


Nature's approach to chemical innovation
Genes, like ideas, move around. Good ones get picked up quickly and spread even faster. If you do not believe it, just try to keep ahead of drug resistance. All those microbes whose very survival depends on solving chemical problems add up to a potent source of chemical novelty. They create new catalysts that we can admire, but cannot design.
Any of the millions, possibly billions, of species that populate our guts, soils, and seas can invent a new catalyst. Four billion years of such evolutionary innovations have generated a mind-boggling diversity: there are myriad enzymes for any given biotransformation and a multitude of transformations catalyzed by any given organism. The biological world is teeming with catalysts, and this cauldron of chemistry is a rich source for yet more. Crowd-sourcing problem-solving is nothing new to the biological world.
But just how are new enzymes created? For one thing, enzymes can be quite versatile; they are often capable of things beyond what nature demands of them. For example, they are catalytically ‘promiscuous’ in various ways, most of which are not beneficial to the organism. But a promiscuous activity can ignite the evolution engine. That enzymes’ serendipitous side capabilities can become the basis for adaptation to a changing environment or new niche opportunities such as using a new food source or exploiting a new reactant has been recognized for years (Copley, Reference Copley2003; O'Brien & Herschlag, Reference O'brien and Herschlag1999). The story of how such processes gave rise to the functionally diverse enzymes that populate the biological world and, increasingly, our databases is being filled in in great detail.
Functionally diverse enzyme superfamilies are collections of chemical novelty whose family trees illustrate how ancestral functions spawned new ones (Bartlett et al. Reference Gerlt and Babbitt2003; Gerlt & Babbitt, Reference Gerlt and Babbitt2009, Reference Gerlt and Babbitt2011). We have learned that evolution is economical: related enzymes that catalyze different reactions share conserved functional elements, including catalytic or metal-binding residues, cofactors, and key residues that modulate the chemistry (Bartlett et al. Reference Bartlett, Borkakoti and Thronton2003). And today's members of functionally diverse families exhibit the catalytic promiscuity, often as a result of their mechanisms, conformational diversity, etc., that presumably fueled their divergence in the past (Babtie et al. Reference Babtie, Tokuriki and Hollfelder2010; Baier & Tokuriki, Reference Baier and Tokuriki2014; Gatti-Lafranconi & Hollfelder, Reference Gatti-Lafranconi and Hollfelder2013; Tokuriki & Tawfik, Reference Tokuriki and Tawfik2009). Experiments lend credence to the proposal that promiscuity underlies much of enzyme innovation. Numerous studies have shown that an enzyme optimized for one reaction can adapt under laboratory mutation and screening/selection to take on the function of a different member of the superfamily, sometimes with efficiency that rivals nature's solutions (Aharoni et al. Reference Aharoni, Gaidukov, Khershonsky, Gould, Roodveldt and Tawfik2005; Gerlt & Babbitt, Reference Gerlt and Babbitt2009; Meier et al. Reference Meier, Rajendran, Malisi, Fox, Xu, Schlee, Barondeau, Höcker, Sterner and Raushel2013).
I am interested in how nature invents new enzymes, because I want to exploit that process to create yet more. I would like to expand the enzyme universe to encompass chemical reactions that are not yet known in the natural world. Since designing enzymes is still beyond our capabilities for anything but the simplest of reactions, it makes sense to look to the very effective evolutionary mechanisms nature has devised and exploited.
Can we use evolution to access non-natural functions?
Research has demonstrated that enzyme functions can sometimes be interconverted when the enzymes share a common ancestor; it is even easier when they still share much of the catalytic machinery and even residual activities left over from the ancestral form. The next step is to evolve functions that are not catalyzed by other family members or, even better, are altogether unknown in the biological world. At first glance, that might seem much more difficult. Directed evolution to engineer enzymes (or ‘molecular breeding’) is regular business in the world of industrial enzymes, where optimization of stability, selectivity, or even activity is reliably routine. But how do we ‘evolve’ novelty? How can we use what we know of nature's inventive mechanisms to step into unknown territory, where nature has not already gone?
Novel function requires novel context, a new ‘niche’ so to speak. Thus to find new reactions, we can explore outside of the limited range of chemicals an enzyme naturally encounters. When enzymes are challenged with substrates that they have not evolved to discriminate against, opportunities arise for serendipitous activities. We the breeders of enzymes must provide these opportunities for innovation.
We must also know what to look for in the parent(s) we choose to start with, just as if we were breeding rabbits or racehorses. And, to be successful, we must be realistic about what we expect to find in the progeny and patient about acquiring and optimizing those features over multiple generations. Because evolution means accumulating improvements brought about by a single or at most a few mutations per generation, the breeder has to start close to the new activity. How do we find an enzyme with the ability to catalyze a chosen reaction, even if at a very low level? Of course, this is where chemical intuition comes in (Kazlauskas, Reference Kazlauskas2005; Renata et al. Reference Renata, Wang and Arnold2015; Toscano et al. Reference Toscano, Woycechowsky and Hilvert2007).
Some enzymes are already teetering on the precipice of novel function–we have to know how to recognize them and push them along. Intuition based on mechanism or chemical and structural similarities can help us identify novel reactions for a given enzyme (or new enzymes for a given reaction), as has been shown in (Hammer et al. Reference Hammer, Marjanovic, Dominicus, Nestl and Hauer2015; Kazlauskas, Reference Kazlauskas2005; Seebeck & Hilvert, Reference Seebeck and Hilvert2003). In addition, we find that appropriate synthetic reagents can occasion new reaction opportunities, for example via their ability to generate novel reactive intermediates. Once a new enzyme activity appears, the enzyme can evolve and diversify into a whole new family. Since Hans Renata, Jane Wang, and I recently reviewed this topic more broadly (Renata et al. Reference Renata, Wang and Arnold2015), I will use only examples from this laboratory to illustrate how we think about evolving new enzymes starting with cytochrome P450s.
New enzymes from old: adding to the cytochrome P450's (already expansive) repertoire
Even a partial listing of cytochrome P450-catalyzed reactions illustrates how effectively nature has molded this versatile protein–iron–heme framework for new chemistry: P450s catalyze aryl–aryl coupling, ring contractions and expansions, S-, N-, and O-dealkylations, decarboxylation, oxidative cyclization, alcohol and aldehyde oxidation, desaturation, sulfoxidation, nitrogen oxidation, epoxidation, C–C bond scission, decarbonylation, and nitration (blue reactions in Fig. 1). A P450 generates multiple reactive species during its catalytic cycle, and these P450-derived oxidants can react with substrates in different ways. We recently outlined the intermediates used by P450 family members to catalyze some of the different reactions shown in Fig. 1 (McIntosh et al. Reference Mcintosh, Farwell and Arnold2014). A particular P450's specificity for one reaction over others is determined by the protein sequence, including active site details, that directs the course of reaction and molds substrate and product selectivity.

Fig. 1. Natural reactions catalyzed by cytochrome P450s (blue) and new, non-natural reactions catalyzed by enzymes derived from cytochrome P450 by protein engineering and evolution (red).
Nature fashioned all this diverse reactivity in one protein scaffold, emerging from one common ancestor. Can we take it even further? Following nature's lead, we looked into expanding the reactive intermediates this enzyme can generate in order to expand its reaction repertoire. The P450's heme cofactor has been a favorite of biomimetic chemists since synthetic porphyrins were first studied as P450 models decades ago. More than a model, however, metalloporphyrins also catalyze a broad range of reactions that do not have enzyme-catalyzed counterparts. A particularly interesting set of reactions proceeds through metal-carbenoid intermediates that had never been exploited for catalysis by heme proteins. The synthetic porphyrin systems relied on synthetic activated reagents such as ethyldiazoacetate (EDA) to generate the metal carbenoid, which could then react with olefins or insert the carbene into various N–H, C–H, or O–H bonds (Baumann et al. Reference Baumann, Mbuvi, Guodong and Woo2007; Chen et al. Reference Chen, Fields and Zhang2004; Wolf et al. Reference Wolf, Hamaker, Djukic, Kodadek and Woo1995). That the metal-carbenoid intermediate resembles compound I in the native P450 catalytic cycle (Fig. 2) spurred us to test whether the P450 and other heme proteins could catalyze cyclopropanation of styrene by EDA (Fig. 1, non-natural reaction 1). We found that a number of proteins, and even free iron–heme, are catalytically promiscuous and can indeed do this, albeit with low efficiency (Coelho et al. Reference Coelho, Brustad, Kannan and Arnold2013a, Reference Coelho, Wang, Ener, Baril, Kannan, Arnold and Brustadb; Heel et al. Reference Heel, Mcintosh, Dodani, Meyerowitz and Arnold2014).
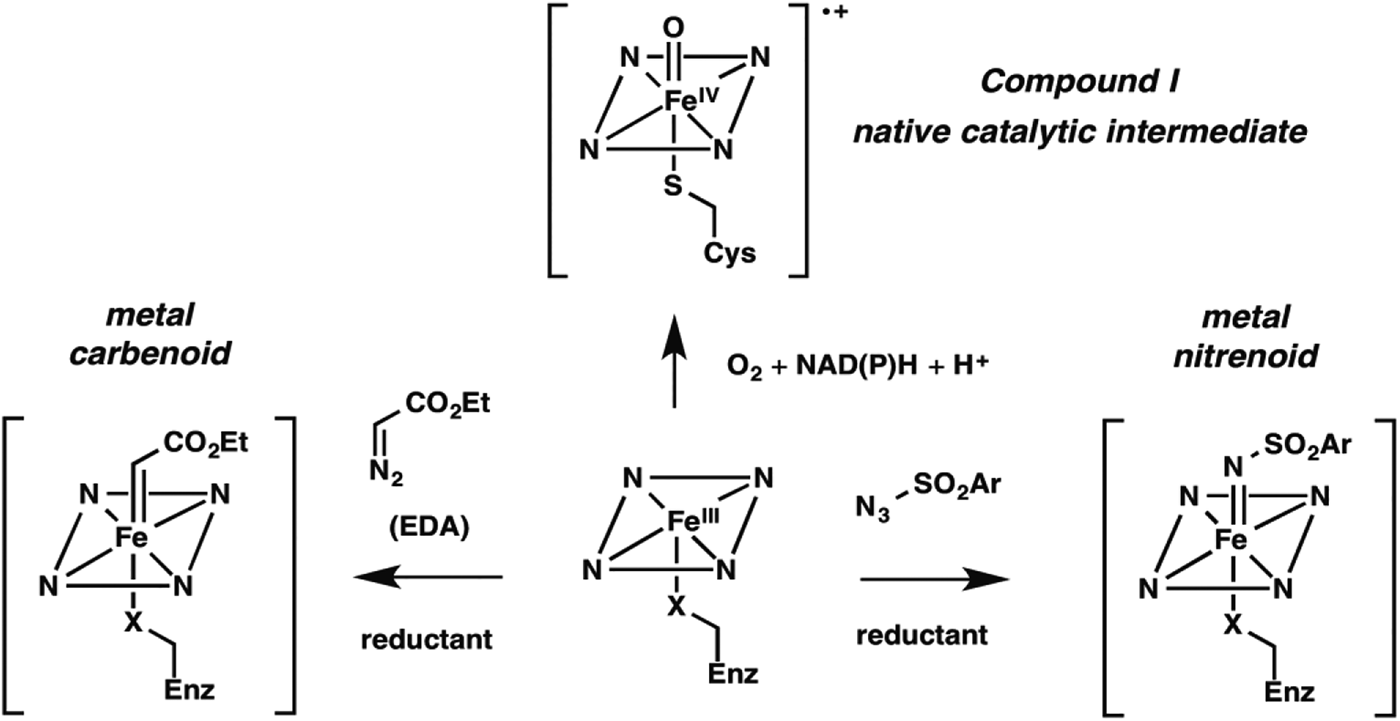
Fig. 2. New enzyme activities use reactive enzyme intermediates generated using synthetic diazo and azide reagents. Metalcarbenoid (middle) and metal-nitrenoid (RHS) intermediates resemble the natural P450 reactive Compound I (LHS) and can transfer the carbene or nitrene to acceptor substrates in the enzyme active site. Synthetic reagents that drive formation of these reactive intermediates can be thought of as providing a new ‘niche’ for evolution of new enzymes.
New enzymes can be expected to start out with low efficiencies – evolution can do the work of optimizing the enzyme for its new role. For example, mutations can reconfigure the active site to provide a stereoelectronic environment that favors productive EDA and olefin binding and increases effective substrate concentration for reaction, promotes formation of the carbenoid intermediate, and assists subsequent reactivity.
Noting that cytochrome P450BM3 catalyzed this non-natural cyclopropanation reaction with selectivity very different from that of free iron–heme (and therefore likely catalyzed the reaction in its malleable active site), we proceeded to explore the effects of mutations. While creating a version that could function inside of a cell, utilizing endogenous reductant NADPH, we discovered that the identity of the iron-ligating amino acid residue (which is always Cys in P450s) is very important: Ser- (Coelho et al. Reference Coelho, Wang, Ener, Baril, Kannan, Arnold and Brustad2013b) and His-ligated (Wang et al. Reference Wang, Peck, Renata and Arnold2014a) enzymes are easily generated and are much better cyclopropanation catalysts. Of course, these mutations obliterate the P450's native monooxygenase activity; they also shift the characteristic Soret peak at 450 nm. The resulting ‘P411’ (for Ser ligation, His ligation shifts the peak to a slightly longer wavelength) is no longer a cytochrome P450 and is in fact a whole new enzyme in function, even though its sequence is 99·8% identical to its monooxygenase ancestor.
Fasan's group recently reported that myoglobin, which naturally uses histidine as the axial ligand to the iron-heme, can also be engineered for cyclopropanation (Bordeaux et al. Reference Bordeaux, Tyagi and Fasan2015), at least in vitro. Both labs have now demonstrated that optimized cyclopropanating enzymes catalyze tens of thousands of turnovers for various olefins, with substrate range and product selectivity that are tunable by protein engineering (Bordeaux et al. Reference Bordeaux, Tyagi and Fasan2015; Coelho et al. Reference Coelho, Wang, Ener, Baril, Kannan, Arnold and Brustad2013b). The P450-derived enzyme functions very well in whole bacterial cells, where a His-ligated variant has been used for preparative-scale synthesis of a pharmaceutical intermediate (Wang et al. Reference Wang, Peck, Renata and Arnold2014a). We have also gone on to demonstrate that other carbene transfer reactions, including N–H insertion (Wang et al. Reference Wang, Renata, Peck, Farwell, Coelho and Arnold2014b) (Fig. 1 reaction 2), can be catalyzed by enzymes derived from a P450.
We reasoned that different synthetic reagents could be used to drive the formation of metal-nitrenoid intermediates in heme proteins (see Fig. 2), possibly enabling them to catalyze nitrene insertion reactions (Fig. 1, reactions 3–5). In choosing our first reaction targets we were inspired by early work of Breslow and Gellman, with iron-tetraphenyl porphyrin complexes (Breslow & Gellman, Reference Breslow and Gellman1983), and then Dawson and coworkers (Svastits et al. Reference Svastits, Dawson, Breslow and Gellman1985), who reported that rabbit liver P450 enzymes catalyze low levels (<5 total turnovers, TTN) of intramolecular nitrene transfer to make benzosultams when provided with iminoiodinane nitrene precursors. As far as we can tell, no one followed up on Dawson's observations until we (McIntosh et al. Reference Mcintosh, Coelho, Farwell, Wang, Lewis, Brown and Arnold2013) and Rudi Fasan (Singh et al. Reference Singh, Bordeaux and Fasan2014) showed that cytochrome P450BM3 catalyzes low levels of intramolecular C–H amination to yield the same benzosultams from the more atom-efficient azide-based nitrene precursors (Fig. 1, reaction 5). Again, we found mutations that strongly modulated C–H amination activity, leading to biocatalysts exhibiting several hundred turnovers in vitro and roughly double that in vivo. Mutation of the conserved axial cysteine to Ser was highly activating for this C–H amination, as it was for cyclopropanation. Mutation of the active site threonine (T268), which in native P450s helps to catalyze O–O bond scission via protonation, also greatly increased activity for both C–H amination and cyclopropanation. The active site can be re-molded to promote the new chemistry, and beneficial mutations can be accumulated in an evolutionary fashion.
In principle, an enzyme provides the opportunity for unprecedented control over reaction selectivity. This is true in practice as well. We showed, for example, that engineering the active site could override the preference for amination at C–H bonds with lower bond dissociation energies that often governs regioselectivity with small molecule catalysts. One P411BM3 variant catalyzed C–H amination of arylsulfonylazide substrates at the homo-benzylic position with high regio- and enantioselectivity, despite the fact that the bond dissociation energy for this C–H is more than 10 kcal mol−1 higher than that of the benzylic C–H (Hyster et al. Reference Hyster, Farwell, Buller, Mcintosh and Arnold2014) (Fig. 3). A second variant was engineered to show strong preference for C–H amination at the benzylic position, also with excellent enantioselectivity.
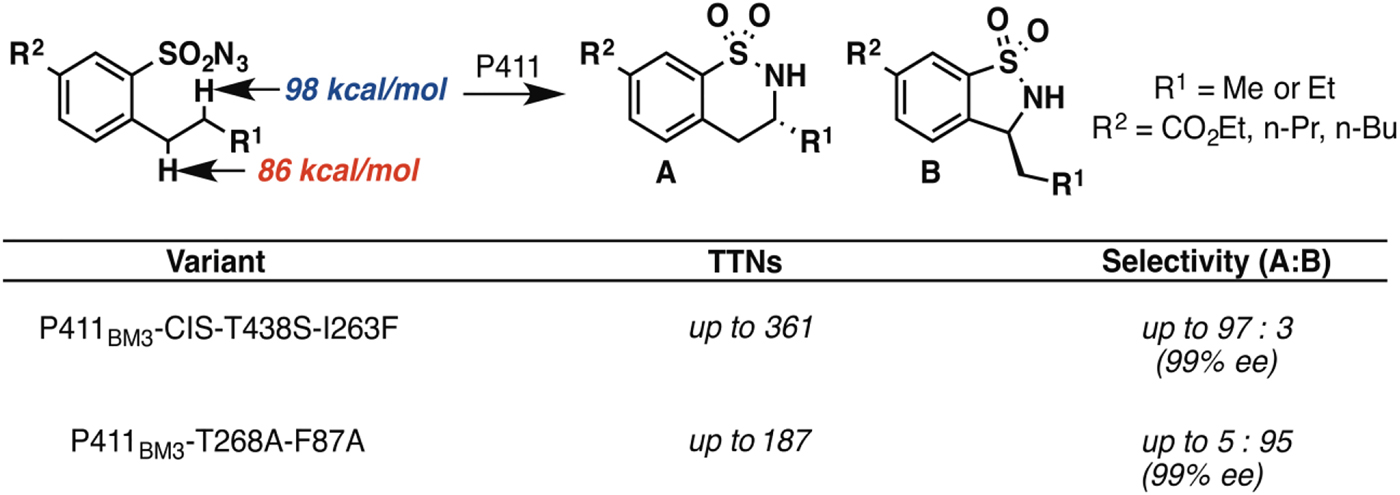
Fig. 3. Engineered P411 variants exhibit regio-divergent selectivities in intramolecular C–H amination (Hyster et al. Reference Hyster, Farwell, Buller, Mcintosh and Arnold2014). One enzyme targets primarily the homo-benzylic C–H position (BDE = 98 kcal mol−1) with high enantioselectivity, whereas another targets the benzylic C–H (BDE = 86 kcal mol−1), also with high enantioselectivity.
P411 variants can also catalyze intermolecular nitrene transfer, as demonstrated recently for sulfimidation (Farwell et al. Reference Farwell, Mcintosh, Hyster, Wang and Arnold2014) and aziridination (Farwell et al. Reference Farwell, Zhang, Mcintosh, Hyster and Arnold2015). In these reactions, sulfides and alkenes serve as nucleophilic acceptors for the nitrene intermediate (Fig. 1, reactions 3 and 4). For example, a P411 enzyme optimized by sequential rounds of mutagenesis and screening exhibited very high enantioselectivity (99% ee) for aziridination of various styrene derivatives (Fig. 4).

Fig. 4. Substrate scope of a P411 variant of cytochrome P450BM3 engineered for aziridination, in whole Escherichia coli cells (Farwell et al. Reference Farwell, Zhang, Mcintosh, Hyster and Arnold2015). (% ee determined as (S – R)/(S + R), all products are S-enriched).
A significant undesired side reaction in these reactions is enzyme-catalyzed azide reduction, particularly with less reactive nitrene acceptors. Engineering the enzyme can significantly reduce side product formation and increase the yield of the desired product, either by increasing the effective concentration of nitrene acceptor and effectively orienting it in the enzyme active site for productive reaction or by reducing the relative rate of competing azide reduction. These studies all demonstrate the critical role that mutations and evolution play in modulating the new enzyme activity and selectivity. Nature could do the same, given a good reason.
Outlook
We have directed the evolution of a cytochrome P450 to display a broad set of new, non-natural activities (Fig. 1), all enabled by the enzyme's ability to generate new reactive intermediates in the presence of reagents not found in the natural environment. The ease with which we could enhance very low levels of promiscuous activities – by accumulating just a few mutations – demonstrates how rapidly this biological catalyst can adapt to new challenges. We simply coaxed nature's evolvable protein framework for catalysis to show off more of its impressive versatility. None of the new reactions I have described here, however, has been fully optimized. It will be fascinating to see what further improvements are possible and the mutational and functional pathways these new enzymes take to achieve those.
The new enzymes catalyze enantioselective carbene and nitrene transfer reactions that are synthetically useful, and they compare favorably to small-molecule catalysts. Furthermore, they can be tuned to exhibit selectivities that may be difficult for the small molecule catalyst to achieve. The vast metalloporphyrin catalysis literature (e.g. Lu & Zhang, Reference Lu and Zhang2011) provides a rich source of inspiration and ideas for future work.
Chemists have invented myriad clever catalytic systems, many of which have no biological counterparts. But they should! Our collective challenge will be to create enzymes to catalyze some of these important reactions, and efforts underway in various laboratories already sketch just a few of the possibilities (e.g. as reviewed by Renata et al. Reference Renata, Wang and Arnold2015). I am sure that many enzyme families can be expanded to encompass new chemistry, and that directed evolution can optimize their functions and generate whole new catalyst families. With new enzymes, we can expect to carry out more reactions under mild, aqueous conditions. We will be able to build highly selective coupled enzyme ‘cascades’ for synthesis. Furthermore, with new enzymes that assemble and function inside a cell, we will be able to expand the scope of metabolic engineering and biosynthesis to create a multitude of new products or find alternative routes to natural products. By coupling the new chemistry to natural and engineered metabolic pathways, we will be able to assemble new routes to utilizing renewable (e.g. plant, algal, methane, CO2) resources to make fuels and chemicals. We have just begun to import human-invented chemistry into biological systems; many opportunities remain to learn from evolution.
Acknowledgments
Our research is supported by the Jacobs Institute for Molecular Engineering for Medicine at Caltech, the Gordon and Betty Moore Foundation through grant GBMF2809 to the Caltech Programmable Molecular Technology Initiative, and the National Science Foundation, Office of Chemical, Bioengineering, Environmental and Transport Systems SusChEM Initiative (grant CBET-1403077). I would like to thank Hans Renata, Chris Prier, and Todd Hyster for preparing the figures and Eric Brustad, Pedro Coelho, Jane Wang, Jared Lewis, John McIntosh, Andrew Buller, Christopher Prier, Todd Hyster, Sheel Dodani, Chris Farwell, Hans Renata, and Ruije Zhang for valuable discussions and scientific contributions.





 Open access
Open access