


Introduction
Heart failure (HF) prevalence in the USA is about 5 million individuals(Reference Mozaffarian and Benjamin1), while it affects more than 23 million individuals worldwide(Reference Liu and Eisen2). HF is the main factor responsible for hospitalisation and disability in the elderly and is the cause of one in nine deaths in the USA(Reference Bui, Horwich and Fonarow3). Consequently, HF imposes a great problem to the healthcare system, amounting to more than $39 billion annually in the USA(Reference Bui, Horwich and Fonarow3). In Europe, the prevalence and incidence of HF, and related costs, are quite similar(Reference Maggioni4,Reference Meyer, Brouwers and Voors5) . Even though there have been relevant improvements in HF prevention and treatment, quality of life (QoL) is often impaired and mortality rates are greater than 10 % per year, reaching 20–50 % in more advanced disease(Reference Kannel6).
HF is attributed mainly to four underlying conditions: hypertension, coronary artery disease, cardiomyopathy and valvular heart disease(Reference Mozaffarian and Benjamin1,Reference Liu and Eisen2) ; however, genetic causes, particularly in dilated cardiomyopathy, also play an immense role(Reference Skrzynia, Berg and Willis7). Still, HF could be partly prevented by improving lifestyle, while an improvement in lifestyle is also suggested when HF has already been diagnosed(Reference Yancy, Jessup and Bozkurt8,Reference McMurray, Adamopoulos and Anker9) . Therapeutic lifestyle changes include adherence to a Mediterranean diet, a reduced-Na diet or Dietary Approaches to Stop Hypertension (DASH) diet as well as exercise training(Reference Rifai and Silver10,Reference Rifai, Pisano and Hayden11) , which are able to reduce the risk of developing HF and improve endothelial function, exercise capacity and QoL in HF patients(Reference Tektonidis, Åkesson and Gigante12). In particular, the DASH diet can protect against HF risk by as much as 29 %(Reference Salehi-Abargouei, Maghsoudi and Shirani13). Moreover, weight loss, moderation of alcohol consumption and increased aerobic exercise are recommended measures(Reference Wexler, Pleister and Raman14).
In recent years, epidemiological studies and clinical trials have investigated the possibility that some dietary supplements and phytochemicals (overall referred to as natural products or nutraceuticals) can contribute to the improvement of HF-related symptoms (Fig. 1).
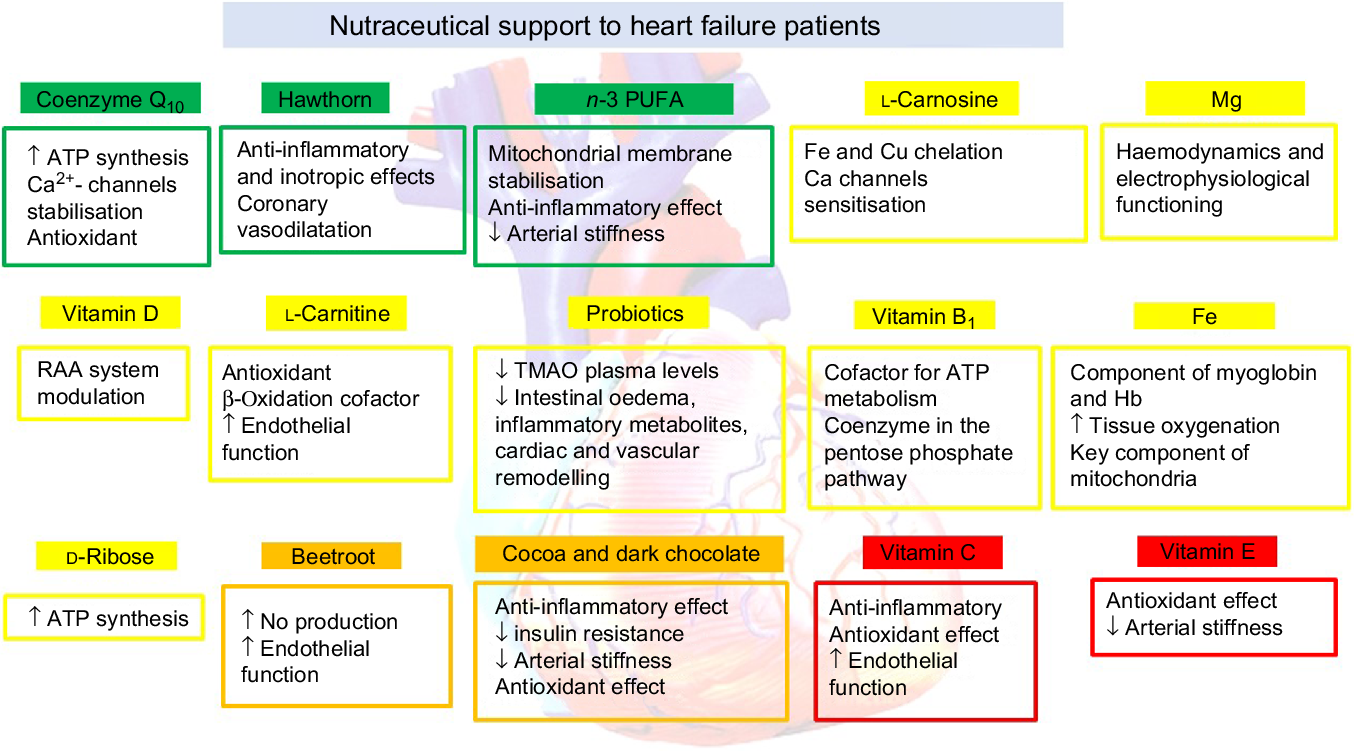
Fig. 1. Nutraceutical support to heart failure patients. RAA, renin–angiotensin–aldosterone; TMAO, trimethylamine N-oxide.
The aim of this position paper is to provide, for the first time, physicians and nutrition experts with a practical tool presenting the scientific evidence on efficacy and safety of nutraceuticals, eventually supporting their use as further HF symptoms improvers, as an add-on to optimal pharmacological treatment.
Methods
A systematic search strategy was developed to identify randomised clinical trials (RCT) and their meta-analyses in Medline/PubMed (January 1970 to June 2019). The terms ‘nutraceuticals’, ‘dietary supplements’, ‘herbal drug’ and ‘heart failure’ were incorporated into an electronic search strategy. The experts discussed and agreed on the recommended levels. For each selected nutraceutical, a short description of the mechanism of action is reported, followed by the clinically observed effects and the most relevant tolerability notes.
The level of evidence of nutraceuticals tested on HF patients has been weighed up and graded according to predefined scales, as outlined in Table 1. Due to the fact of the limited data the experts did not decide to evaluate each selected nutraceutical with the class of the evidence. The experts of the writing and reviewing panels completed declaration of interest forms where real or potential sources of conflicts of interest might be perceived.
Table 1. Classification of the level of evidence

Nutraceuticals
Coenzyme Q10
Coenzyme Q10 (CoQ10) is an organic molecule, which is composed of a lipophilic core (benzoquinone) and an isoprenoid side chain; it was identified in 1940 and isolated for the first time from beef heart mitochondria by Frederick Crane of Wisconsin (USA), in 1957(Reference Cicero and Colletti15). CoQ10 (10 refers to the number of isoprene repeats) is synthesised endogenously, starting from tyrosine and acetylcoenzyme A; it is universally present in the cells of the body, particularly concentrated in the mitochondria, in both reduced form (ubiquinol) and oxidised form (ubiquinone). The level of CoQ10 is highest in organs with high rates of metabolism such as the heart, kidney and liver (114, 66·5 and 54·9 µg/g tissue, respectively), where it functions as an energy transfer molecule(Reference Saini16). The overall body content of CoQ10 is only about 500–1500 mg and decreases with age. It is naturally contained in oily fish (such as salmon and tuna), organ meats (such as liver and heart) and whole grains, but it can be consumed also as a dietary supplement(Reference Menke, Niklowitz and de Sousa17).
By functioning as a reducing equivalent transfer agent between cytochromes in the mitochondrial electron transport chain, CoQ10 play a crucial role in oxidative phosphorylation (i.e. ATP biosynthesis). CoQ10 is also the only lipid-soluble antioxidant that slows lipid peroxidation in the circulation (by maintaining the reduced state of α-tocopherol and ascorbic acid)(Reference Littarru and Tiano18). Other functions of CoQ10 include stabilisation of Ca-dependent channels, cell signalling and cell growth through local regulation of cytosolic redox intermediates such as reduced nicotinamide adenine dinucleotide phosphate (NADPH)(Reference Hernández-Camacho, Bernier and López-Lluch19). Since CoQ10 is an essential cofactor for ATP biosynthesis, it is not surprising that the highest concentration, compared with other tissues, is focused in myocardial mitochondria(Reference Saini16). Thus, it has been proposed that CoQ10 deficiency could play an aetiopathogenic role in the development and progression of HF.
CoQ10 bioavailability is extremely variable depending on dosage, particle size, formulation, the release method and the mode of administration (for example with or without water, before or after meals)(Reference Weis, Mortensen and Rassing20). In particular, ubiquinol (the reduced form) seems to be the most available form of CoQ10, especially if conveyed through micelles, liposomes, nano-emulsions, polymeric or solid lipid nanoparticles, solid and aqueous dispersions, or nanostructured lipid carriers supplemented in the fed state(Reference Miles, Horn and Miles21,Reference Kumar, Rao and Kumar22) . However, because of its hydrophobicity and large molecular weight, absorption of dietary CoQ10 is slow and limited. The time taken to reach the maximum concentration (Tmax) is about 6 h, with an elimination half-life of about 33 h. The reference intervals for plasma CoQ10 range from 0·40 to 1·91 μmol/l in healthy adults(Reference Bhagavan and Chopra23,Reference Bentinger, Dallner and Choknacki24) . Finally, in a recent paper by López-Lluch et al. (Reference López-Lluch, del Pozo Cruz and Sánchez-Cuesta25), the level of bioavailability of seven different formulations of CoQ10 was measured over 48 h after ingestion of 100 mg. The two best absorbable formulations were soft-gel capsules containing ubiquinone or ubiquinol. The increase in plasma levels (at 4 and 8 h) was higher after intake of 100 mg ubiquinone when compared with all other formulations(Reference López-Lluch, del Pozo Cruz and Sánchez-Cuesta25).
Clinical evidence
The lowest levels of myocardial CoQ10 have been observed in patients of New York Heart Association (NYHA) class IV and the highest levels in patients of NYHA class I(Reference Weber, Bysted and Hłlmer26,Reference Onur, Niklowitz and Jacobs27) . The Q-SYMBIO trial (Q10-SYMptoms, BIomarker status [Brain-Natriuretic Peptide], and long-term Outcome [hospitalizations/mortality])(Reference Mortensen, Rosenfeldt and Kumar28) was a multicentre, randomised placebo-controlled trial, which assessed the impact of the daily intake of CoQ10 on total mortality and not just on the surrogate endpoints. A group of 420 patients, with moderate or severe HF for a period of 2 years, were treated with 300 mg of CoQ10 (ubiquinone) (n 202) or placebo (n 218). At the end of treatments they benefited from a significant reduction in major adverse cardiac events (15 % of the patients in the CoQ10 group v. 26 % in the placebo group; hazard ratio (HR) 0·50; 95 % CI 0·32, 0·80; P = 0·003), cardiovascular mortality (9 v. 16 %; P = 0·026), all-cause mortality (10 v. 18 %; P = 0·018) and incidence of hospital stays for HF (P = 0·033)(Reference Mortensen, Rosenfeldt and Kumar28). A recent meta-analysis of fourteen RCT including 2149 patients has shown that administration of CoQ10 reduces mortality risk (pooled risk ratio = 0·69; 95 % CI 0·50, 0·95; P = 0·02; I 2 = 0 %) and helped in improving exercise capacity (standardised mean difference (SMD) 0·62; 95 % CI 0·02, 0·30; P = 0·04; I 2 = 54 %) compared with placebo. Moreover, left ventricular (LV) ejection fraction (LVEF) also improved in CoQ10-treated subjects compared with controls (SMD 0·62; 95 % CI 0·02, 1·12; P = 0·04; I 2 = 75 %)(Reference Lei and Liu29), partially in contrast with what was reported in a previous meta-analysis including fewer studies(Reference Fotino, Thompson-Paul and Bazzano30).
The heterogeneity of results obtained on EF may therefore be partly explained by many factors such as the diversity of CoQ10 supplemented through different pharmaceutical forms and dosages (CoQ10 plasma concentrations are very variable and were reported in few RCT)(Reference Belardinelli, Mucaj and Lacalaprice31-Reference Keogh, Fenton and Leslie33), diversity of HF grade of patients enrolled (NYHA I–IV), duration of treatment and co-treatment with conventional therapies. In particular, it has been suggested that blood CoQ10 concentrations should be >2 μg/ml to improve EF in subjects with more severe HF(Reference Langsjoen34). Moreover, it seems that the CoQ10 effect on LVEF could be more relevant in patients untreated with statins and/or angiotensin-converting enzyme inhibitors (+6·7 %) compared with the subgroup of patients treated with these drugs (+1·2 %)(Reference Sander, Coleman and Patel35). Finally, CoQ10 lowers the need for inotropic drugs and reduces the appearance of ventricular arrhythmias after surgery in the prevention of complications in patients undergoing cardiac surgery(Reference de Frutos, Gea and Hernandez-Estefania36) (Table 2).
Table 2. Nutraceuticals with clinical effects on heart failure (HF): level of evidence, tested dosages, effects on symptoms, effects on laboratory or instrumental parameters and effects on hard outcomes
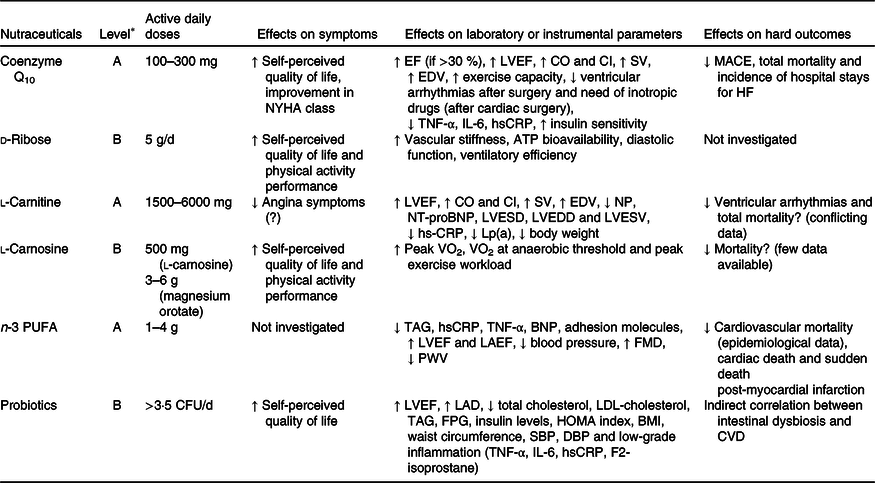
BNP, brain natriuretic peptide; CFU, colony-forming units; CI, cardiac input; CO, cardiac output; DBP, diastolic blood pressure; EDV, end-diastolic volume; EF, ejection fraction; FMD, flow-mediated dilation; FPG, fasting plasma glucose; HOMA, homeostatic model assessment; hsCRP, high-sensitivity C-reactive protein; LAD, left atrial diameter; LAEF, left atrial emptying function; Lp(a), lipoprotein (a); LVEDD, left ventricular end diastolic diameter; LVEF, left ventricular ejection fraction; LVESD, left ventricular end systolic diameter; LVESV, left ventricular end systolic volume; MACE, major adverse cardiac events; NP, natriuretic peptide; NT-proBNP, N-terminal pro–B-type natriuretic peptide; NYHA, New York Heart Association; PWV, pulse wave velocity; SBP, systolic blood pressure; SV, stroke volume.
* See Table 1 for classification of the level of evidence.
Based on the available studies, CoQ10 has a high safety profile and at doses ranging from 60 to 600 mg/d does not cause clinically relevant adverse events(Reference Mazidi, Kengne and Banach37,Reference Banach, Serban and Sahebkar38) .
Expert opinion
Available meta-analyses support that supplementation with CoQ10 (especially with doses ≥200 mg/d) can be of benefit in patients with chronic HF, in particular in early stage of HF, and might effect a reduction of major adverse cardiac events and total mortality.
Hawthorn flavonoid fraction
Hawthorn extract from Crataegus monogyna and oxyacantha is a flavonoid-rich herbal remedy with known anti-inflammatory, antioxidant, inotropic and coronary vasodilator effects(Reference Ford, Adams and Graves39). The most studied dry ethanol (45 %) extract of hawtorn leaves with flowers is WS 1442 (drug:extract ratio 4–6·6:1) which contains 17·3–20·1 % oligomeric procyanidins and several flavonoids, including hyperoside, vitexin-rhamnoside, rutin and vitexin as well as triterpenoids and phenol carboxylic acids(Reference Koch and Malek40). In vitro experiments with human myocardial tissue demonstrated a positive inotropic effect of hawthorn with a concentration-dependent increase of myocardial contractility accompanied by a transient rise in intracellular Ca(Reference Wang, Xiong and Feng41). The effect is probably mediated by cyclic AMP-independent inhibition of Na-K-ATPase and is accompanied by an improved energy turnover of myocytes(Reference Schwinger, Pietsch and Frank42,Reference Münch, Brixius and Frank43) ; however, in contrast to cardiac glycosides, hawthorn prolongs the potential action and the refractory period, thus possessing pronounced anti-arrhythmic properties which have been especially evaluated in animal models(Reference Krzeminski and Chatterjee44). Hawthorn extract WS 1442 has also been shown to raise endothelial Ca levels by inhibition of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) and activation of the inositol trisphosphate (IP3) pathway, protecting against thrombin-induced vascular barrier dysfunction and subsequent oedema formation(Reference Willer, Malli and Bondarenko45).
Clinical evidence
The Survival and Prognosis: Investigation of Crataegus Extract WS 1442 in congestive heart failure (SPICE) trial has investigated the effect of 900 mg/d of WS 1442 on mortality and hospitalisation rate in NYHA class II–III HF with reduced EF (HFrEF) patients, in a 24-month, randomised, placebo-controlled trial. A total of 2681 patients (WS 1442: n 1338; placebo: n 1343) were randomised and the primary endpoint was time till the first cardiac event. The authors showed that there was an insignificant trend towards cardiac mortality reduction with WS 1442 (11 % at month 24; HR 0·89; 95 % CI 0·73, 1·09; P = 0·269). However, in the subgroup with LVEF ≥25 %, WS 1442 reduced sudden cardiac death by 41 % (HR 0·59; 95 % CI 0·37, 0·94) at month 24; P = 0·025). Current data suggest that WS 1442 can potentially reduce the incidence of sudden cardiac death, at least in patients with less compromised LV function(Reference Holubarsch, Colucci and Meinertz46,Reference Holubarsch, Colucci and Meinertz47) .
Some meta-analyses of RCT investigated the efficacy of hawthorn extract on different HF parameters. One investigated the effects of hawthorn supplementation (900 and 1800 mg/d groups) on maximum workload; the results showed a statistically significant increase over placebo by a weighted mean difference (WMD) of 5·4 (95 % CI 0·7, 10·0) W (P = 0·024). Moreover, at the end of the treatment phase, the treatment group differences compared with placebo for score change (four ‘typical’ HF symptoms, i.e. general capability, lassitude, early fatigability and effort dyspnoea) v. baseline were both significant for WS 1442 doses 900 mg/d (P = 0·04) and 1800 mg/d (P = 0·004)(Reference Tauchert48). A Cochrane meta-analysis of RCT concluded that treatment with hawthorn compared with placebo was more beneficial for the physiological outcome of maximal workload (WMD = 5·35 (95 % CI 0·71, 10·00) W; P<0·02; n 380), exercise tolerance (WMD = 122·76 (95 % CI 32·74, 212·78) W × min; n 98) and the pressure–heart rate product, an index of cardiac oxygen consumption (WMD = –19·22 (95 % CI –30·46, –7·98) mmHg/min; n 264). Furthermore, shortness of breath and fatigue were also significantly improved (WMD = –5·47, 95 % CI –8·68, –2·26; n 239)(Reference Pittler, Guo and Ernst49). These results were confirmed by a more recent meta-analysis of RCT that evaluated the data of >600 patients treated with quantified Crataegus extract or placebo; the subjects of the active group showed improvements in physiological outcome parameters, in particular in maximal workload, LVEF and pressure–heart rate product increase (PHRPI) at 50 W ergometric exercise. Moreover, the results on LVEF were independent from baseline data, while maximal workload and PHRPI were demonstrated to be related to baseline severity. Typical symptoms of HF patients, like reduced exercise tolerance, exertional dyspnoea, weakness, fatigue and palpitations, were improved more with active treatment and in subjects with more severe symptoms at baseline(Reference Eggeling, Regitz-Zagrosek and Zimmermann50) (Table 3).
Table 3. Botanicals with clinical effects on heart failure (HF): level of evidence, tested dosages, effects on symptoms, effects on laboratory or instrumental parameters and effects on hard outcomes
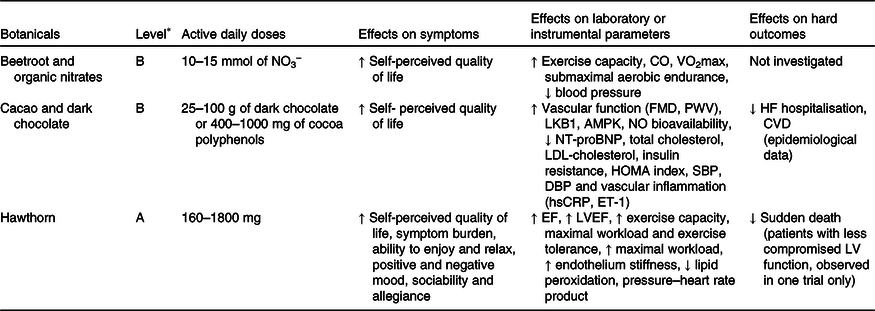
AMPK, AMP-activated protein kinase; CO, cardiac output; hsCRP, high-sensitivity C-reactive protein; DBP, diastolic blood pressure; EF, ejection fraction; ET-1, endothelin 1; FMD, flow-mediated dilation; HOMA, homeostatic model assessment; LKB1, liver kinase B1; LV, left ventricular; LVEF, left ventricular ejection fraction; NO3 −, inorganic nitrate; NT-proBNP, N-terminal pro–B-type natriuretic peptide; PWV, pulse wave velocity; SBP, systolic blood pressure.
* See Table 1 for classification of the level of evidence.
The recommended daily dose of hawthorn extract is 320–900 mg (and active daily doses 160–1800 mg) to be taken in two or three doses per d. Adverse events reported were mild, transient and infrequent and in general comparable with placebo, including mild rash, headache, sweating, dizziness and gastrointestinal symptoms(Reference Daniele, Mazzanti and Pittler51).
Expert opinion
According to clinical evidence, Crataegus extracts have proven benefits regarding functional capacity, symptom control and health-related QoL in both HFrEF and HF with preserved EF (HFpEF). However, further studies are required, as a large proportion of the positive data has been obtained in patients not pharmacologically treated with the current standards of HF management.
n-3 PUFA
Accumulating evidence suggests that supplementation with n-3 PUFA could exert some positive effects in HF patients, especially during the early stages of the disease. Among the possible mechanisms of action that might be responsible for this is the possible role of the fatty acids EPA and DHA that have a direct action on the mitochondrial membrane, modifying its structure and function(Reference Khairallah, Sparagna and Khanna52-Reference O’Shea, Khairallah and Sparagna54). In particular, dietary supplementation with DHA at a clinically relevant dose increases DHA incorporation into phospholipids of the mitochondrial membrane and decreases the susceptibility of isolated cardiac mitochondria to undergo mitochondrial permeability transition induced by Ca2+ and stress(Reference Galvao, Khairallah and Dabkowski55). DHA could in fact decrease viscosity of the membrane and a greater ease of movement of membrane proteins(Reference Stanley, Khairallah and Dabkowski56,Reference Chrysohoou, Metallinos and Georgiopoulos57) .
Clinical evidence
In the meta-analysis of seven prospective epidemiological studies involving 176 441 subjects with 5480 incident cases of HF, the pooled relative risk for HF comparing the highest with the lowest category of fish intake was 0·85 (95 % CI 0·73, 0·99; P = 0·04); the corresponding value for marine n-3 PUFA was 0·86 (95 % CI 0·74, 1·00; P = 0·05)(Reference Djoussé, Akinkuolie and Wu58). In the large GISSI-HF trial the investigators enrolled patients with chronic HF of NYHA class II–IV, irrespective of cause and LVEF, and randomly assigned them to 1 g n-3 PUFA daily (n 3494) or placebo (n 3481). Of the patients, 27 % (955) died from any cause in the n-3 PUFA group and 1014 (29 %) in the placebo group (HR 0·91; 95 % CI 0·833, 0·998; P = 0·041). It was also reported that 57 % of patients (1981) in the n-3 PUFA group and 2053 (59 %) in the placebo group died or were admitted to hospital for cardiovascular reasons (HR 0·92; 99 % CI 0·849, 0·999; P = 0·009). In absolute terms, fifty-six patients needed to be treated for a median duration of 3·9 years to avoid one death or forty-four to avoid one event like death or admission to hospital for cardiovascular reasons(Reference Tavazzi, Maggioni and Marchioli59). In the same trial, baseline LVEF increased with n-3 PUFA by 8·1 % at 1 year, 11·1 % at 2 years, and 11·5 % at 3 years v. 6·3 % at 1 year, 8·2 % at 2 years, and 9·9 % at 3 years in the placebo group (P = 0·005)(Reference Ghio, Scelsi and Latini60). A meta-analysis of RCT also highlighted a significant reduction in cardiac death in the active group compared with control, in particular in subgroup analysis with EPA+DHA dosages >1 g/d (12·9–29·1 % lower risks; P<0·05)(Reference Maki, Palacios and Bell61). Based on these data it seems that the effect of n-3 PUFA on HF seems to be dose- and time-related(Reference Moertl, Hammer and Steiner62).
A further trial enrolled >200 patients with ischaemic HF or dilated cardiomyopathy, NYHA class I–III on optimal medical treatment, who were divided into two groups: the first one received supplementation with 1000 mg of n-3 PUFA for 14 weeks while the second group took a placebo. At the end of the 14 weeks, the results showed a reduction in end-diastolic and end-systolic LV dimensions by 2·5 % (P = 0·047) and 3·7 % (P = 0·01), an improvement of LVEF by 3·6 % (P = 0·021) and a reduction of brain natriuretic peptide (BNP) levels by 34·6 % (P = 0·001) compared with placebo(Reference Dabkowski, O’Connell and Xu63). Then, more recently, in a double-blind, placebo-controlled, cross-over trial, treatment with 2 g/d of n-3 PUFA, compared with placebo, for 8 weeks in thirty-one patients with ischaemic HF was shown to improve LVEF (by 4·7 v. 1·7 %), E:E’ ratio (early ventricular filling (E) to early annular mitral (E’) velocities; decreased by –9·47 v. –2·1 %), ST2 levels (decreased by –4·5 v. –2·4 %), flow-mediated dilation (increased by 44 v. 11 %) and high-sensitivity C-reactive protein (hsCRP) levels (decreased by –6·1 v. 4·3 %) (P<0·05 for all)(Reference Oikonomou, Vogiatzi and Karlis64). These results confirm those from previous investigations suggesting beneficial effects of EPA/DHA on haemodynamics, LV indices and inflammation(Reference Mehra, Lavie and Ventura65,Reference Pepe and McLennan66) . Of course, the prognostic significance of these changes has yet to be clarified (Table 2).
Based on the available evidence, the 2016 guidelines of the European Society of Cardiology suggest that PUFA supplementation may be considered in symptomatic HF patients to reduce the risk of cardiovascular hospitalisation and cardiovascular death(Reference Ponikowski, Voors and Anker67).
n-3 PUFA are well tolerated beyond some mild gastrointestinal adverse events(Reference Cicero, Reggi and Parini68). However, even in the recently published REDUCE-IT trial(Reference Omar, Vande Hei and Battisha69) that demonstrated a statistically significant absolute risk reduction of 4·8 % in its primary endpoint (cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, coronary revascularisation, or unstable angina), the use of high-dose (2 × 2 g) highly purified EPA was associated with increased incidence of atrial fibrillation.
Expert opinion
The available evidence supports the supplementation of EPA and DHA to improve HF prognosis, especially in patients after myocardial infarction.
Levo-carnitine (l-carnitine)
l-Carnitine, a chemical analogue of choline, is a hydrophilic quaternary amine and is involved in several physiological activities such as lipid metabolism and mitochondrial defence. In particular, it plays an important role in lipid metabolism by acting as an obligatory cofactor for the oxidation of fatty acids and facilitating the transport of long-chain fatty acids from the cytosol to the mitochondrial matrix for β-oxidation(Reference El-Hattab and Scaglia70). In agreement with the ‘energy starvation’ hypothesis, suggesting that insufficient ATP supply (in addition to increased oxidation, inflammation and fibrosis) underlies the contractile dysfunction presenting in HF(Reference Katz71), it seems that l-carnitine might improve energy metabolism in cardiomyocytes and contribute to the improvement of clinical symptoms and cardiac function. Furthermore, l-carnitine exerts cardioprotective effects through the reduction of oxidative stress(Reference Flanagan, Simmons and Vehige72) and cardiac fibrosis(Reference Blanca, Ruiz-Armenta and Zambrano73,Reference Omori, Ohtani and Sakata74) .
Clinical evidence
A meta-analysis of seventeen RCT enrolling 1625 HF subjects showed a considerable improvement in overall cardiac function (evaluated in term of decreased NYHA class) (OR 3·47 (95 % CI 2·49, 4·82); P<0·01), LVEF (WMD = 4·1 (95 % CI 2·34, 5·93) %; P = 0·01), stroke volume (WMD = 8·20 (95 % CI 6·41, 10·01) ml; P = 0·01), cardiac output (WMD = 0·9 (95 % CI 0·76, 1·01) litres/min; P<0·01) and ratio of the early (E) to late (A) ventricular filling velocities (E/A) (WMD = 0·2 (95 % CI 0·11, 0·35); P<0·01). In addition, treatment with l-carnitine also resulted in significant decreases in serum levels of BNP (WMD = –124·6 (95 % CI −220·49, −28·71) pg/ml; P = 0·01), serum levels of N-terminal pro–B-type natriuretic peptide (NT-proBNP) (WMD = –510·4 (95 % CI −785·42, −235·30) pg/ml; P<0·01), LV end-systolic diameter (WMD = –4·1 (95 % CI −6·57, −1·55) mm; P<0·01), LV end-diastolic diameter (WMD = –4·8 (95 % CI −7·08, −2·49) mm; P<0·01) and LV end-systolic volume (WMD = –20·2 (95 % CI –35·6, –4·7) ml; P<0·01). No significant differences were reported in all-cause mortality, 6-min walk, and adverse events between l-carnitine and control groups(Reference Song, Qu and Yang75). These results were obtained with l-carnitine doses ranging from 1·5 to 6 g/d and follow-up length ranging from 7 d to 3 years. In a further meta-analysis of ten RCT enrolling 925 patients, l-carnitine consumption was associated with significant reductions in serum CRP (–0·60 mg/l (5·71 nmol/l); 95 % CI –0·87, –0·32 mg/l) and TNF-α concentrations (–0·36 (95 % CI –0·56, –0·15) pg/ml)(Reference Mazidi, Rezaie and Banach76). Then, l-carnitine supplementation could have a mild but significant impact on body weight(Reference Pooyandjoo, Nouhi and Shab-Bidar77) and plasma level of lipoprotein (a)(Reference Serban, Sahebkar and Mikhailidis78). The prognostic importance and clinical relevance of these data still need to be clarified (Table 2).
l-Carnitine is in general well tolerated; dry mouth, rash and mild gastrointestinal problems could infrequently occur.
Expert opinion
l-Carnitine treatment might be effective for congestive HF patients as an adjuvant to conventional therapy, improving clinical symptoms and cardiac function, decreasing serum levels of BNP and NT-proBNP. Further research is required to more accurately assess the clinical relevance of l-carnitine administration for supporting HF care.
Thiamine (vitamin B1)
Thiamine (vitamin B1) is an essential water-soluble vitamin required for cellular energy production. Thiamine pyrophosphate is the key coenzyme in the pentose phosphate pathway for transketolation of glucose-6-phosphate to ribose-5-phosphate(Reference Kattoor, Goel and Mehta79). Thiamine pyrophosphate is needed for the functioning of the pyruvate dehydrogenase complex (converting pyruvate to acetyl CoA) and α-ketoglutarate dehydrogenase (converting α-ketoglutarate to succinate) in the Krebs cycle, thus being essential for ATP metabolism(Reference Lonsdale80). In general, the recommended daily allowance of thiamine for adults is about 1·2 mg for men and 1 mg for women(Reference Anderson, Vickery and Nicol81). Deficiency of thiamine is far more common in underdeveloped and developing countries (in particular in individuals with chronic alcohol use, patients on total parenteral nutrition or who have undergone weight loss surgery) due to high incidence of poor nutritional status(Reference Kerns, Cherinne Arundel and Chawla82). Direct impairment of myocardial energy production has been proposed as a possible basis for the development of the HF state seen in beriberi, the disease related to chronic dietary deficiency of thiamine(Reference Oliveira, Guatimosim and Castro83).
Clinical evidence
Multiple studies have shown that thiamine deficiency is more prevalent in HF patients than in the general population. In a meta-analysis of nine observational studies, the incidence of thiamine deficiency in patients with HF has been reported to be 2·5 times higher compared with that of control subjects without HF (OR 2·53, 95 % CI 1·65, 3·87). The incidence of thiamine deficiency has ranged from 3 to 91 % in various studies conducted in both in-patient and out-patient settings(Reference Jain, Mehta and Al-Ani84). The main mechanisms for thiamine deficiency in HF are the reduction of thiamine intake and its poor absorption due to cardiac cachexia and splanchnic congestion; however, a main cause could be the increased urinary excretion determined by the treatment with high doses of loop diuretics(Reference Katta, Balla and Alpert85). In a cross-sectional prospective observational analysis on thirty-two male NYHA II HF patients on prolonged diuretic therapy, sixteen patients received 300 mg/d of thiamine for 28 d: a 13·5 % increase of LVEF was observed in thiamine recipients (P = 0·021) when compared with control(Reference Jikrona, Suharjono and Ahmad86). Shimon et al. (Reference Shimon, Almog and Vered87) randomised thirty hospitalised HF patients secondary to myocardial ischaemia and administered intravenous thiamine for 7 d or placebo in a double-blind manner followed by 6 weeks of oral thiamine at 200 mg/d in all patients. At the end of complete treatment patients experienced a 22 % increase in LVEF as compared with the baseline value (P<0·01). Similar results were obtained in a small RCT carried out of nine symptomatic patients with HF with an increase in LVEF from 29·5 to 32·8 % (P = 0·024) after supplementation with oral thiamine at 300 mg/d for 28 d(Reference Schoenenberger, Schoenenberger-Berzins and der Maur88). However, a recent RCT (fifty-two patients with HF and LVEF <40 % treated with 300 mg/d of thiamine or placebo for a period of 1 month) showed no significant difference in either systolic or diastolic echocardiographic parameters and dyspnoea between the two groups, even if patients in the thiamine group showed a significant improvement in peripheral oedema (34·6 v. 3·8 %; P = 0·005)(Reference Mousavi, Namazi and Avadi89). In this study, however, the proportion of patients who were on furosemide was incredibly low (ten out of fifty-two). Second, spironolactone was prescribed to most (twenty-two out of twenty-six) patients in the thiamine group, which may have caused a decrease in the incidence of thiamine deficiency in this cohort of patients(Reference Rocha, Silva and de Albuquerque90).
In general, at dosages between 25 and 300 mg/d, thiamine is confirmed to have an excellent profile of safety.
Expert opinion
Given the small sample size and inherent limitations of the available studies, long-term RCT with large samples are needed to confirm or not the positive effects of thiamine in patients with HF(Reference Kattoor, Goel and Mehta79).
Cocoa and dark chocolate
Cocoa (Theobroma cacao) is a rich source of polyphenols, generally varying from 12 to 18 % of dry weight depending on variety, growing region and processing operations of the beans(Reference Fernández-Murga, Tarín and García-Perez91). Among polyphenols, cocoa is particularly rich in flavonoids, in particular flavanols that are present as aglycones both in the monomeric and polymerised form. Among the monomeric flavanols the flavan-3-ols (37 % of total monomeric flavanols form), with (–)-epicatechin occurring in the largest quantities, represent 35 % of the total content of phenolic compounds in cocoa beans (reaching concentrations of 16·5 mg/g dry weight in Costa Rica cacao)(Reference Rusconi and Conti92). A rich source of cocoa polyphenols is dark chocolate, even if the treatment of cocoa beans through fermentation, roasting, drying and alkalisation processes is the cause of a significant loss of polyphenol content and of the antioxidant, metabolic and vascular effects. Therefore, the concentration of polyphenols in the cocoa bean is significantly different from that of cocoa powder and chocolate(Reference Ortega, Romero and Macià93). The mechanisms of action through which cocoa polyphenols could act on HF have not been yet fully clarified – the most reliable hypotheses concern the improvement of the endothelium-dependent vasodilator responses, mediated by increase in NO synthesis, suppression of endothelin-1 synthesis and reduction of NT-proBNP(Reference Schroeter, Heiss and Balzer94,Reference Heiss, Jahn and Taylor95) .
Clinical evidence
In a prospective cohort study of 31 917 Swedish men aged 45–79 years, moderate chocolate consumption was associated with a lower rate of HF hospitalisation or death: the multivariable-adjusted rate ratio was 0·88 (95 % CI 0·78, 0·99) for those consuming 1–3 servings per month, 0·83 (95 % CI 0·72, 0·94) for those consuming 1–2 servings per week, 0·82 (95 % CI 0·68, 0·99) for those consuming 3–6 servings per week, and 1·10 (95 % CI 0·84, 1·45) for those consuming ≥1 serving per d (P = 0·001)(Reference Steinhaus, Mostofsky and Levitan96). However, a higher than moderate intake was not associated with a decreased risk. This effect may be attributable to the high energy content of commercially available chocolate, and therefore the risk to increase weight gain. However, in the large Physicians’ Health Study, the association between chocolate consumption and incident HF was stronger in lean than in overweight/obese subjects(Reference Petrone, Gaziano and Djousse97), making the interpretation of this relationship more complex, however, on the other hand supporting in a way the obesity paradox hypothesis(Reference Alagiakrishnan, Banach and Ahmed98). In a recent small RCT, thirty-two patients with chronic HF, stable on guideline-directed medical therapy, were randomised to consume 50 g/d of high-flavanol dark chocolate (HFDC; 1064 mg of flavanols/d) or low-flavanol dark chocolate (LFDC; 88 mg of flavanols per d) for 4 weeks and then crossed over to consume the alternative dark chocolate for a further 4 weeks. At the end of treatment with HFDC, plasma NT-proBNP level was significantly reduced compared with baseline (−44 (sd 69) %), and and in the follow-up (−41 (sd 77) %) values, but also compared with LFDC (−33 (sd 72) %) treated patients. HFDC also significantly reduced diastolic blood pressure compared with the values after LFDC (−6·7 (sd 10·1) mmHg)(Reference De Palma, Sotto and Wood99). In addition, it was recently shown that flavanol-rich chocolate also improves vascular function in patients with congestive HF(Reference Flammer, Sudano and Wolfrum100). Dark chocolate consumption in patients with HF was also related to an improvement in maximum work (W) (P = 0·026), with no changes with placebo. In addition, a significant increase in protein levels was observed for liver kinase B1 (LKB1), adenosine monophosphate-activated protein kinase (AMPK) and PPAR-γ coactivator 1-α (PGC1α) and in their active forms (phosphorylated AMPK and LKB1) as well as in citrate synthase(101). However, the prognostic significance of these changes has yet to be clarified (Table 3).
Cocoa is usually very well tolerated, both as functional food and as a dietary supplement (flavanols). The main risk with commercial chocolate is the increase in body weight, if consumption is not quantitatively controlled.
Expert opinion
More data are needed due to study limitations and inconsistency: the incidence of HF in epidemiological studies was often reported as a cumulative incidence in years without knowing the HF-free survival curves; although the results suggest that chocolate consumption may play a role in preventing HF, it is still unclear how much chocolate consumption is needed to achieve this and whether the effect is linked to preventing or only delaying the development of HF. Finally, it is difficult to evaluate the risk of HF following consumption of different types of chocolate or different amounts of energy intake.
l-Carnosine
l-Carnosine (β-alanyl-l-histidine) is a dipeptide produced primarily in the liver (from β-alanine, through carnosine synthase) and stored especially in the skeletal muscle and in the heart(Reference Boldyrev, Aldini and Derave102). The rate-limiting step in the synthesis of l-carnosine is represented by the tissue concentration of β-alanine (produced as a result of uracil hepatic metabolism), and the oral administration of β-alanine has been shown to increase the levels of l-carnosine in the heart(Reference Sale, Saunders and Harris103). However, the intake of β-alanine at the active dose of 800 mg at a single time can give ‘pins and needles’ paraesthesia that can last 1 h, during the peak of plasma β-alanine(Reference Artioli, Gualano and Smith104). l-Carnosine should exert a positive effect on HF patients through different mechanisms of action: the antioxidant and peroxyl radical-trapping ability at physiological concentrations due to its imidazole ring(Reference Kohen, Yamamoto and Cundy105), the prevention of hydroxylradicals production via chemical Fenton catalysis by Fe and Cu chelation(Reference Pavlov, Revina and Dupin106), as well as the inhibition of the fragmentation and inactivation of Cu,Zn-superoxide dismutase by peroxyl radicals(Reference Kang, Kim and Choi107). The antioxidant properties of histidine derivatives have also been supposed to be responsible for the cardioprotective effect against ischaemia–reperfusion injury demonstrated in vitro (Reference Seddon, Looi and Shah108). Moreover, l-carnosine plays a sensitising action in cardiac contraction, probably related to an up-regulation of Ca release from the sarcoplasmic reticulum(Reference Zaloga, Roberts and Black109). This effect could lead to a procontractile action(Reference Bokeriya, Boldyrev and Movsesyan110), potentially useful in HF patients.
Clinical evidence
In an RCT, the effect of the oral administration of 500 mg of l-carnosine for 6 months on QoL and exercise performance has been tested in fifty patients with stable HF and impaired LV function. Patients who received orodispersible l-carnosine experienced an improvement in 6-min walking test (6MWT) distance (P = 0·014) and an improvement in self-perceived QoL (assessed with a visual analogue scale score; P = 0·039) between baseline and follow-up. In addition, in an l-carnosine group compared with controls, at the end of 6 months of treatment the authors showed an improvement in peak VO2 (P<0·0001), VO2 at anaerobic threshold, 6MWT, peak exercise workload and self-perceived QoL assessed by the EuroQoL five dimensions questionnaire (EQ-5D test) and the visual analogue scale score(Reference Lombardi, Carubelli and Lazzarini111). A potential alternative to direct l-carnosine administration could be supplementation with magnesium orotate, which seems also to be clinically useful in HF(Reference Rosenfeldt112). The orotate is in fact a precursor of uridine, a pyrimidine nucleoside used from cardiac tissue as a source of β-alanine and subsequently of l-carnosine. A placebo-controlled study evaluating the effects following the administration of magnesium orotate (6 g/d for 1 month, 3 g/d for 11 months) in patients with severe congestive HF, reported a survival rate of 75·7 % in patients treated with magnesium orotate, compared with 51·5 % survival for those treated with placebo (P<0·05)(Reference Stepura and Martynow113). Orotate can be considered as a ‘delayed release’ form of β-alanine, which can be better tolerated than β-alanine itself, probably because the evolution of β-alanine proceeds gradually after orotate intake. Consequently, the intake of orotates or orotic acid could be a strategy for boosting the level of carnosine in the body and in the heart tissues(Reference McCarty and Di Nicolantonio114). Timed-release β-alanine preparations may also be employed to prevent the above-cited paraesthesia(Reference Décombaz, Beaumont and Vuichoud115), but this formulation has not yet been tested in HF patients (Table 2).
Carnosine is overall usually well tolerated, and no specific concerns have been raised till now as regards its use as a dietary supplement
Expert opinion
Given the small sample size and inherent limitations of the available studies, long-term RCT with large samples are needed to confirm or not the positive effects of l-carnosine in patients with HF.
Vitamin D
Vitamin D (vitD; commonly referred to as D2 and D3, respectively, ergocalciferol and cholecalciferol) is a collection of fat-soluble steroids, obtained via endogenous production or from dietary intake, with the latter accounting for nearly 10–20 % of our total supply(Reference Holick116). Cholecalciferol is produced from its precursor 7-dehydrocholesterol, in the skin, after exposure to the UVB spectrum of sunlight. In its original state, vitamin D3 is inactive and requires two separate hydroxylation reactions (the first in the liver and the second in the kidney) to become biologically active (calcitriol). Calcitriol exerts its effects via the vitD receptor and it is responsible for the regulation of gene transcription and protein synthesis, with the known effect on Ca homeostasis and bone metabolism(Reference Wang, Zhu and DeLuca117). Nevertheless, in recent years, the biological influence of vitD has been significantly expanded beyond just Ca regulation. VitD receptor has been isolated from a number of different tissues not traditionally involved in Ca homeostasis, such as the myocardium(Reference O’Connell and Simpson118) and fibroblasts(Reference Simpson, Thomas and Arnold119). Despite emerging evidence supporting a pathophysiological relationship between vitD and HF, the exact mechanism by which vitD deficiency leads to poor clinical outcome has not been clearly established. One of the hypotheses is the relationship between the heart and kidney: these two organs are interrelated and the impairment of one system can induce pathological processes within the other, thus accelerating the progressive failure of both. This interrelationship serves as the pathophysiological basis of the clinical entity called cardiorenal syndrome(Reference Pourdjabbar, Dwivedi and Haddad120). The progression of cardiorenal syndrome involves the renin–angiotensin–aldosterone system and sympathetic nervous system overactivation, as well as systemic inflammation, which could alter electrolyte and fluid regulation, cause endothelial dysfunction, and both LV remodelling and fibrosis along with renal fibrosis and failure(Reference Sun, Zhang and Zhang121,Reference Mann122) . These changes set up a vicious cycle, resulting in further systemic dysfunction until organ failure occurs. VitD is particularly low in individuals with cardiorenal syndrome, probably due to reduced α-1-hydroxylase activity and the depletion of vitD-binding proteins secondary to proteinuria(Reference Li123). Similarly, with those HF patients, they are also associated with increased cardiovascular events(Reference Levin, Bakris and Molitch124). In a study of patients with diabetic nephropathy, paricalcitol supplementation significantly reduces proteinuria(Reference de Zeeuw, Agarwal and Amdahl125), a known predictor of cardiovascular events(Reference Keane and Eknoyan126,Reference Brenner, Cooper and de Zeeuw127) . VitD deficiency in HF patients could also be linked to an overactivation of the renin–angiotensin–aldosterone system(Reference Liu, Voors and van Veldhuisen128), and to increased production and release of inflammatory cytokines, which can have a direct negative effect on the myocardium or indirectly affect other vital organs, causing myocardial apoptosis, hypertrophy, fibrosis, LV remodelling, negative ionotropic effects(Reference Gullestad, Ueland and Vinge129-Reference Haudek, Taffet and Schneider131), increased renal fibrosis and renal failure(Reference Upadhyay, Larson and Guo132,Reference Yu, Yang and Yu133) . In vitro studies suggest that vitD suppresses proinflammatory cytokines such as TNF-α and IL-6 and up-regulates anti-inflammatory cytokines such as IL-10(Reference Mora, Iwata and von Andrian134), while the lack of vitD is associated with increased myocardial matrix metalloproteinase expression, increased collagen deposition and fibrosis(Reference Li, Feng and Kadokami135,Reference Gunja-Smith, Morales and Romanelli136) .
Clinical evidence
Many observational studies have suggested a possible relationship between vitD status and prevalence, incidence and severity of HF(Reference Bielecka-Dabrowa, Sakowicz and Pietrucha137,Reference Faridi, Lupton and Martin138) . In a North-American study carried out on 8351 participants, individuals in the lowest tertile were approximately twice as likely to have HF compared with those in the highest tertile (OR 2·1; 95 % CI 1·2, 3·6)(Reference Kim, Sabour and Sagar139). Similar results were confirmed in the larger Intermountain Heart Collaboration Study in a cohort of 41 504 patients(Reference Anderson, May and Horne140). In the German Ludwigshafen Risk and Cardiovascular Health Study with 3299 patients referred for coronary angiography, a significant negative correlation was observed between serum 25-hydroxyvitamin D (25(OH)D) and NT-proBNP concentration (r –0·2; P<0·001) and mean serum 25(OH)D concentration was positively correlated with LVEF and negatively correlated with NYHA functional class(Reference Pilz, Marz and Wellnitz141). 25(OH)D levels were negatively associated with N-terminal pro-A-type natriuretic peptide in NYHA II–IV patients(Reference Zittermann, Schleithoff and Tenderich142). In addition, individuals with a serum 25(OH)D concentration <10·0 ng/ml (<25·0 nmol/l) had significantly larger LV volumes compared with those with a serum 25(OH)D concentration >10·0 ng/ml (>25·0 nmol/l), and mean fractional shortening was significantly lower in those with a serum 25(OH)D concentration <10·0 ng/ml (<25·0 nmol/l) (31·6 v. 37·1 %; P<0·05)(Reference Ameri, Ronco and Casu143). In general, mean serum 25(OH)D concentration is usually lower in patients with HF compared with controls(Reference Iqba, Ducharme and Desai144-Reference Arroyo, Laguardia and Bhattacharya146). VitD status may be related to physical functioning in HF: there seems to be a negative correlation between cardiopulmonary stress test performance and serum 25(OH)D concentration ≤ 9·0 ng/ml (≤ 22·5 nmol/l) compared with those with levels ≥10·0 ng/ml (≥ 25·0 nmol/l)(Reference Shane, Mancini and Aaronson147). A significant positive correlation between serum 25(OH)D concentration, 6MWT distance (r 0·4; P<0·05)(Reference Boxer, Dauser and Walsh148) and maximal oxygen uptake during cardiopulmonary stress testing was observed in HF patients(Reference Boxer, Kenny and Cheruvu149). VitD status may also be related to prognosis in HF: in the above-cited Ludwigshafen Risk and Cardiovascular Health Study (patients were followed-up for a median of 7·7 years), individuals with severe vitD deficiency (serum 25(OH)D concentration <10·0 ng/ml (<25·0 nmol/l)) were approximately four times more likely to die from HF than those with a serum 25(OH)D concentration in the optimal range (>30·0 ng/ml (>75·0 nmol/l)) (HR 4·1; 95 % CI 1·8, 9·6). Even patients waiting for heart transplantation in the highest 25(OH)D tertile were approximately twice as likely to survive in comparison with those in the lowest tertile (HR 0·4; 95 % CI 0·3, 0·5)(Reference Zittermann, Schleithoff and Gotting150).
In an RCT, 123 patients with HF treated with vitD (2000 IU/d) + Ca (500 mg/d) for 9 months experienced significantly increased IL-10 and reduction of TNF-α compared with placebo (administered with at Ca 500 mg/d only)(Reference Schleithoff, Zittermann and Tenderich151). In paediatric HF patients, daily vitD supplementation with 1000 IU of cholecalciferol resulted in significant improvement in LV end diastolic and systolic diameters, LVEF and myocardial performance index, together with reductions in inflammatory cytokines(Reference Shedeed152). VitD supplementation (between 800 and 1000 IU/d) in HF patients with low 25(OH)D levels was associated with a significant reduction in mortality, independent of the baseline 25(OH)D levels(Reference Gotsman, Shauer and Zwas153). However, supplementation with a very high dosage (100 000 IU/week) was not associated with improvement of 6MWT distance after 20 weeks of treatment, despite a significant reduction in BNP levels(Reference Witham, Crighton and Gillespie154), probably because in this study the obtained serum 25(OH)D levels were not optimised.
In general, vitD supplementation is believed to be safe because it rarely raises serum vitD levels to the toxic range even after repeated intravenous ingestion of extremely high doses of synthetic vitD. However, prolonged consumption of vitD supplementation may induce hypercalcaemia, hypercalciuria and hyperphosphataemia, which are considered the initial signs of vitD intoxication(Reference Razzaque155). Use of thiazide diuretics in combination with Ca and vitD supplements may cause hypercalcaemia in the elderly or those with compromised renal function or hyperparathyroidism(Reference Robien, Oppeneer and Kelly156).
Expert opinion
It seems that vitD might be useful in the supporting HF therapy and improve prognosis. However, large-scale randomised, multicentre clinical trials are still needed before routine vitD supplementation can be recommended as part of clinical care of HF patients. VitD should be supplemented in vitD-deficient subjects, and especially in those with heart diseases.
Magnesium
Mg plays a role in many enzymic processes, contributing to stable cardiovascular haemodynamics and electrophysiological functioning: it is an important component in mitochondrial function, modulating cellular K permeability and affecting Ca uptake and its distribution(Reference Rude157,Reference Gattlieb158) . In respect of other electrolyte alterations, the pathophysiology of hypomagnesaemia (serum Mg <1·5 mg/dl (0·62 mmol/l)) remains less studied even if is not infrequently observed in HF patients: the prevalence is about 7 % of well-compensated ambulatory subjects to 52 % in more advanced HF patients who are aggressively treated with diuretics(Reference Ralston, Mumane and Unverferth159). To date, it is rather clear that the effective correction of Mg disturbances is favourable in HF subjects, in particular in preventing potentially life-threatining arrhythmias(Reference Douban, Brodsky and Whang160). In HF, Mg depletion stems from several factors such as reduced dietary intake, altered distribution of the ion, renal losses, oedematous states (involving the intestinal mucosa that might interfere with the absorption of microelements), respiratory alkalosis and excessive catecholamine release(Reference Wester161). Moreover, diuretics produce most of renal Mg loss, especially in the volume-expanded setting of HF and in associated hyperaldosteronism and it has been demonstrated that K depletion inhibits the renal reabsorption of Mg, leading to hypermagnesiuria and hypomagnesaemia(Reference Wu, Ackermann and Sonnenberg162). Hypomagnesaemia seems to have vasoconstrictor properties due to the inhibition of PG-induced relaxation and to the enhancement of the activity of the vasoconstrictor neurohormones through alterations in Ca uptake. In HF patients, the presence of adequate total-body Mg stores has been associated with a reduction in the risk of arrhythmias, digitalis toxicity and haemodynamic abnormalities(Reference Douban, Brodsky and Whang160).
Clinical evidence
Most of the evidence supporting Mg supplementation in HF patients comes from observational data. Small RCT carried out in HF patients have underlined that Mg supplementation improves LVEF(Reference Witte, Nikitin and Parker163) and heart rate variability(Reference Almoznino-Sarafian, Sarafian and Berman164). On the other hand, observational studies have linked low serum Mg to more adverse CVD risk factor profiles(Reference He, Liu and Daviglus165-Reference Lee, Miller and Guallar167) and greater risk of CVD events(Reference Misialek, Lopez and Lutsey168). In the Atherosclerosis Risk in Communities (ARIC) study, participants in the lowest quintile of Mg levels were at 2·5 times greater risk of incident HF after adjustment for demographic factors, and with additional adjustment for behaviours and CVD risk factors(Reference Lutsey, Alonso and Michos169). HF worsening secondary to very severe hypomagnesaemia has been described, and, in some cases, Mg supplementation determines the reversal of HF(Reference Altura and Altura170). Overall, among patients with HF, low serum Mg has been associated with increased all-cause mortality(Reference Adamopoulos, Pitt and Sui171). The British Heart Regional Study (a prospective study of 3523 men aged 60–79 years with no prevalent HF or myocardial infarction followed up for a mean period of 15 years) showed that serum Mg was inversely related to risk of incident HF (after adjustment for conventional CVD risk factors and incident myocardial infarction). In addition, the benefit of high serum Mg on HF risk was most evident in men with electrocardiogram evidence of ischaemia (HR 0·29; 95 % CI 0·13, 0·68; P<0·05)(Reference Wannamethee, Papacosta and Lennon172). However, a meta-analysis of seven eligible prospective studies did not observe a clear correlation between hypomagnesaemia and HF, if not in elderly HF patients with reduced LVEF(Reference Angkananard, Anothaisintawee and Eursiriwan173). However, in the Jackson Heart Study cohort, Mg intake <2·3 mg/kg was related to increased risk of subsequent HF hospitalisations(Reference Taveira, Ouellette and Gulum174), and, in a more recent study, Mg deficiency independently predicted poor QoL and earlier cardiac event-free survival in HF patients(Reference Song and Kang175). Finally, a recent meta-analysis showed a clear and significant link between Mg supplementation and reduction of inflammation(Reference Mazidi, Rezaie and Banach176).
Low serum Mg seems to be associated with an increased risk of incident atrial fibrillation(Reference Lopez, Agarwal and Grams177,Reference Khan, Lubitz and Sullivan178) that is closely related with HF(Reference Wang, Larson and Levy179,Reference Chamberlain, Redfield and Alonso180) . In particular, many studies have highlighted the correlation between serum Mg deficiency and atrial fibrillation risk after cardiac surgery: in that setting, Mg is sometimes used prophylactically to prevent atrial fibrillation events(Reference Miller, Crystal and Garfinkle181-Reference Banach, Kourliouros and Reinhart183). In addition, Mg is believed to be linked to CVD risk through a broad range of physiological roles; low serum concentrations have been associated with impaired glucose homeostasis and insulin action, elevated blood pressure, chronic inflammation, impaired vasomotor tone and peripheral blood flow, and electrocardiogram abnormalities(Reference Rude, Coates, Betz, Blackman and Cragg184). A recent meta-analysis of forty prospective cohort studies totalling more than 1 million participants has shown that increasing dietary Mg intake is associated with a reduced risk of stroke, HF, diabetes, and all-cause mortality, but not CHD or total CVD(Reference Fang, Wang and Han185).
Mild diarrhoea, stomach upset, nausea and heartbeat are the most common dose-dependent side effects after the use of Mg. Mg toxicity rarely occurs except in patients with renal dysfunction: severe kidney failure and heart block are the two relative contraindications(Reference Urso, Brucculeri and Caimi186).
Expert opinion
The available data suggest that we should avoid hypomangnesaemia in patients with HF. However, the prognostic significance of serum Mg concentration in HF patients as well as clinical relevance of its supplementation still needs to be investigated. Additional work is needed to elucidate whether the association between HF and Mg deficiency is causal and to clarify the specific mechanisms that underlie the association.
Beetroot and inorganic nitrates
NO is not only an endothelium-derived relaxing factor, but is also a key cellular signalling molecule with pleiotropic effects in many tissues. For example, in skeletal muscle NO helps to modulate contractile function, through the nitrosation or S-nitrosylation of various proteins(Reference Lima, Forrester and Hess187). In addition, during concentric activity, NO significantly increases the rate of force development, maximal shortening velocity, and maximal power of both single muscle fibres and isolated muscles via the activation of the classic NO soluble guanyl cyclase-cycle GMP (NO-sGC-cGMP) pathway(Reference Coggan and Peterson188). In failing cardiac muscle, however, the increased production of reactive oxygen species leads to more rapid destruction of NO, and hence reduced NO-sGC-cGMP signalling, which in turn is thought to contribute to reduced contractility in HF(Reference Hare and Stamler189). It is possible therefore that NO bioavailability is diminished in the skeletal muscles of patients with HF, thus contributing to their reduced muscle function(Reference Coggan and Peterson188). It is also well established that HF leads to endothelial dysfunction in various tissues, including skeletal muscle, as a result of reduced NO production via endothelial NO synthase(Reference Katz190). Emerging evidence suggests that supplementation of dietary inorganic nitrate (NO3 −) has beneficial effects on vascular health, blood pressure, exercise capacity and oxygen metabolism though targeted NO production(Reference Kapil, Weitzberg and Lundberg191). After ingestion, NO3 − is reduced to bioactive NO2 − by bacteria found in the oral cavity. NO2 − is then taken up by the plasma from the digestive system and can be converted to NO, particularly under hypoxic or acidic conditions(Reference Cosby, Partovi and Crawford192), which can occur in HF and during exercise. Inorganic nitrates slowly produce NO and determine more mild but sustained vasodilation while organic nitrates rapidly release relatively large amounts of NO(Reference Lundberg, Carlstrom and Larsen193,Reference Omar, Artime and Webb194) , with potentially adverse effects on myocardial functionality in HF patients(Reference Redfield, Anstrom and Levine195). Compared with placebo, either a single, acute dose or 1 week of daily dosing of beetroot juice (BRJ) significantly increased plasma NO2 − similarly, by 138 and 129 % compared with placebo, respectively(Reference Miller, Marsh and Dove196).
Clinical evidence
In some RCT, BRJ has been shown to increase time to exhaustion during high-intensity exercise and to reduce oxygen consumption (VO2) during submaximal exercise (i.e. reduce oxygen cost at a given submaximal work rate)(Reference Lansley, Winyard and Fulford197-Reference Hoon, Johnson and Chapman199). These results were obtained not only in healthy individuals but also in HF patients. In these latter subjects, a single dose of BRJ (12·9 mmol NO3 –) was shown to increase total work performed and cardiac output compared with placebo, while decreasing systemic vascular resistance and reducing aortic augmentation index during a maximal exercise test(Reference Zamani, Rawat and Shiva-Kumar200). In healthy older adults, BRJ supplementation has been shown to improve VO2 kinetics(Reference Kelly, Fulford and Vanhatalo201). Daily supplementation with BRJ containing 11·2 mmol of NO3 – increased NO production and muscle function with an improvement of physical exercise in HF patients(Reference Coggan, Leibowitz and Spearie202). In another RCT, 1 week of daily dosing with BRJ containing 6·1 mmol NO3 – significantly improved submaximal aerobic endurance (+24 %; P = 0·02 compared with placebo) and blood pressure (P<0·001) in elderly patients with HFpEF(Reference Eggebeen, Kim-Shapiro and Haykowsky203). Acute dietary NO3 – intake was also related to an increase of VO2 peak and improved physical performances in HFrEF patients(Reference Coggan, Broadstreet and Mahmood204). BRJ may have positive effects combined with an aerobic exercise training regimen in HFrEF patients, even if data are still limited and inconsistent(Reference Shaltout, Eggebeen and Marsh205). BRJ has also been shown to improve exercise capacity and oxygen metabolism in older patients with peripheral arterial disease(Reference Kenjale, Ham and Stabler206). Data on EF and hard outcomes are still lacking; however, a meta-analysis of RCT including 1248 patients showed that BRJ is associated with significant reductions in blood pressure (systolic blood pressure: –3·55 (95 % CI –4·5, –2·5) mmHg; diastolic blood pressure: –1·3 (95 % CI –1·9 to –0·7) mmHg) compared with controls, thus reducing the afterload. The mean difference of SBP was larger between BRJ-supplemented and control groups in the longer than in the shorter (≥14 compared with <14 d) study durations (–5·1 compared with –2·7 mmHg) and the highest compared with the lowest (500 compared with 70 and 140 ml/d) doses of BRJ (–4·8 compared with –2·4 mmHg)(Reference Bahadoran, Mirmiran and Kabir207). Long-term effects on EF and hard outcomes are lacking (Table 3).
In general, BRJ supplementation seems to be tolerable and safe after short-term administration. However, most available clinical trials are short term (<30 d). Long-term data are lacking.
Expert opinion
Taking into account relatively limited data, mainly short term, on the possible administration of beetroot and NO3 – in HF patients it is difficult to draw any recommendations of its usefulness. Therefore, large-scale randomised, multicentre clinical trials are still needed.
d-Ribose
d-Ribose is a pentose carbohydrate that plays many physiological roles. Myocardial ATP levels are important to maintain cell integrity and function: ATP has a key role in the interaction between Ca and the sarcoplasmic reticulum (important for ventricular relaxation)(Reference Pauly and Pepine208). In fact, deficient ATP levels are found in IHD and HF(Reference Kriett, Ward and Bianco209) and can be responsible for diastolic dysfunction and a non-compliant ventricle. Exogenous supplementation with d-ribose enhances the regeneration of ATP levels by bypassing rate-limiting, slow enzymic steps in glycolysis through the pentose phosphate pathway(Reference St Cyr, Bianco and Schneider210,Reference Zimmer211) . In an experimental model of ischaemic injury, supplementation with d-ribose might quickly increase immediately the levels of ATP, improving diastolic dysfunction and substantially shortening the lengthy time recovery that normally occurs following ischaemia(Reference Schneider, St Cyr and Mahoney212).
Clinical evidence
Preliminary results in HF patients confirm what was observed in experimental models: in a prospective feasibility study, d-ribose improved diastolic dysfunction, self-perceived QoL and physical function(Reference Omran, Illien and MacCarter213), while in a pilot trial d-ribose improved ventilatory efficiency(Reference Vijay, MacCarter and Shecterle214). In an RCT, the effects of the administration of 5 g daily of d-ribose for 6 weeks were evaluated in patients with NYHA II–IV. At the end of the treatment, the results showed an improvement of tissue Doppler velocity (E’) (in 75 % of the patients) what was also maintained in the follow-up visit at 9 weeks (i.e. 3 weeks after the treatment with d-ribose was stopped). In addition, half of the patients achieved an improvement in their ratio of E:E'(Reference Bayram, St Cyr and Abraham215). At least part of these effects could be related to the anti-ischaemic properties of d-ribose: supplementation with this pentose carbohydrate enabled patients with stable coronary artery disease to exercise longer without developing angina or electrocardiographic changes(Reference Pliml, von Arnim and Stablein216). Moreover, improvement in heamodynamic parameters has also been recorded perioperatively, in patients undergoing off-pump coronary artery bypass(Reference Perkowski, Wagner and Marcus217) and in individuals following aortic valve replacement(Reference Vance, Einzig and Kreisler218) (Table 2).
Side effects were negligible.
Expert opinion
d-Ribose might offer an energetic benefit in patients with ischaemic CVD, including HF (particularly; with the improvement of diastolic dysfunction). Even if the preliminary data are encouraging, clinical studies conducted to date are still few, small and relatively short.
Probiotics
Recent clinical and preclinical studies underline the key role of intestinal microbiota in cardiovascular health and in HF prognosis in particular(Reference Nagatomo and Tang219). Intestinal eubiosis is important in regulation of the function of the intestinal barrier, together with mucosal immunity, Na and water homeostasis and the functionality of tight junctions. In particular, subjects with HF manifest gastrointestinal disorders of absorption, motility, tissue perfusion and oedema, which determine alterations of the intestinal bacterial flora that in long term are responsible for an increase in translocation of endotoxins in the blood, an increase in preload and afterload and an aggravation of the clinical picture(Reference Sandek, Bjarnason and Volk220,Reference Krack, Sharma and Figulla221) .
Clinical evidence
A strong correlation appears to exist between HF severity and the severity of intestinal dysbiosis, measured through the serum levels of trimethylamine N-oxide (TMAO), an amine produced by the metabolism of choline and phosphatidylcholine from intestinal microbiota (especially from opportunistic/pathogenic micro-organisms)(Reference Organ, Otsuka and Bhushan222). It is hypothesised that vascular remodelling and progressive coronary atherogenesis may occur in the context of high levels of TMAO(Reference Wang, Klipfell and Bennett223). The aetiopathogenetic mechanism is not clear yet; however, it is evident that there is a direct proportionality between the blood levels of TMAO and an increase in intestinal oedema, inflammatory metabolites and cardiac and vascular remodelling(Reference Koeth, Wang and Levison224). In a recent prospective study, the potential pathophysiological role of intestinal microbiota in HF and its relationship to mortality from all causes was examined; in particular, the authors studied (in 720 subjects and for a duration of 5 years of follow-up) the role of TMAO, measured through fasting blood samples. The study found that the highest TMAO levels were reported in patients with HF (mean TMAO levels: 5·0 μm) compared with healthy subjects (mean TMAO levels: 3·5 μm; P<0·001), with a risk of mortality increased by 3·4 times(Reference Wilson, Zeneng and Yiying225). Finally, it has been shown that elevated TMAO levels modify lipid metabolism through changes in the functionality of reverse cholesterol transport, sterol metabolism and modification of the quality and quantity of bile acids(Reference Shih, Wang and Lee226,Reference Hartiala, Bennett and Tang227) . An RCT conducted in patients with NYHA class II or III and LVEF <50 %, treated for 3 months with a preparation containing 1000 mg/d of probiotics (Saccharomyces boulardii), evaluated the efficacy of this supplementation on haemodynamic parameters. At the end of 3 months of treatment, the group treated with probiotics benefited from a significant reduction in uric acid levels (–1·08 mg/dl (–64·24 μmol/l), P = 0·014 v. placebo: –0·01 mg/dl (–0·59 μmol/l), P = 0·930), total cholesterol (–7·63 mg/dl (–0·20 mmol/l), P = 0·010 v. placebo: –2·02 mg/dl (–0·05 mmol/l), P = 0·603), hsCRP (–0·23 mg/dl (–2·3 mg/l), P = 0·116 v. placebo: +0·44 mg/dl (+4·4 mg/l), P = 0·011), improvement in LVEF (+6·6 %, P = 0·005 v. placebo: +4·2 %, P = 0·173) and of the left atrial diameter (–0·29 cm, P = 0·044 v. placebo: +0·2 cm, P = 0·079)(Reference Costanza, Moscavitch and Faria Neto228). Preclinical data have shown results comparable with Lactobacillus rhamnosus (Reference Gan, Ettinger and Huang229). A recent study by Maier et al. (Reference Maier, Pruteanu and Kuhn230) showed that about one-quarter of non-antibiotic drugs are able to inhibit the growth of at least one main strain of intestinal bacterial flora. Among the cardiovascular drugs, the most interesting ones appear to be Ca antagonists and numerous anti-arrhythmics. Therefore, the microbiome dysbiosis–drug interaction in HF patients is particularly evident compared with the general population and so greater attention should be paid to possible alterations and new aetiopathogenetic mechanisms caused to the microbiome(Reference Wilson, Zeneng and Yiying225). The ongoing Gut-Heart trial has randomised 150 patients with stable HF and a LVEF<40 % to receive the antibiotic rifaximin, the probiotic yeast S. boulardii (ATCC 74012) or no treatment in a 1:1:1 fashion. The primary endpoint is EF at 3 months. The outcomes of the trial will shed some light into the possible therapeutic avenues in the future targeting the gut microbiome (Table 2).
In general, supplementation with probiotics in cardiovascular prevention has proved to be safe and free of any relevant side effects.
Expert opinion
Some probiotic strains (in particular lactobacilli, bifidobacterial, in addition to S. boulardii) could be applied as an adjuvant to conventional therapy in HF treatment. Further long-term clinical trials (including the results from the Gut-Heart trial) are necessary to investigate the effects of probiotic supplementation on cardiovascular outcomes.
Iron
Fe deficiency is defined in the general population as serum ferritin <30 ng/ml and transferrin saturation below 20 %; however, different cut-off values are used in the presence of inflammatory co-morbidities (ferritin is an acute-phase protein that increases during inflammation) such as inflammatory bowel disease (ferritin <100 ng/ml), chronic kidney disease (ferritin <500 ng/ml plus transferrin <30 %) and chronic kidney disease (transferrin <100 ng/ml or <100–299 ng/ml plus transferrin <20 %) to diagnose Fe deficiency(Reference Jimenez, Kulnigg-Dabsch and Gasche231). The burdening prevalence of this co-morbid condition is illustrated by the findings of such deficit in 30–50 % in chronic stable disease(Reference Jankowska, Rozentryt and Witkowska232,Reference Tkaczyszyn, Comín-Colet and Voors233) and –80 % in acute HF(Reference Van Aelst, Abraham and Sadoune234,Reference Cohen-Solal, Damy and Hanon235) . Furthermore, Fe deficiency independently predicts more severe symptomatic burden, higher morbidity, as noted by markedly increased hospitalisations and readmission rates, and mortality(Reference Okonko, Mandal and Missouris236), which underscores its importance in HF. Fe deficiency in HF patients (as in the general population) may be due to either reduced Fe intake, increased Fe body losses and/or impaired Fe absorption(Reference Ganz and Nemeth237). Briefly, anaemia is often the result of compromised Fe stores and/or impaired transportation. In addition to Fe deficiency, multiple other factors can contribute to anaemia, including inflammation, renal dysfunction and haemodilution(Reference Cunha, Rocha and Menezes Falcão238).
Clinical evidence
A small study noted that Fe intake was markedly decreased in HF patients, aggravating with increased disease severity(Reference Hughes, Woodside and McGartland239). However, the Iron Repletion effects ON Oxygen UpTake in Heart Failure (IRONOUT HF) multicentre double-blind RCT, that included 255 patients with symptomatic Fe deficiency and HFrEF treated for 16 weeks with oral intake of 150 mg twice daily of Fe polysaccharide or placebo, found no significant differences between groups regarding natriuretic peptide levels, symptomatic score, 6MWT or peak VO2. This RCT also does not support a role for oral Fe in Fe deficiency in HFrEF patients(Reference Lewis, Malhotra and Hernandez240). Today, no trial has formally tested the role of oral Fe in patients with HF and mid-range EF (HFmrEF) or HF with HFpEF. According to 2016 European Society of Cardiology guidelines for the diagnosis and treatment of acute and chronic HF, intravenous ferric carboxymaltose should be considered in symptomatic patients with HFrEF and Fe deficiency (serum ferritin <100 µg/l, or ferritin between 100 and 299 µg/l and transferrin saturation <20 %) in order to alleviate HF symptoms, and improve exercise capacity and QoL(Reference Maki, Palacios and Bell61). While intravenous Fe administration in stable symptomatic (NYHA II–III) chronic HFrEF is supported by several clinical trials, no convincing evidence is available regarding oral Fe supplementation(Reference Drozd, Jankowska and Banasiak241).
Whether low bioavailability due to intestinal wall oedema, adverse events and subsequent low rates of adherence, and polypharmacy hinder the benefits of oral Fe in these subpopulations is currently unknown. Moreover, oral Fe supplementation is often associated with multiple unwanted events, particularly gastrointestinal adverse effects(Reference McDonagh and Macdougall242), which outweigh the null benefit of such dietary supplement in HFrEF.
Expert opinion
The authors of this Position Paper support the recent European Society of Cardiology guidelines as for potential Fe supplementation in HFrEF patients. Further studies are necessary as for potential benefits of oral supplementation and in other types of HF (HFpEF and HFmrEF).
Vitamin C
Ascorbic acid (vitamin C) protects against oxidative stress by reducing levels of free oxygen radicals and inhibiting LDL oxidation and oxidative cell damage(Reference Bruckdorfer243). In addition, it is responsible to improve arterial stiffness and immune function, and to reduce inflammatory markers responsible for systemic inflammation(Reference Wilcox, Curb and Rodriguez244).
Clinical evidence
Vitamin C is contained mostly in fruits and vegetables and some(Reference Djoussé, Driver and Gaziano245,Reference Levitan, Wolk and Mittleman246) , but not all the studies(Reference Wang, Tuomilehto and Jousilahti247) have underlined that a diet high in fruit and vegetables, rich in antioxidants, is associated with a reduced risk of HF. Moreover, only a few studies have examined the association between plasma vitamin C levels and incidence of HF, suggesting a positive correlation between elevated plasma vitamin C levels and reduction of the HF risk(Reference Pfister, Sharp and Luben248). In a prospective study including 3919 men aged 60–79 years with no HF at baseline and followed up for a mean period of 11 years, a higher plasma vitamin C level was associated with a significantly lower risk of incident HF in both men with and without previous myocardial infarction after adjustment for lifestyle characteristics, diabetes mellitus, blood lipids, blood pressure and heart rate (HR 0·81 (95 % CI 0·70, 0·93) and 0·75 (95 % CI 0·59, 0·97) for 1 sd increase in log vitamin C, respectively)(Reference Wannamethee, Bruckdorfer and Shaper249). In a recent study, 200 HF patients completed a 3 d food diary to determine vitamin C intake: 39 % of patients had vitamin C deficiency that was associated with an hsCRP level higher than 3 mg/l in the hierarchical logistic regression (OR 2·40; 95 % CI 1·13, 5·10; P = 0·023). In addition, both vitamin C deficiency and hsCRP level higher than 3 mg/l predicted shorter cardiac event-free survival in hierarchical Cox regression and produced a 2·3-fold higher risk for cardiac events (P = 0·002) in moderation analysis. Higher levels of hsCRP predicted shorter cardiac event-free survival only in patients with vitamin C deficiency (P = 0·027), but not in those with vitamin C adequacy(Reference Song and Kang250). In addition, a meta-analysis of forty-four RCT showed a significant positive effect of vitamin C on endothelial function (SMD 0·50, 95 % CI 0·34, 0·66; P<0·001), in particular in the HF subgroup (SMD 0·48; 95 % CI 0·08, 0·88; P<0·02)(Reference Ashor, Lara and Mathers251). A possible mechanism of correlation between plasma vitamin C levels and its beneficial effects may be related to the arterial dilation mediated by the modulation of NO release(Reference Plantinga, Ghiadoni and Magagna252). However, the association between vitamin C and HF seems to involve plasma vitamin C rather than dietary vitamin C, as reported also by the European Investigation into Cancer and Nutrition (EPIC) Norfolk study(Reference Bingham, Welch and McTaggart253).
In general, supplementation with vitamin C at dosages between 500 mg and 3 g/d is considered safe and well tolerated(Reference Spoelstra-de Man, Elbers and Oudemans-Van Straaten254).
Expert opinion
Taking into account the available data in HF settings, no recommendation can be given on the potential usefulness of vitamin C supplementation. To date no study has also directly investigated the impact of chronic vitamin C supplementation on HF incidence or prognosis.
Vitamin E
Preclinical models suggest that oxidative stress characterised by the excessive production of reactive oxygen species and reduction of antioxidant defence capacity may play an important role in the pathophysiology of HF(Reference Sawyer255). Vitamin E includes eight distinct chemical entities: α-, β-, γ- and δ-tocopherol and α-, β-, γ- and δ-tocotrienol. The most studied is α-tocopherol, while other forms are poorly understood(Reference Sen, Khanna and Rink256). Vitamin E can protect against oxidative stress by reducing levels of free oxygen radicals and inhibiting LDL oxidation and oxidative cell damage(Reference Lewis, Malhotra and Hernandez240). In addition, lipid-lowering activity has been documented through PPAR (PPAR-α, PPAR-β and PPAR-γ) activation and HMG-CoA reductase inhibition(Reference Li, Tan and Kang257).
Clinical evidence
In clinical trials, vitamin E (at doses between 50 and 200 mg) showed an improvement of endothelial function (reducing the serum levels of hsCRP, advanced glycation endproducts, metalloproteinases and cell adhesion molecules) and arterial stiffness (improving pulse wave velocity, pulse volume and augmentation index)(Reference Rasool, Rahman and Yuen258,Reference Prasad259) . The Physician’s Health Study II and the Women’s Health Study reported no association between vitamin E supplements and HF in a primary prevention population(Reference Chae, Albert and Moorthy260,Reference Sesso, Buring and Christen261) while other studies reported increased risk of HF with vitamin E supplements in those with CVD(Reference Marchioli, Levantesi and Macchia262,Reference Lonn, Bosch and Yusuf263) , raising concern about the use of vitamin E supplements. In addition, a prospective study of 3919 men aged 60–79 years with no prevalent HF followed up for a mean period of 11 years highlighted that high intake of dietary vitamin E may be associated with increased HF risk(Reference Wannamethee, Bruckdorfer and Shaper249). The reason for an increased risk of HF esepcially in older men(Reference Finkel and Holbrook264) is still unclear, but it may be related to the fact that α-tocopherol may become a pro-oxidant in a pre-existing environment with increased oxidative stress, thereby depressing myocardial function. Moreover, α-tocopherol may suppress other fat-soluble antioxidants, such as γ-tocopherol (more powerful antioxidant than α-tocopherol), disrupting the natural balance of antioxidant systems and increasing vulnerability to oxidative damage(Reference Deveraj and Jialei265). In contrast to dietary vitamin E, no association was observed between plasma vitamin E and risk of HF(Reference Hodge, Simpson and Fridman266) – it is well established that plasma concentrations of vitamin E increase with the amount of total plasma lipids(Reference Thurnham, Davies and Crump267), and particularly with LDL-cholesterol, the principal carrier of tocopherol in the circulation. Thus, vitamin E absorption may be influenced by factors other than diet and seems to be an unreliable marker of dietary vitamin E (α-tocopherol) intake. This may explain the differences in association between dietary and plasma vitamin E and HF.
No specific concern has been raised for vitamin E supplementation in the range of the reccomended daily allowance.
Expert opinion
Available epidemiological data and results from RCT do not suggest any benefits from the supplementation of vitamin E in prevention or as an adjuvant to conventional therapy in HF.
Other minerals and vitamins
Emerging evidence suggests a pathophysiological role of Zn and Cu dyshomeostasis in HF, including promotion of adverse remodelling and clinical deterioration.
Copper
Cu is a redox-active metal, implicated as an enzymic cofactor in various processes including antioxidant defence mechanisms (Cu/Zn-superoxide dismutase, caeruloplasmin), cellular respiration (cytochrome c oxidase), Fe transport (caeruloplasmin), and cross-linking of collagen and elastin (protein-lysine-6-oxidase)(Reference Didonato, Aulak and Huang268).
Zinc
Zn, the second most abundant trace element in the body, is involved in gene expression, cell growth and differentiation as a catalytic and structural cofactor. It is implicated in the antioxidant defence and regulation of various metalloproteases (for example, angiotensin-converting enzyme, matrix metalloproteinases, and Cu/Zn-superoxide dismutase) associated with the healing process, playing a pivotal role in counteracting stress-induced detrimental changes(Reference Holmberg and Laurell269). In a study carried out on 125 HF patients and twenty-one healthy volunteers, serum Cu levels were significantly higher in HFpEF individuals compared with control (P<0·05). Additionally, serum Cu in patients with LVEF <40 % was significantly higher compared with both controls (P<0·001) and HFpEF patients (P = 0·003). Serum Zn was significantly lower in acute HF (P<0·001) and HF (P = 0·039) compared with control. Serum Cu was increased both in acute chronic HF and correlated with LV systolic and diastolic function. Serum Zn, in contrast, was decreased both in acute HF and chronic HF and independently predicted by clinical status and LV diastolic function(Reference Alexanian, Parissis and Farmakis270).
Selenium
Severe Se deficiency is an accepted cause of reversible HF, a condition known as Keshan disease(Reference Ge and Yang271). A number of studies have suggested an association between less severe Se deficiency or suboptimal Se status and HF; the majority suggest that patients with HF tend to have lower circulating levels when compared with individuals free from the condition. De Lorgeril et al. (Reference De Lorgeril, Salen and Accominotti272) observed a significant positive correlation between blood Se concentration and bicycle exercise stress test maximal oxygen uptake in twenty-one patients with HF (r 0·9; P = 0·0005), suggesting an association between Se status and physical functioning in HF. In addition, in the study by Alehagen et al. (Reference Alehagen, Alexander and Aaseth273) regarding healthy elderly Swedish municipality members, cardiovascular mortality was higher in the subgroup with the lower Se concentrations <65 μg/l in comparison with those having an Se concentration >85 μg/l. A 5-year prospective randomised double-blind placebo-controlled trial among Swedish citizens aged 70 to 88 years was performed in 443 participants given combined supplementation of Se and CoQ10 or a placebo. During a follow-up time of 5·2 years a significant reduction in cardiovascular mortality was found in the active treatment group v. the placebo group (5·9 v. 12·6 %; P = 0·015). NT-proBNP levels were significantly lower in the active group compared with the placebo group (mean values: 214 v. 302 ng/l at 48 months; P = 0·014). In echocardiography a significant better cardiac function score was found in the active supplementation compared with the placebo group (P = 0·03)(Reference Alehagen, Johansson and Björnstedt274).
In summary, patients with HF tend to have lower circulating levels of Se and Zn and higher levels of Cu, when compared with healthy individuals; however, the prognostic importance and clinical relevance still need to be investigated(Reference Da Cunha, Albanesi Filho and da Cunha Bastos275,Reference Malek, Dvorak and Jiresova276) .
Riboflavin and pyridoxine
Riboflavin and pyridoxine levels were measured in 100 patients admitted to a hospital with HF, and fifty age- and sex-matched controls. The percentage of individuals with evidence of riboflavin deficiency (erythrocyte glutathione reductase activity coefficient >1·2) was significantly higher in patients with HF compared with controls (27·0 v. 2·2 %; P<0·001). At the same time, the percentage of patients with pyridoxine deficiency (plasma pyridoxine concentration <4·9 ng/ml (<20 nmol/l)) was also significantly higher in patients with HF (38·0 v. 19·0 %; P = 0·02)(Reference Keith, Walsh and Darling277). The clinical relevance of these findings has yet to be clarified.
Vitamin B12
Observational studies investigating vitamin B12 status in the setting of HF have demonstrated conflicting results. Some authors suggest no correlation between LVEF and vitamin B12 or folate concentration, in 349 patients undergoing coronary angiography and left ventriculography(Reference Herzlich, Lichstein and Schulhoff278,Reference Witte, Desilva and Chattopadhyay279) . In a larger study carried out on 987 HF patients, vitamin B12 levels were weakly negatively correlated with LVEF (r –0·1; P = 0·015) and weakly positively correlated with NT-proBNP (r 0·1; P = 0·004). In addition, mean serum vitamin B12 concentration increased significantly with increasing NYHA class. No relationship was observed between folate status and markers of HF severity(Reference Herrmann, Muller and Kindermann280). An open study without placebo control by Andersson et al. (Reference Andersson, Edvinsson and Edvinsson281) evaluated treatment with pyridoxine (3 mg/d), folate (0·8 mg/d) and vitamin B12 (0·5 mg/d) for 6 weeks in fourteen elderly patients with HF. Treatment was associated with a significant increase in mean plasma folate concentration, but this was not associated with any significant change in levels of NT-proBNP. However, a significant decrease in mean arterial blood pressure and heart rate was observed (95·8 to 90·2 mmHg and 75·0 to 70·0 beats per min, respectively; P<0·05 for both)(Reference Andersson, Edvinsson and Edvinsson281). Further studies are still necessary in HF settings.
Discussion and future directions
In the times of a huge number of supplements and natural products available on the market, it is necessary and critical to present data only for those that have any evidence-based medical data on efficacy and safety in different conditions, such as lipid disorders(Reference Cicero, Colletti and Bajraktari282,Reference Banach, Patti and Giglio283) , hypertension(Reference Sosnowska, Penson and Banach284), the metabolic syndrome(Reference Patti, Al-Rasadi and Giglio285) and CVD, including its complication – HF. Only then, remembering that nutraceuticals might be considered only as an addition to optimal therapy, we will avoid overusing these, very often useless preparations; on the other hand, we might help in optimising the applied pharmacotherapy.
Based on the available data it was suggested that a low intake of some nutraceuticals, vitamins and minerals has been associated with an increased risk of developing HF, and supplementation with some nutraceuticals has been reported to improve different parameters related to HF, even on top of guideline-driven conventional treatment. Nutraceutical components with a potential impact on HF have not yet been deeply investigated. For instance, amino acids (AA) are important constituents of muscle proteins and exert many activities as regulators of both myocardium protein turnover(Reference Bing, Siegel and Ungar286,Reference Young, McNulty and Morgan287) and energy metabolism(Reference Rosenkranz, Okamoto and Buckberg288). The heart’s reliance on AA increases during HF because of high myocardium anabolic activity and cardiomyocyte energy shortage(Reference Neubauer, Horn and Cramer289). Thus, it has been supposed that the lack of AA intake or absorption could further impair myocardium metabolism in HF patients. In a study involving forty-one patients with clinically stable HF (NYHA class II to IV), compared with controls, HF patients had reduced arterial AA levels, of which both their number and reduced rates were associated with HF severity. Arterial aspartic acid correlated with stroke volume index (r 0·626; P<0·0001) and cardiac index (r 0·424; P = 0·0028). The content of arterial aspartic acid (µmol/l) multiplied by the cardiac index was associated with LVEF (r 0·377; P = 0·0076). All NYHA groups had adequate protein intake (≥1·1 g/kg per d) and inadequate energy intake (kJ < resting energy expenditure (REE) × 1·3) was found only in class IV patients. The study showed that HF patients had reduced arterial AA levels directly related to clinical disease severity and LV dysfunction(Reference Aquilani, La Rovere and Corbellini290). However, no intervention trial, similarly to other studies with nutraceuticals in HF settings, has yet demonstrated that AA supplementation could have a positive impact on HF.
It has also to be investigated if a combination of nutraceuticals could exert a more relevant impact on HF-related parameters compared with supplementation with a single dietary factor. To date, two studies have measured the effect of a combination multivitamin–multimineral supplement on patients with HF. In the first study, forty-one patients (LVEF ≤40 %) undergoing coronary artery bypass graft were randomised to receive a nutritional supplement containing vitamins, minerals, CoQ10 (150 mg/dose) and carnitine (3 g/dose) or placebo. Treatment with the nutritional supplement was associated with a significant fall in mean LV end-diastolic volume (170·5 ml to 158·9 ml; P<0·05)(Reference Jeejeebhoy, Keith and Freeman291). Witte et al. (Reference Witte, Nikitin and Parker163) observed similar results in a trial involving thirty-three HF subjects randomised to receive a multivitamin–multimineral supplement containing coenzyme Q10 (150 mg/dose) or placebo. After 1 year of treatment, serum levels of folate and vitamin B12 increased significantly in the micronutrient group. This was associated with a significant increase in mean LVEF (25·6 to 30·9 %; P<0·05) and a significant increase in mean QoL HF-related tool score (64·4 to 73·9 %; P<0·05), suggesting a significant QoL improvement(Reference Witte, Nikitin and Parker163). Both studies were, however, very small, and so it cannot be concluded that this approach would be a positive suggestion. In fact, this is another main limitation of available data on nutraceutical application in HF patients (relatively small studies, mainly with short-term follow-up), so in most of the cases it is impossible to draw any reliable recommendations.
In conclusion, a growing body of clinical evidence suggests that the intake of adequate dosages of some nutraceuticals (hawthorn extract, CoQ10, l-carnitine, d-ribose, carnosine, vitD, probiotics, n-3 PUFA and beet nitrates) might be associated with improvements in self-perceived QoL and/or functional parameters such as EF, stroke volume and cardiac output in HF patients, with minimal or no side effects. Those benefits tended to be greater in earlier HF stages. Efficacy on at least one major cardiovascular event is documented in larger RCT for CoQ10 (Q-SYMBIO trial, 2 years, 420 patients), hawthorn (SPICE trial, 1 year, 2681 patients), n-3 PUFA (GISSI-HF, 3·9 years; 6975 patients). For the rest of the above-mentioned nutraceuticals, we still require longer studies and data on their efficacy from the Cardiovascular Outcome Trial. In no case can the use of nutraceuticals be substituted for the consolidated pharmacological treatment of HF.
Acknowledgements
There is no funding to declare.
A. F. G. C. has given talks, furnished scientific consultancies, and/or participated in trials sponsored by Amgen, Angelini, Boehringer Ingelheim, Meda, Menarini, Merck Sharp & Dohme, and Sanofi-Synthelabo.
S. v. H. has been a paid consultant to Vifor Pharma, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Brahms, Chugai Pharma, Roche and Novartis.
D. V. has given talks and attended conferences sponsored by Bristol-Myers Squibb/Pfizer, Novartis, Servier, Amgen, Bayer and AstraZeneca; and has received speaker fees from Pfizer, Novartis, Servier, Bayer, AstraZeneca and Terapia.
Tomasz Tomasik has received personal fees from Eli Lilly Polska, Boehringer Ingelheim, Novaris and Shire.
Ž. R. has received honoraria from Sanofi-Aventis.
Peter Penson owns four shares in AstraZeneca PLC. He has received honoraria and/or travel grants.
M. B. has served on the speakers’ bureau of Abbott/Mylan, Abbott Vascular, Actavis, Akcea, Amgen, Biofarm, KRKA, Merck Sharp & Dohme, Sanofi, Servier and Valeant. He has served as a consultant to Abbott Vascular, Akcea, Amgen, Daiichi-Sankyo, Esperion, Lilly, Merck Sharp & Dohme, Resverlogix and Sanofi, and and has received grants from Sanofi and Valeant.
This review has the official endorsement of: Argentina Lipid Society; Association of Cardiologists of the Republic of Uzbekistan; Association of Preventive Pediatrics of Serbia; Baltic Society of Atherosclerosis; Chinese Atherosclerosis Society; College of Family Physicians in Poland; Croatian Atherosclerosis Society; Czech Society for Atherosclerosis; Egyptian Association for Endocrinology, Diabetes and Atherosclerosis (EAEDA); Egyptian Association of Vascular Biology and Research (EAVA); Estonian Society of Hypertension; Emirates Cardiac Society; Emirates Society of Cardiology; French Atherosclerosis Society; German Atherosclerosis Society; Hellenic Atherosclerosis Society; Hellenic Lipidology Society; Hellenic Society of Lipidology, Atherosclerosis and Vascular Disease; Hungarian Atherosclerosis Society; International Natural Product Sciences Taskforce (INPST); Israeli Society for Treatment and Prevention of Atherosclerosis; Italian Nutraceuticals Society (SINut); Italian Society for Cardiovascular Prevention; Kosovo Society of Cardiology; Kyrgyz Atherosclerosis Society; Latvian Society of Cardiology; Lipid and Atherosclerosis Society of Southern Africa; Lipid and Blood Pressure Meta-analysis Collaboration (LBPMC) Group; Lipid Association of India; Lithuanian Heart Association; Mighty Medic; New French Atherosclerosis Society (NSFA); Polish Lipid Association (PoLA); Romanian National Forum for Prevention; Romanian Society of Cardiology; Russian National Atherosclerosis Society; Saudi Group for Cardiovascular Prevention and Rehabilitation; ScreenPro-FH; Serbian Association for Arteriosclerosis, Thrombosis and Vascular Biology Research (SAATVBR); Slovak Atherosclerosis Association; Slovenian Society of Cardiology; Society of Endocrine and Metabolic Disease of South Africa; Society on Sarcopenia, Cachexia and Wasting Disorders; South African Heart Association; Swiss Society for Familial forms of Hypercholesterolemia (SSFH); Taiwan Society of Lipids and Atherosclerosis; Tunisian Association of Study and Research on Atherosclerosis; Ukrainian Atherosclerosis Society; Venezuelan Society of Atherosclerosis; Very Large Database of Lipids (VLDL).
Appendix
International Lipid Expert Panel experts (alphabetically) are:
Fahad Alnouri (Cardiovascular Prevention Unit, Adult Cardiology Department, Prince Sultan Cardiac Center, Riyadh, Saudi Arabia)
Fahmy Amara (Unit of Diabetes & Metabolism, Alexandria University, Alexandria, Egypt)
Atanas G. Atanasov (Institute of Genetics and Animal Breeding of the Polish Academy of Sciences, Jastrzebiec, Poland; Department of Pharmacognosy, University of Vienna, Vienna, Austria; Institute of Neurobiology, Bulgarian Academy of Sciences, Sofia, Bulgaria)
Gani Bajraktari (Institute of Public Health and Clinical Medicine, Umeå University, Umeå, Sweden; Clinic of Cardiology, University Clinical Centre of Kosova, Prishtina, Kosovo; Medical Faculty, University of Prishtina, Prishtina, Kosovo)
Marcin A. Bartlomiejczyk (Department of Hypertension, Medical University of Lodz, Poland; Polish Mother’s Memorial Hospital Research Institute (PMMHRI), Lodz, Poland)
Bojko Bjelakovic (Clinic of Pediatrics, Clinical Center, Nis, Faculty of Medicine, University of Nis, Serbia)
Eric Bruckert (Pitié-Salpêtrière Hospital and Sorbonne University, Cardio Metabolic Institute, Paris, France)
Alberto Cafferata (Facultad de Medicina, Instituto Universitario de Ciencias de la Salud, Fundación H.A. Barceló, Argentina)
Richard Ceska (Third Department of Medicine, Department of Endocrinology and Metabolism of the First Faculty of Medicine, Charles University and General University Hospital, Prague, Czech Republic)
Xavier Collet (Institute of Metabolic and Cardiovascular Diseases, Inserm, Toulouse, France)
Olivier Descamps (Department of Internal Medicine, Centres Hospitaliers Jolimont, Haine Saint-Paul, Belgium; Department of Cardiology, Cliniques Universitaires Saint-Luc, Bruxelles, Belgium)
Nair Devaki (Department of Clinical Biochemistry, the Royal Free London NHS Foundation Trust, Pond Street, London, UK)
Dragan Djuric (Institute of Medical Physiology “Richard Burian”, Faculty of Medicine, University of Belgrade, Belgrade, Serbia)
Ronen Durst (Cardiology Department, Hadassah Hebrew University Medical Center, Ein Kerem, Jerusalem, Israel)
Marat V. Ezhov (National Cardiology Research Center, Moscow, Russia)
Zlatko Fras (Preventive Cardiology Unit, Department of Vascular Medicine, Division of Medicine, University Medical Centre Ljubljana, Slovenia; Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia)
Dan Gaita (Institutul de Boli Cardiovasculare, Universitatea de Medicina si Farmacie Victor Babes din Timisoara, Romania)
Adrian V. Hernandez (Health Outcomes, Policy, and Evidence Synthesis (HOPES) Group, University of Connecticut/School of Pharmacy, Storrs, CT, USA; Vicerrectorado de Investigación, Universidad San Ignacio de Loyola (USIL), Lima, Peru)
Steven R. Jones (The Johns Hopkins Ciccarone Center for the Prevention of Heart Disease, Baltimore, MD, USA)
Jacek Jozwiak (Department of Family Medicine and Public Health, Faculty of Medicine, University of Opole, Opole, Poland)
Nona Kakauridze (Department of Internal Medicine, Faculty of Medicine, Tbilisi State Medical University, Tbilisi, Georgia)
Niki Katsiki (Second Department of Propaedeutic Internal Medicine, Medical School, Aristotle University of Thessaloniki, Hippocration Hospital, Thessaloniki, Greece)
Karam Kostner (Mater Hospital, University of Queensland, St Lucia, QLD, Australia)
Raimondas Kubilius (Department of Rehabilitation, Medical Academy, Lithuanian University of Health Sciences, Kaunas, Lithuania)
Gustavs Latkovskis (Institute of Cardiology and Regenerative Medicine, Faculty of Medicine, University of Latvia, Riga, Latvia; Pauls Stradins Clinical University Hospital, Riga, Latvia)
G. B. John Mancini (Department of Medicine, Division of Cardiology, University of British Columbia, Vancouver, British Columbia, Canada)
A. David Marais (Chemical Pathology Division of the Department of Pathology, University of Cape Town Health Science Faculty, Cape Town, South Africa)
Seth S. Martin (Ciccarone Center for Prevention of Heart Disease, Division of Cardiology, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA)
Julio Acosta Martinez (Medico Cardiologo de la Policlinica Metropolitana, Caracas, Venezuela)
Mohsen Mazidi (Department of Twin Research and Genetic Epidemiology, King’s College London, St Thomas’ Hospital, Strand, London, UK)
Erkin Mirrakhimov (Kyrgyz State Medical Academy, named after Akhunbaev I.K., Bishkek, Kyrgyzstan)
Andre R. Miserez (Diagene Research Institute, Reinach, Switzerland; President of Swiss Society of Familial Forms of Hypercholesterolemia (SSFH), Breitenbach, Switzerland; University of Basel, Basel, Switzerland)
Olena Mitchenko (Dyslipidaemia Department, Institute of Cardiology AMS of Ukraine, Ukraine; Ukrainian Atherosclerosis Society)
Patrick R. Moriarty (Division of Clinical Pharmacology, Division of Internal Medicine, University of Kansas Medical Center, Kansas City, KS, USA)
Demosthenes B. Panagiotakos (School of Health Science and Education, Department of Nutrition and Dietetics, Harokopio University of Athens, Athens, Greece)
György Paragh (Department of Internal Medicine, Faculty of Medicine, University of Debrecen, Debrecen, Hungary)
Daniel Pella (1st Department of Internal Medicine, Faculty of Medicine, Pavol Jozef Safarik University, Košice, Slovakia)
Peter Penson (School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Liverpool, UK)
Zaneta Petrulioniene (Vilnius University Faculty of Medicine, Vilnius, Lithuania; Vilnius University Hospital Santaros Klinikos, Vilnius, Lithuania)
Matteo Pirro (Department of Medicine, University of Perugia, Perugia, Italy)
Arman Postadzhiyan (Bulgarian Society of Cardiology, Medical University of Sofia, Sofia, Bulgaria)
Raman Puri (Indraprastha Apollo Hospital, New Delhi, India)
Ashraf Reda (Menoufia University NLI of FHSC, President of EAVA)
Jemaa Riadh (Laboratory of Biochemistry, Faculty of Medicine of Tunis, Rabta Hospital, University of Tunis El Manar, Tunis, Tunisia)
Dimitri Richter (Cardiac Department, Euroclinic, Athens, Greece)
Manfredi Rizzo (Biomedical Department of Internal Medicine and Medical Specialties, University of Palermo, Palermo, Italy)
Massimiliano Ruscica (Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy)
Naveed Sattar (Institute of Cardiovascular and Medical Sciences, University of Glasgow, Glasgow, UK)
Maria-Corina Serban (Department of Functional Sciences, Discipline of Pathophysiology, “Victor Babes” University of Medicine and Pharmacy, Timisoara, Romania)
Abdulla M. A Shehab (Medical Education Department, United Arab Emirates University, Al Ain, United Arab Emirates)
Aleksandr B. Shek (Republican Specialised Center of Cardiology, Tashkent, Uzbekistan)
Cesare R. Sirtori (Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy)
Claudia Stefanutti (Department of Molecular Medicine, Sapienza University of Rome, Rome, Italy)
Tomasz Tomasik (Department of Family Medicine, Chair of Internal Medicine and Gerontology, Jagiellonian University Medical College, Krakow, Poland)
Margus Viigimaa (Tallinn University of Technology, North Estonia Medical Centre, Tallinn, Estonia)
Branislav Vohnout (Institute of Nutrition, Faculty of Nursing and Health Professional Studies and Coordination Centre for Familial Hyperlipoproteinemias, Slovak Medical University in Bratislava, Bratislava, Slovakia; Institute of Epidemiology, School of Medicine, Comenius University, Bratislava, Slovakia)
Michal Vrablik (1st Faculty of Medicine, Charles University and General University Hospital, Prague, Czech Republic)
Nathan Wong (Department of Medicine, School of Medicine University of California, Irvine, CA, USA; Heart Disease Prevention Program, Division of Cardiology, University of California, Irvine, CA, USA)
Hung-I Yeh (Department of Medicine, Mackay Medical College, Taipei, Taiwan; Cardiovascular Division, Department of Internal Medicine, MacKay Memorial Hospital, Taipei, Taiwan)
Jiang Zhisheng (Institute of Cardiovascular Disease, University of South China, Hengyang, Hunan, China)
Andreas Zirlik (University Heart Centre Freiburg University, Department of Cardiology and Angiology I, Faculty of Medicine, University of Freiburg, Freiburg, Germany)





 Open access
Open access