



Introduction
Gastroenteritis causes the second greatest burden of all infectious diseases worldwide. The brunt of severe outcomes of diarrhoeal disease falls in low-income countries [Reference Lozano1]. Globally, almost one-fifth of all cases of acute gastroenteritis are norovirus-induced and an estimated 212 000 annual deaths are norovirus-related [Reference Pires2]. Similarly to noroviruses, sapoviruses are detected worldwide and are ranked second to fourth as the major viral pathogens among patients with sporadic gastroenteritis [Reference Oka3].
Sapoviruses and noroviruses belong to the family of Caliciviridae, naked viruses with positive-sense single-stranded RNA genome. The genome of sapoviruses is organised into two open reading frames (ORF1 and ORF2). The large ORF1 encodes the non-structural proteins and the major virus capsid protein of the virus (VP1). The small ORF2 encodes the minor capsid protein (VP2) [Reference Oka3]. The genome of noroviruses is organised into three ORFs (ORF1–ORF3). The large ORF1 encodes the non-structural proteins. The smaller ORF2 and ORF3 encode the VP1 and the VP2, respectively [Reference Robilotti, Deresinski and Pinsky4].
Both caliciviruses affect humans and animals and show extensive genetic diversity. Based on the VP1 gene, sapoviruses and noroviruses are phylogenetically classified into 15 (GI–GXV) and nine (GI–GIX) genogroups, respectively. Each of which can be subdivided into genotypes [Reference Oka3, Reference Oka5, Reference Vinje6]. Human pathogenic sapovirus strains are assigned to four genogroups and belong to at least 18 genotypes (GI.1–GI.7, GII.1–GII.8, GIV.1, GV.1 and GV.2) [Reference Oka5]. Norovirus genogroup GI, GII, GIV, GVIII and GIX (formerly GII.15) infect humans. Within the most frequent human pathogenic norovirus genogroups, GII and GI, at least 22 and 9 genotypes have been detected, respectively [Reference Vinje6–Reference Pietsch8].
Several norovirus vaccines are currently in development [Reference Lucero, Vidal and O'Ryan9]. As acquired norovirus immunity is mainly genotype-specific [Reference Malm10], the success of vaccine formulations will substantially depend on the accurate selection of included norovirus genotypes. Thus, solid knowledge of norovirus diversity in the different areas of the world is indispensable. Infants and young children in low- and middle-income countries should be among the primary targets for anti-norovirus vaccination [Reference Lopman11]. However, information regarding the impact and epidemiology of noroviruses in African countries remains limited [Reference Mans12, Reference Magwalivha13]. As the clinical symptoms of norovirus and sapovirus gastroenteritis are indistinguishable [Reference Oka3], estimates of the impact of sapovirus infections are important to precisely define the role of norovirus in severe paediatric gastroenteritis.
To further our knowledge on the epidemiology of both caliciviruses in Sub-Saharan Africa, the present study aimed to exploit the stool positivity rate and genetic diversity of sapoviruses and noroviruses among outpatient infants and young children with diarrhoea in Northwest Ethiopia.
Materials and methods
Sample collection
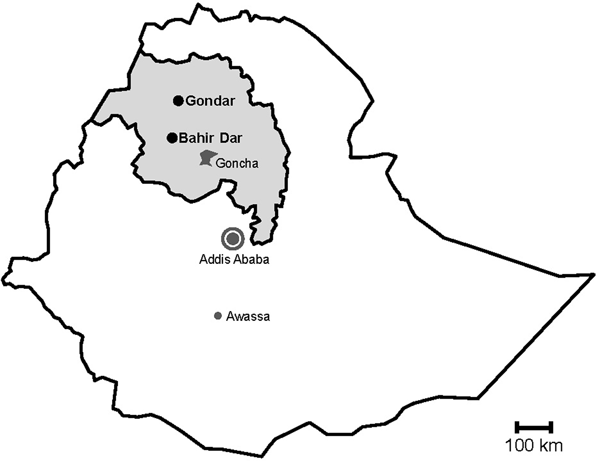
During rotavirus surveillance, a total of 450 faecal specimens were collected from outpatient children of <5 years of age with diarrhoea at two sampling sites, Gondar and Bahir Dar, in Northwest Ethiopia from November 2015 to April 2016 (Fig. 1) [Reference Gelaw, Pietsch and Liebert14].
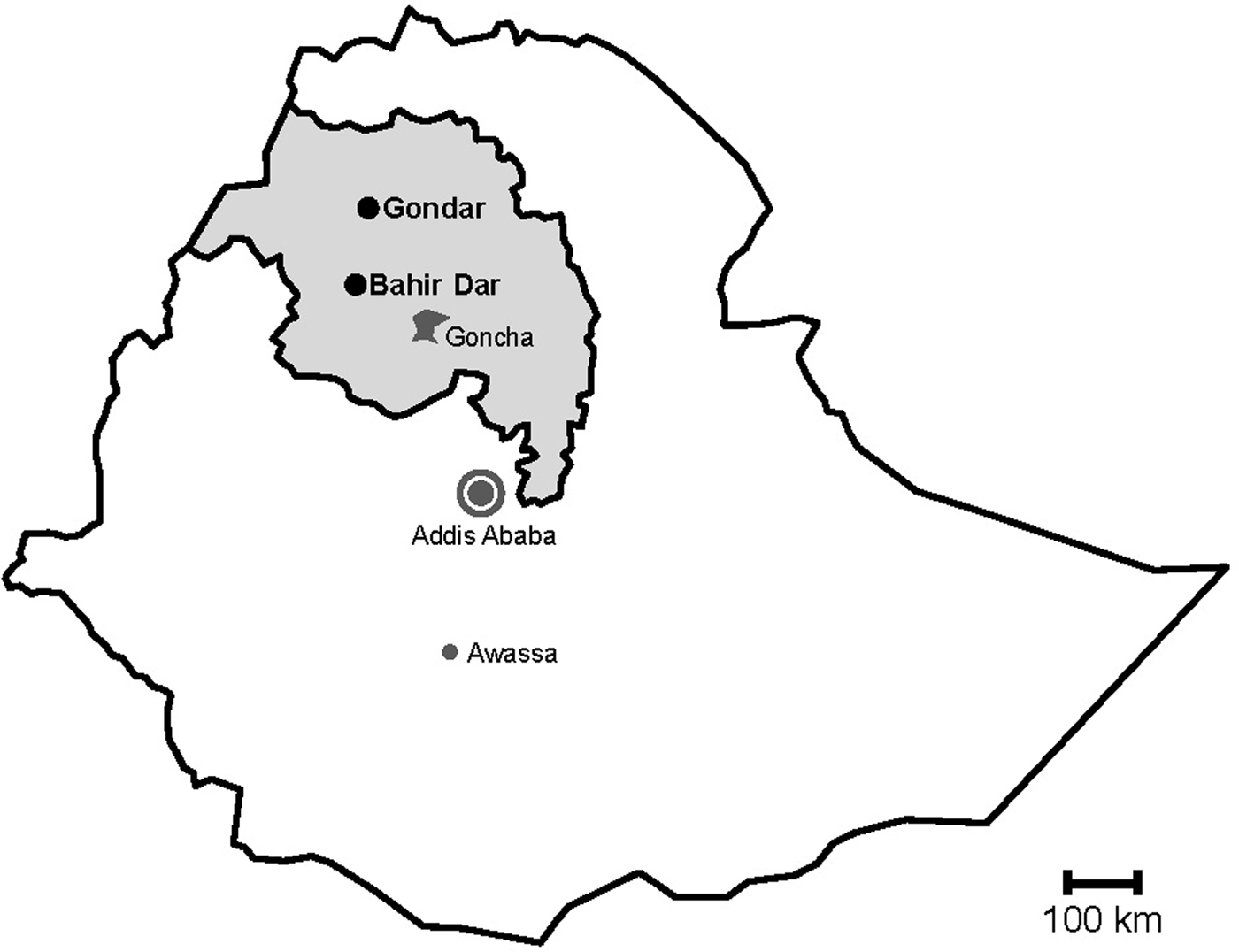
Fig. 1. Map of sampling sites. Stools were sampled in the cities Gondar and Bahir Dar, which are both situated in the Amhara National Regional State (grey) in Northwest Ethiopia. Previous data on sapoviruses and/or noroviruses are available from the nearby rural Goncha Siso Enese region, the capital Addis Ababa and the city of Awassa.
Viral detection and molecular characterisation
Nucleic acids were extracted from 10% stool suspensions in phosphate buffered saline (MagNA Pure 96 DNA and Viral NA Small Volume Kit, Roche, Germany) and screened for the presence of GI, GII, GIV and GV sapovirus and GI and GII norovirus RNA by real-time RT-PCR [Reference Pietsch8, Reference Hoehne and Schreier15–Reference Pietsch and Liebert18].
For sapovirus and norovirus capsid genotyping, partial VP1 genes were amplified (One-Step RT-PCR Kit, Hilden, Germany), purified (Wizard SV Gel and PCR Clean-Up System, Promega, Mannheim, Germany) and sequenced (BigDye Terminator v1.1 Cycle Sequencing kit and ABI 3500 Genetic Analyzer, PE Applied Biosystems, Foster City, CA) using different primer sets. A detailed overview of the primers and probes used in this study can be found in the online Supplementary Table S1. Sapovirus capsid genotypes were identified by phylogenetic analysis of 675–712 nucleotides of the N-terminal VP1 gene region following the recommended classification and nomenclature [Reference Oka19]. Norovirus capsid genotypes were identified based on 273–285 nucleotides of VP1 gene region C using Norovirus Typing Tool Version 2.0 [Reference Kroneman20]. Phylogenetic trees for sapoviruses and noroviruses were constructed based on nucleic acid sequence alignments with available reference GenBank accessions using Maximum Likelihood method in MEGA5 software. Bootstrap analysis was performed with 1000 replicates. The obtained sequences of the present sapovirus and norovirus strains have been deposited in GenBank database (GenBank accessions MK246651-MK246752).
Statistical analysis
Data were entred and analysed using the Statistical Package for the Social Sciences (SPSS) software version 16. Categorical variables were compared using chi-square and Fisher's exact tests. P values of <0.05 were regarded as statistically significant.
Ethical clearance
The Ethical Review Board of the University of Gondar approved the research protocol. Written informed consent was obtained from the parents or guardians of the children.
Results
Sapovirus and norovirus RNA stool positivity
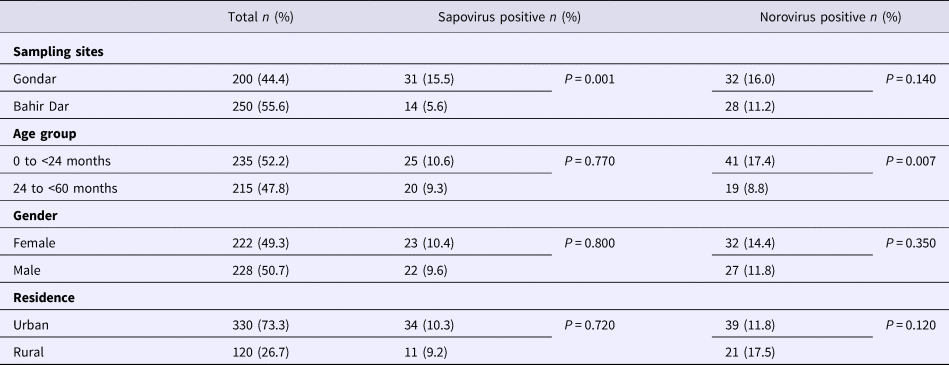
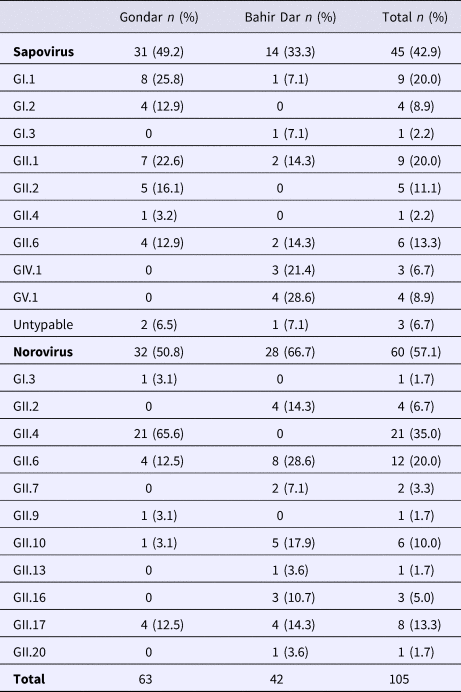
Real-time RT-PCR showed sapovirus RNA in 45 (10.0%, 95% CI 7.6–13.1%) and norovirus RNA in 60 (13.3%, 95% CI 10.5–16.8%) stool samples, respectively. In three stool samples, co-presence of both caliciviruses was shown. Mixed infections with wildtype rotavirus A were identified in 13 (27.2%) sapovirus and 11 (18.3%) norovirus positive stool samples. Whereas sapovirus RNA was more frequently detected in samples from Gondar (15.5%) than from Bahir Dar (5.6%) (P = 0.001), the norovirus stool positivity rate was not significantly different between the two sampling sites. Regarding the age of children, norovirus stool positivity rate was higher in children aged <2 years (17.4%, P = 0.007) than in children within the third to the fifth year of age (8.9%). In contrast, sapovirus stool positivity rate did not vary across age. There was neither association between sapovirus or norovirus RNA stool positivity and children's sex, nor residence, nor co-infection with wildtype rotaviruses (Table 1).
Table 1. Sapovirus and norovirus positivity of stool samples
Genotypic distribution and phylogenetic analysis of sapoviruses
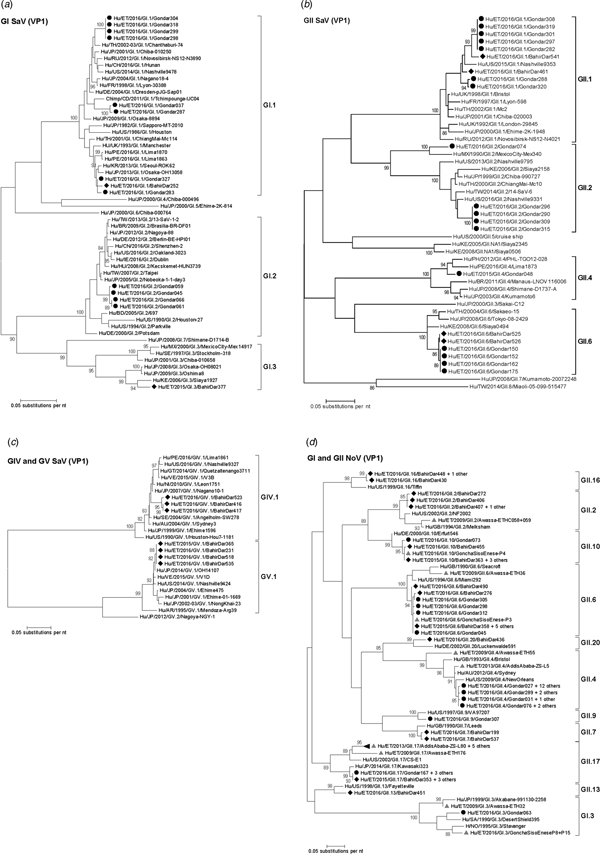
Amplification of VP1 and subsequent genotyping was successful in 42 (93.3%) sapovirus positive stool samples. Overall, sapovirus RNA was detected throughout the 6 months of the sampling period (Fig. 2A). Sapovirus strains of all four known human pathogenic genogroups, GI, GII, GIV and GV, were present in Bahir Dar. In contrast, only GI and GII sapoviruses were detected in Gondar. At genotype level, diversity was high at both sampling sites and included nine different genotypes (Table 2). In GI.1, GII.1 and GII.2 genotypes, an additional intra-genotypic diversity was shown by phylogenetic analysis (Fig. 3A and B). In contrast, only a single strain or very closely related strains were detected for GI.2, GI.3, GII.4, GII.6, GIV.1 and GV.1 sapoviruses (Fig. 3).
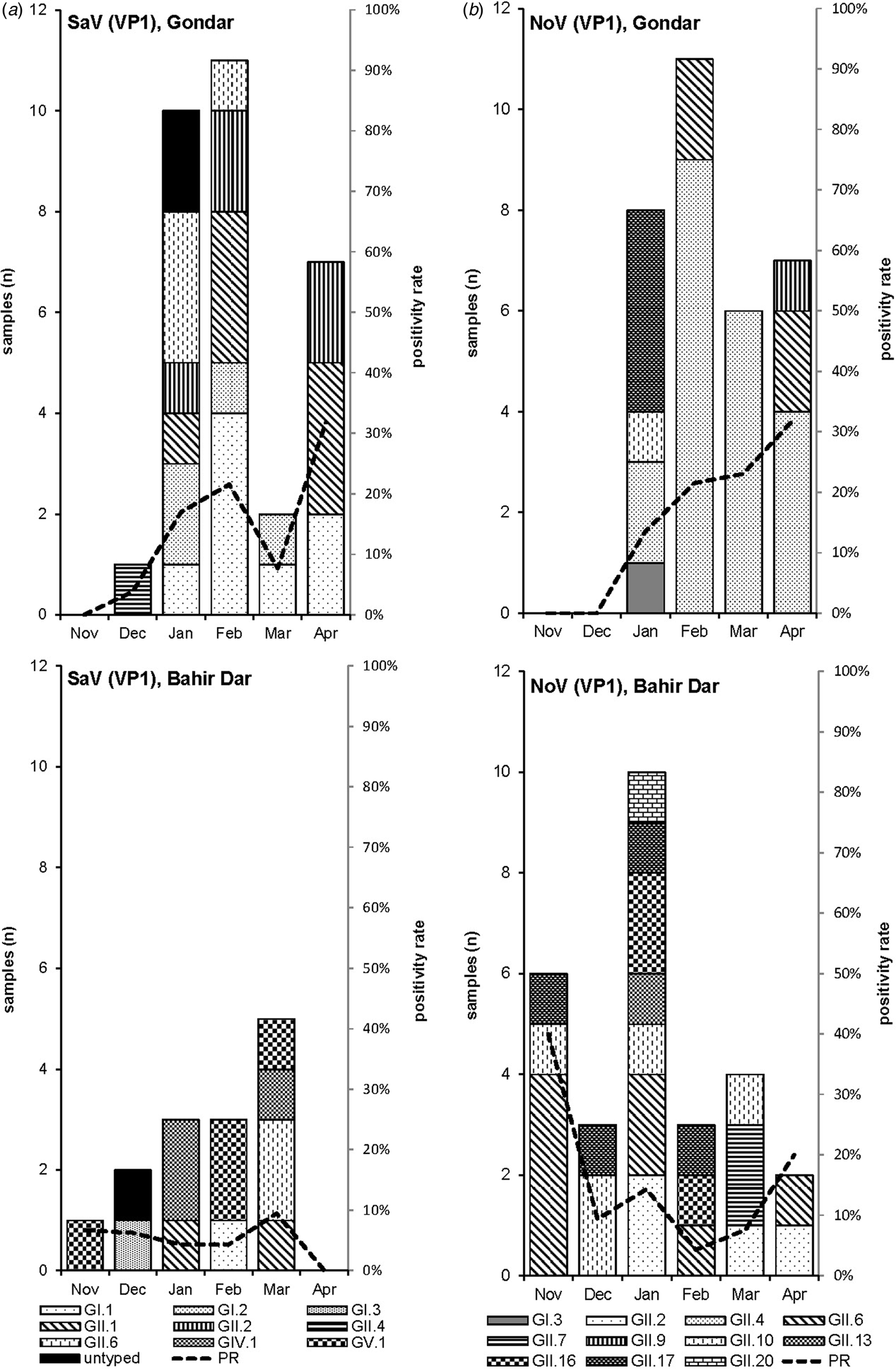
Fig. 2. Monthly positivity rate of stool samples (PR) and distribution of sapovirus (A) and norovirus (B) genotypes in Gondar and Bahir Dar.
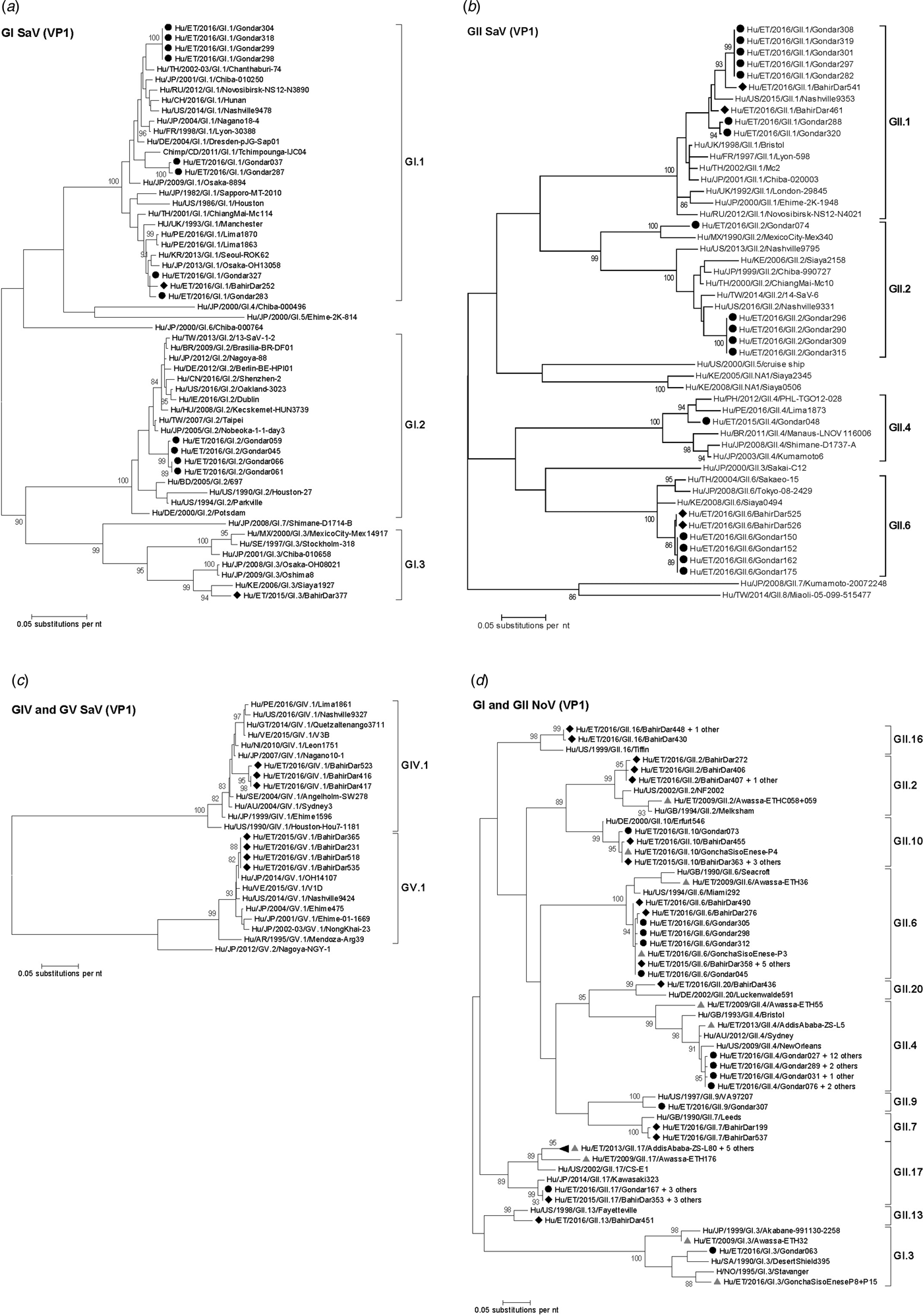
Fig. 3. Phylogenetic analysis of sapoviruses (A, B and C) and noroviruses (D) based on Maximum Likelihood estimations of partial VP1 gene nucleic acid sequences. Numbers at each node represents bootstrap values obtained with 1000 replicates (>80 are shown). Strains from the present study are labelled with a black circle (Gondar) and black diamond (Bahir Dar). Grey triangles indicate previously reported strains from Ethiopia.
Table 2. Sapovirus and norovirus genotypes per sampling site
The intra-genotypic analysis of the different GII.2 strains revealed phylogenetic clustering of one of the present Ethiopian GII.2 strains, Hu/ET/2016/GII.2/Gondar074, with strain Hu/MX/1990/GII.2/MexicoCity-Mex340 and 93.2% nucleic acid identity between the two strains (Fig. 3B). The cluster of the present Ethiopian and the Mexican strain was clearly separated from all other available GII.2 sequences with sufficient lengths and showed low nucleic acid identity with all of them (82.6–84.2%). In comparison to shorter GII.2 sequences, the highest nucleotide identity reached a similar level (85.1%) and was shown for sapovirus strain Hu/RU/2002/GII.2/Moscow-2240-RF.
Only four sufficiently long VP1 sequences of African sapovirus strains with the same genotypes as detected in the present study were available. Three of the strains clustered very closely with present Ethiopian strains. First, strain Chimp/CD/2011/GI.1/Tchimpounga from a chimp living in close contact to humans in Republic of Congo clustered with two present GI.1 sequences from Gondar. Second, strain Hu/KE/2006/GI.3/Siaya1927 from Kenya was phylogenetically related to the present GI.3 strain from Bahir Dar (Fig. 3A). Third, strain Hu/KE/2008/GII.6/Siaya0494 from Kenya was related to all present GII.6 strains. The fourth strain from the African region, Hu/KE/2006/GII.2/Siaya2158 from Kenya, fell into the same cluster as most GII.2 strains of the present study, but was not very closely related (Fig. 3B). The two available VP1 sequence fragments of GII.1 sapovirus strains from Ethiopia, Hu/ET/2016/GII.1/GonchaSisoEnese-P5 and -P19, were too short to be included into phylogenetic analyses and were most similar (97.5% and 98.2%) to the present strain Hu/ET/2016/GII.1/BahirDar461.
Genotypic distribution and phylogenetic analysis of noroviruses
Genotyping was successful in all norovirus positive stool samples. Within noroviruses, GII genogroup strains prevailed (98.3%). In total, eleven distinct norovirus genotypes were identified, including rare GII.9, GII.10 and GII.20 strains. At both sampling sites, norovirus genotypes were diverse. Different noroviruses, but no GII.4 strains, were detected in Bahir Dar. In Gondar, norovirus detections were only detected from January to April 2016 and were predominantly GII.4 genotypes (Fig. 2B, Table 2).
All present GII.4 sequences were identified as GII.4 Sydney 2012 variants by Norovirus Typing Tool and formed a phylogenetic cluster distinct from GII.4 strains detected in 2009 and 2013 in the Ethiopian cities Addis Ababa and Awassa (Fig. 3D). Comparison to a more recent GII.4 norovirus, Hu/ET/GII.4/2016/GonchaSisoEnese-P11, was not possible, as the available sequencing data of the VP1 gene region C was too short. The same applied for GII.9. Similarly to GII.4, the present GII.2 and GII.17 strains showed no relatedness to noroviruses detected in 2009 and 2013 in Ethiopia. Neither did the present GI.3 norovirus strain cluster with GI.3 sequences reported from Awassa in 2009 or Northwest Ethiopia in 2016. However, GII.6 and GII.10 sequences obtained during a recent study in Northwest Ethiopia were closely related to GII.6 and GII.10 strains of the present stool samples (Fig. 3D).
Discussion
The present study underlines the role of norovirus and sapovirus infections in acute gastroenteritis in Eastern Africa and shows calicivirus RNA positivity in about one-quarter of stool samples from Ethiopian infants and young children with diarrhoea. This value is similar to the lately assessed positivity rate of wildtype rotaviruses in the same sample set [Reference Gelaw, Pietsch and Liebert14]. Due to the recent introduction of anti-rotavirus vaccines in Ethiopia, the relative role of norovirus and sapovirus-related diarrhoea cases is likely to increase in the future [Reference Pitkanen, Vesikari and Hemming-Harlo21, Reference Santos22]. The high number of calicivirus and rotavirus co-infections indicates frequent exposure to viral enteric pathogens, which may be shed for weeks after acute infection [Reference Atmar23–Reference Mukhopadhya25].
The present study provides the first comprehensive human sapovirus genotyping data from Ethiopia. Consistent with previous reports from Sub-Saharan Africa [Reference Shioda26–Reference Page28], sapovirus RNA was detected in one out of ten present stool samples. Interestingly, sapovirus stool positivity rate significantly differed between Gondar and Bahir Dar. The detected sapovirus strains belonged to a variety of genotypes at both sampling sites and showed a fluctuating monthly distribution. These results point to local co-circulation of different sapovirus strains and frequent sporadic infections in the area. Notably, the observed genetic diversity may well remain underestimated as (i) three detected sapoviruses could not be typed and (ii) the used real-time RT-PCR approach has very recently been shown to fail to detect some sapovirus genotypes [Reference Oka29].
In line with the present results, diversity of sapoviruses in Sub-Saharan Africa was shown to be high in previous studies [Reference Matussek30, Reference Murray31]. But sapovirus sequencing data from the African region remains limited. Accordingly, previously reported human sapovirus sequences from Ethiopia are restricted to one partial polymerase gene [Reference Sisay32] and GII.1 and GII.2 sapovirus genome fragments obtained from three stool sample pools [Reference Altan33]. As many published VP1 sequences from the African region are short or scattered within the VP1 gene, the present phylogenetic analysis was mainly based on VP1 sequencing data from other continents and additionally included strains from Kenya and Republic of Congo. Notably, wherever data of the present genotypes from the African region was available, at least one of the present sapoviruses from Ethiopia clustered very closely or nearby. Whereas most of the present sapovirus variants were similar to recent strains, sapovirus Hu/ET/2016/GII.2/Gondar074 was phylogentically distant and related to an orphan GII.2 strain detected in Mexico in 1990 [Reference Farkas34]. Apparently, this GII.2 sapovirus variant is rare, or may alternatively be rarely identified by commonly used detection and/or genotyping approaches.
Noroviruses were detected at a similar frequency as sapoviruses in the present stool samples. Their stool positivity rate was highest in infants and children aged <2 years, which is consistent to recently reviewed data from other low-income countries [Reference Mans12, Reference Shioda35, Reference O'Ryan, Riera-Montes and Lopman36]. In agreement with previous reports from Ethiopia [Reference Sisay32, Reference Yassin37] and reviewed data from Sub-Saharan Africa [Reference Munjita38], genotype diversity was high and GII noroviruses prevailed. High norovirus diversity and recurrent norovirus infections early in life have also been shown in a recent seroepidemiological study from an Eastern African country Uganda, which identified almost 95% positivity and responses to multiple human norovirus genotypes by age 2 years [Reference Thorne39].
For Ethiopia, nucleic acid sequences of six genogroup GI (GI.1, GI.3, GI.4, GI.5, GI.6 and GI.7) and nine genogroup GII genotypes (GII.2, GII.3, GII.4, GII.6, GII.9, GII.10, GII.12, GII.14 and GII.17) have been reported in human stool samples [Reference Sisay32, Reference Altan33, Reference Yassin37]. The present study confirmed seven of those genotypes (GI.3, GII.2, GII.4, GII.6, GII.9, GII.10 and GII.17) and furthermore adds GII.7, GII.13, GII.16 and GII.20 noroviruses. Notably, GII.3 strains considered epidemiologically important in Ethiopia previously [Reference Yassin37], were not detected in the present stool samples. Strain diversity might be even higher, as polymerase genotypes and therefore norovirus recombinant strains were not assessed in the present study. Intragenotypic phylogenetic relatedness of the present strains with noroviruses published earlier from the country was low, except for GII.6 and GII.10 genotypes reported from Goncha Siso Enese region in 2016 [Reference Altan33], i.e. reported during the same time at a nearby sampling site.
These results point to a dynamic and diverse norovirus population within Ethiopia, which possibly reflects an ongoing global circulation of human noroviruses resulting in a frequent import of pandemic strains. Such a rapid global spread was recently described for a novel GII.17 strain [Reference de Graaf40], which was also detected in stools from both present sampling sites. It is important to be aware, that studies with a limited number of sampling sites, short sampling periods and small sample sets may only capture fragmentary snapshots of the true genetic diversity in a country. Local circulation patterns may not reflect the nation-wide circulation of the strains. The possible bias by selection of sampling sites can be well illustrated by the results from the present two proximate sampling sites Gondar and Bahir Dar. Whereas noroviruses in stool samples from Gondar were predominated by the globally most prevalent genotype GII.4, this genotype was not detected at all in samples from Bahir Dar.
Similarly to the geographical location, the sampling period influences the results. Norovirus seasonality is less obvious in the African regions as in the Northern hemisphere and not well understood in East Africa [Reference Mans12]. The present study was restricted to 6 months sampling during the dry and warm season. Norovirus positivity rate and genotypes changed during that period and may well be different again during the cooler wet season. Due to inconsistent sampling sites, sampling periods and sample numbers, comparative analysis of the data of different studies from one country may fail to estimate the true genetic diversity and dynamics of norovirus epidemiology in time. Standardised continuous monitoring of circulating norovirus strains in an increased number of samples at multiple sampling sites would be necessary to reduce the bias.
In conclusion, sapovirus and norovirus stool positivity in young children with diarrhoeal disease is high in Northwest Ethiopia. Genetically diverse caliciviruses are co-circulating and may vary in time and between proximate sites. Continuous monitoring will be important to provide profound data on the dynamic calicivirus epidemiology and diversity in Ethiopia.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0950268819001031.
Author ORCIDs
A. Gelaw, 0000-0001-9239-1616; C. Pietsch, 0000-0001-6029-3368
Acknowledgements
Our special thanks go to the study participants and the data collectors involved in this study, especially for Mr. Eyuel Kassa and Mr. Derese Hailu. We are also grateful to Sandra Bergs for excellent technical assistance. This research was partially supported by the Association of Sponsors and Friends of Leipzig University (Vereinigung von Förderern und Freunden der Universität Leipzig) and received funding from the German Academic Exchange Service (DAAD).
Conflict of interest
None.
Author contributions
The manuscript contains data that have not previously been published or submitted elsewhere; it has not been submitted for publication elsewhere. AG and UGL designed the study. CP, AG and PM carried out the laboratory experiments. AG and CP performed the data analysis and drafted the manuscript. UGL revised it. All authors read and approved the final manuscript.






 Open access
Open access