partial but prominent improvement in motor and sensory nerve conduction findings (reported separately as a letter to the editor).Reference Erdener, Nurlu, Gocmen, Erdem-Ozdamar and Kurne 14 Case 3 was given cyclophosphamide and oral prednisolone as maintenance and continued to have his treatment in another country and died after approximately 1 year. Case 4 received IVMP treatment for CNS and PNS symptoms and recovered well with this treatment. For the long-term treatment of PNS symptoms, she received azathioprine and she progressed very slowly. Case 5, who had a long, insidious history of disease, received IVMP treatment for 5 days; however, no improvement was observed. The patient was lost to follow-up.
Discussion
In this article, five patients eventually diagnosed with both MS and CIDP are presented with their clinical, radiological, and laboratory characteristics. Three patients in group I were initially diagnosed with MS and experienced a severe relapse involving both the CNS and PNS shortly after the onset of disease activity. Multiple contrast-enhancing parenchymal lesions in CNS, diffuse enhancement, and thickening in the cranial nerves, spinal nerve roots, and even plexuses were detected by MRI (Figure 2, Table 2). OCB was negative and conduction block was not found in electrophysiological studies of peripheral nerves. Plasma exchange was more effective than IVMP. On the other hand, patients 4 and 5 did not have a fulminant episode involving both CNS and PNS at the same time. Rather, the relapses were separated by time. Both the MS and CIDP of these patients showed a milder, more insidious course and did not necessitate aggressive treatment. In addition, they had classic CSF findings of MS (normal CSF protein level, OCB positivity). These findings suggest that a distinct pathological process involving both the CNS and PNS that is different from classical MS or CIDP may have caused disease in patients of the first group. By contrast, in the second group, the stochastic co-occurrence of both CNS and PNS demyelination appears to be coincidental (Table 1).
Simultaneous demyelination of the CNS and PNS in the same episode is seen in an even rarer subset of patients with combined central and peripheral demyelination (CCPD) and as in situ demonstration of these episodes was not previously available in the literature. Our patients’ MRI findings are unique for three reasons. First, active demyelination of brain, spinal cord, and cranial and spinal nerves during the same episode is shown for the first time. Second, presence of widespread cranial and spinal nerve enhancement and thickening/hypertrophy at early stages of disease is atypical even for pure CIDP.Reference Duggins, McLeod and Pollard 15 Although spinal root hypertrophy is relatively more frequent in the later stages of CIDP, cranial nerve hypertrophy has been reported as case reports in CIDP literature.Reference Guibord, Chalk, Wein, Richardson, Snipes and Del Carpio 16 - Reference McMillan and Miller 18 Simultaneous involvement of both is even rarer and three of our patients had this rare finding.Reference Duarte, Martinez, Rodriguez, Mendoza, Sempere and Claveria 19 In the first case, enhancement of CNs persisted for 2 years during the follow-up period. The MRI demonstrated contrast enhancement of intercostal nerves in patients 1 and 3, a quite rare finding of CIDP.Reference Oguz, Oguz, Cila and Tan 20 Third, this study demonstrates the anatomical extent of the involvement of peripheral nerves in situ, for the first time. As seen in Table 2, trunks, divisions, and cords in addition to intra- and extraforaminal nerve roots are involved in disease process.
Four recent articles discussed the clinical and laboratory features of patients with demyelination of both CNS and PNS.Reference Quan, Pelak, Tanabe, Durairaj and Kleinschmidt-Demasters 1 , Reference Kawamura, Yamasaki and Yonekawa 3 , Reference Sharma, Saadia, Facca, Bhatia, Ayyar and Sheremata 10 , Reference Zephir, Stojkovic and Latour 12 Zephir et al reported five patients in whom CNS was affected before PNS, oligoclonal band was negative, and conduction block was not detected with electromyelograph.Reference Zephir, Stojkovic and Latour 12 Authors postulated that these features may present a distinct dysimmune entity different from classic MS and CIDP. Two patients presented by Quan et al were more similar to our patients, but had a limited number of hypertrophic cranial and spinal nerves.Reference Quan, Pelak, Tanabe, Durairaj and Kleinschmidt-Demasters 1 Likewise, two of the five cases whom Sharma et al presented had a few contrast-enhancing and thickened spinal and cranial nerve roots.Reference Sharma, Saadia, Facca, Bhatia, Ayyar and Sheremata 10 Last, Kawamura et al presented seven cases with CCPD, six of whom were positive for anti-neurofascin antibodies. Three of the patients had simultaneous presentation of CNS and PNS demyelination and one had hypertrophic cauda equine.Reference Kawamura, Yamasaki and Yonekawa 3 Yet, none of these patients had extensive demyelination of the nervous system and prominent thickening of peripheral nerves during the same relapse.
To find similar patients with CCPD and hypertrophic nerve roots, we reviewed the literature using the keywords “central and peripheral demyelination,” “hypertrophic neuropathy,” “nerve root hypertrophy,” and “onion bulb formation” and found 22 cases (Table 3).Reference Quan, Pelak, Tanabe, Durairaj and Kleinschmidt-Demasters 1 , Reference Arias, Requena, Pereiro and Cabello 2 , Reference Ketelaer, Leruitte and Perier 4 - Reference Thomas, Walker and Rudge 11 Only four of these patients are documented with MRI and the remainder belong to the pre-MRI era and were examined by nerve biopsy and/or postmortem examination. In particular, two articles providing postmortem examination data of 11 patients strikingly demonstrated the extent of hypertrophic spinal and cranial nerve roots in these patients.Reference Rosenberg and Bourdette 7 , Reference Schoene, Carpenter, Behan and Geschwind 9 Median age at onset of symptoms in these patients was 26.0 years (interquartile range [IQR]=20.3-33.8, n=18) and the period between first CNS and PNS relapses was 5.0 years (IQR=1.5-6.0, n=11). Supplementary Table 3 lists remaining patients in the literature with MS plus CIDP and without any reported hypertrophic nerve roots. Median age of these patients is 36.0 years (IQR=30.5-56.5, n=15) and the period between first CNS and PNS symptoms is 7.0 years (IQR=3.0-15.5, n=12).Reference Sharma, Saadia, Facca, Bhatia, Ayyar and Sheremata 10 , Reference Zephir, Stojkovic and Latour 12 , Reference Falcone, Scalise, Minisci, Romito, Cancelli and Gigli 21 - Reference Sato, Nakamura, Iwahashi, Morita, Hashimoto and Ikeda 24 Some of these patients presented initially with CIDP.Reference Mendell, Kolkin, Kissel, Weiss, Chakeres and Rammohan 22 These features are common with our patients in group II who presented at a later age with a more progressive and insidious clinical course. Although statistical comparison cannot be made because of the low number of patients, we noted that patients with CCPD and hypertrophic nerve roots present relatively at a younger age.
Table 3 Review of the literature for patients with a demyelinating disease involving both the CNS and PNS and hypertrophic neuropathy

CN, cranial nerve; e, enhancement; ms, multisegmental; ND, not done; OB, onion bulb; t, thickness.
CCPD is supposed to be a distinct entity rather than a coincidence by most authors in the literature. It was argued that spreading of disease activity from central to peripheral myelin antigens may be responsible.Reference Falcone, Scalise, Minisci, Romito, Cancelli and Gigli 21 Alternatively, a common disease mechanism effective from the beginning of disease may be responsible for that syndrome. MS diagnosis precedes CIDP in most of the patients. As the duration between the first CNS and PNS relapses is short and spinal nerves are found to be hypertrophic in the first CIDP relapse, it is still possible that both CNS and PNS demyelination have been ongoing from the beginning. Neurofascin has been identified as a target for autoantibodiesReference Mathey, Derfuss and Storch 25 ; the presence of antibodies to neurofascin in patients with CCPD suggested that this might be a separate disease entity.Reference Kawamura, Yamasaki and Yonekawa 3 In addition, inadequate response of many patients’ PNS symptoms to IVMP therapy, but remission after IVIG or plasma exchange strongly favors the antibody-mediated disease mechanism theory. Yet, not all the patients, including ours, are positive for this antibody. This finding may be caused by difference of the testing methods, or other antibodies than anti-neurofascin may be involved in disease process.
Management of this syndrome is challenging. IVMP was inadequate and IVIG and plasmapheresis were required. We used plasmapheresis as rescue therapy for all three patients in group I, and all of them responded well. Both IVIG and plasmapheresis are effective in severe cases of CIDP.Reference Dalakas 26 , Reference Vallat, Sommer and Magy 27 In the first patient, IVIG/monthly and later methotrexate was initially used as maintenance therapy. This patient with longer follow-up showed a severe and progressive course (Figure 1A). In the second case, we used intermittent plasmapheresis as maintenance therapy. He had three more relapses under plasma exchange maintenance in 1 year with good recovery (Figure 1B).
Certain limitations of our study merit consideration. First, this is an observational study. Second, because of the low number of cases in our cohort, which is actually because of the rarity of the disease, statistical comparison is not possible. We performed a review of literature to overcome that obstacle. Also, although the recently defined anti-neurofascin antibody was tested, further research for other possible antibodies could not be made. We think that the CCPD phenomenon is not as rare as previously thought: some cases are not diagnosed at all and many of them are diagnosed at later disease stages, causing opportunities for clinical and basic research to be missed. It would be beneficial for both clinical and research purposes to investigate such cases by dedicated CNS and PNS MRI methods in addition to serological testing.
Conclusion
We propose that patients with CCPD do not have the same “one” disease but rather form a spectrum. In addition to a recent description of anti-neurofascin antibodies in many of these patients,Reference Ketelaer, Leruitte and Perier 4 our patients in group I in addition to the similar cases in the literature provide strong evidence for a separate inflammatory diffuse demyelination syndrome with hypertrophic nerve roots and a probably distinct immunological mechanism. Our patients did not recognize defined targets, and this finding implies that further research for other antibody specificities is warranted.
Statement of Authorship
A.V. and R.G. contributed equally to this work.
Disclosures
The authors have nothing to disclose.
Supplementary Material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/cjn.2015.238









