


Introduction
The Sea of Okhotsk belongs to the seasonal sea-ice zone and is at the lowest latitude at which sea ice is found (besides the Bohai Sea), with maximum ice extent of 50–90% in late winter (Reference Granskog, Kaartokallio and KuosaGranskog and others, 2010). Most of the sea ice is produced in the northern continental shelf region (Reference Martin, Drucker and YamashitaMartin and others, 1998). Sea-ice extent is largely controlled by wind, and sea ice is transported from north to south (Reference Granskog, Kaartokallio and KuosaGranskog and others, 2010, and references therein). The northern Sea of Okhotsk also receives significant terrestrial inputs of dissolved organic matter (DOM) from the Amur River, which during ice formation are injected to deeper waters, together with brine, and transported in the subsurface into the southern Sea of Okhotsk and further to the Pacific (e.g. Reference Nakatsuka, Toda, Kawamura and WakatsuchiNakatsuka and others, 2004). Further south, water masses are composed of a mixture of waters entering the region, where the Soya current brings warmer waters from the Japan Sea, Pacific waters enter through the Kuril Straits, and the Sakhalin current transports waters from the northern shelf to the south.
In contrast, the water masses in the eastern part of Saroma-ko lagoon, located adjacent to the Sea of Okhotsk on the northern shore of Hokkaido, Japan, consist mainly of adjacent Okhotsk Sea water, the freshwater input from the Saromabetsu River and seasonally of ice melt (Reference Shirasawa and LeppärantaShirasawa and Leppäranta, 2003; Reference Nomura, Takatsuka, Ishikawa, Kawamura, Shirasawa and Yoshikawa-InoueNomura and others, 2009). Almost the entire surface of Saroma-ko lagoon is generally covered with sea ice from early January through early April, with large year-to year variability.
Chromophoric DOM (CDOM) is ubiquitous in marine environments and can make up a substantial proportion of the DOM pool in aquatic systems (Reference ThurmanThurman, 1985), representing up to 60% of the bulk dissolved organic carbon (DOC) pool (Reference Ertel, Hedges, Devol, Richey, de Nazaré and RibeiroErtel and others, 1986). DOM is a major source of energy for heterotrophs, and thus CDOM plays an important role in the biogeochemistry of natural waters (Reference HillHansell and Carlson, 2002). CDOM exerts a significant influence on the optical properties of ocean water and sea ice. For example, it affects the penetration of ultraviolet radiation (known to inhibit phytoplankton productivity), the light available for primary production, and the distribution of radiant heating in sea ice and surface ocean and thereby surface stratification (e.g. Reference Mopper and KieberMopper and Kieber, 2002; Reference Ehn, Granskog, Reinart and ErmEhn and others, 2004; Reference Granskog, Macdonald, Mundy and BarberGranskog and others, 2007; Reference HillHill, 2008; Reference Uusikivi, Vähätalo, Granskog and SommarugaUusikivi and others, 2010). Further, exposure to light can have a significant effect on the cycling of DOM, as the absorption of ultraviolet and visible light initiates the formation of a number of reactive products, including biologically labile ammonium and phosphate, and the remineralization of carbon (e.g. Reference Moran and ZeppMoran and Zepp, 1997; Reference Obernosterer and BennerObernosterer and Benner, 2004; Reference Vähätalo and ZeppVähätalo and Zepp, 2005). The optical properties of CDOM in Baltic Sea ice differ from those of the underlying sea water (Reference Ehn, Granskog, Reinart and ErmEhn and others, 2004) as a result of in situ CDOM production (Reference Stedmon, Thomas, Granskog, Kaartokallio, Papadimitriou and KuosaStedmon and others, 2007), most likely by sympagic (ice-associated) organisms, resulting in protein-like compounds that dominate over humic-like fluorescence, contrary to parent sea water. In Antarctic sea ice, where marine DOM dominates, the CDOM composition is also markedly different from that in underlying sea water, suggesting autochthonous CDOM production and accumulation in sea-ice brines (Reference Stedmon, Thomas and PapadimitriouStedmon and others, 2011), potentially making sea ice an important source of labile CDOM during sea-ice melt (Reference NormanNorman and others, 2011).
Sea-ice studies in the Sea of Okhotsk have mainly focused on understanding the physical characteristics of sea ice (Reference Ukita, Kawamura, Tanaka, Toyota and WakatsuchiUkita and others, 2000; Reference Toyota, Kawamura, Ohshima, Shimoda and WakatsuchiToyota and others, 2004, 2007). The amount of particulate impurities in sea ice has been reported by Reference Granskog and Ser.Granskog (2000), but studies on the biogeochemistry of sea ice are limited (Reference McMinn, Hattori, Hirawake and IwamotoMcMinn and others, 2008; Reference NomuraNomura and others, 2010, 2011). Since sea ice is mainly produced in the northern region of the Sea of Okhotsk and transported to the south where it melts (e.g. Reference Kimura and WakatsuchiKimura and Wakatsuchi, 2004), besides fluxes of heat and fresh water, sea ice is also potentially important for the supply of any material accumulated in, or transported with, the ice (Reference NomuraNomura and others, 2010). In this case, the southern Sea of Okhotsk is the main region receiving inputs from melting sea ice. In contrast to the Sea of Okhotsk, several studies on Saroma-ko lagoon report on the biogeochemistry and sympagic assemblages in sea ice (e.g. Reference Robineau, Legendre, Kishino and KudohRobineau and others, 1997; Reference McMinn and HattoriMcMinn and Hattori, 2006; Reference Nomura, Takatsuka, Ishikawa, Kawamura, Shirasawa and Yoshikawa-InoueNomura and others, 2009, 2011).
We hypothesize that there are contrasting regimes in DOM composition, both spatially between Okhotsk and Saroma-ko lagoon, but also between sea water and sea ice, which might result in a flux of compositionally different DOM by sea-ice melt (cf. Reference NormanNorman and others, 2011). With this in mind, we examine the composition of CDOM in sea ice and under-ice waters in the southern Sea of Okhotsk and the adjacent Saroma-ko lagoon in late winter and spring. We present the first data on CDOM absorbance and fluorescence of sea ice in the region, as well as in underlying surface waters. The main objective of this study is to describe the optical characteristics and possible origin of CDOM, and discuss the potential role of sea ice as a source of CDOM in the region.
Materials and Methods
Study area
Samples were collected from pack ice in the southern Sea of Okhotsk during two cruises on board the Japanese Coast Guard vessel Soya on 11–13 February 2008 and 9–12 February 2009, from landfast ice during land-based sampling in Saroma-ko lagoon (hereafter Saroma) on 3–4 March 2008 and 20 February 2009 (Fig. 1, Saroma), and from nearshore drift ice during land-based sampling off the Utoro Peninsula in the southeastern Sea of Okhotsk on 7 March 2008 (Fig. 1, site U). At each location sea ice and under-ice water (1-3 m from the ice underside) were collected. In total 14 sites were sampled (Table 1), with 14 sea-water samples and 51 ice sections (see below).
Table 1. Sampling sites and conditions


Fig. 1. Location of sampling sites in 2008 (squares; station number below symbol) and 2009 (circles; station number above symbol) in the southern Sea of Okhotsk. Saroma-ko lagoon (Saroma) and Utoro (U) sampling sites are also indicated.
Field sampling and sample processing
Sea-ice samples were collected using an ice corer (Mark II, Kovacs Enterprises, Inc., USA). One core was used to measure ice temperature (Testo 110 NTC, Brandt Instruments, Inc., USA); a second core was used for the analyses. In 2009, pancake and nilas ice (Table 1) were sampled from a basket lowered from the ship to the sea surface. One half of the core or part of the collected ice was used for immediate processing and subsampling. The other half of the core was placed in plastic tubing or bags and stored in an on-board freezer at -15°C.
For pigment analysis, ice samples were cut into sections, usually every 5 or 10cm, and melted directly on board the ship with the addition of artificial sea water (salinity 40). Samples were melted at room temperature in the dark in acid-washed lidded Teflon cups.
The second half of the ice-core samples from the Soya expeditions were placed in polyethylene tubing immediately after collection and kept horizontal in a freezer (at - 15°C) during the cruise. These half-core samples from Soya for CDOM, salinity and δ18O analyses were processed in the freezer room at Hokkaido University shortly after each cruise. The core half was cut into a 4 cm x 3 cm rectangular cross section and then cut into sections. To avoid contamination during sampling and handling processes, 3 mm of the outside of the ice sections were removed with a clean stainless-steel plane. The remaining ice was placed in an acid-washed Teflon cup for melting at room temperature in the dark.
Sea-ice samples from land-based sampling in Saroma and Utoro were processed directly on site (pigment analysis). Immediately after retrieval the samples were cut into 5-10cm sections and placed in pre-cleaned lidded polyethylene cups for melting. Samples for pigment analysis were melted as for the samples from Soya. Frozen cores were taken to Hokkaido University in Sapporo and processed for salinity, δ18O and CDOM analyses as described for the samples from Soya.
Under-ice water samples were collected on all occasions through the ice-core holes using a 500 mL Teflon water sampler (GL Science Inc., Japan), usually at 3 m depth (Table 1), ∼15min after drilling of ice cores to avoid disturbance caused by drilling.
Melted sea ice and under-ice water were subsampled for salinity and oxygen isotopic ratio (δ18O) measurements into 10mL glass vials, and 40 mL pre-combusted amber glass vials for CDOM. Salinity and δ18O samples were kept at room temperature (+15°C). CDOM samples were filtered through PALL Acrodisk® membrane filters (Supor® membrane, pore size 0.2 m, with 0.8m Supor® pre-filter) using acid-washed all-plastic syringes. Ultrapure water treated as samples showed no trace of leaching of CDOM into the samples during melting, filtering and storage. CDOM samples were stored in the dark at +4°C. Samples of known volume were filtered onto Whatman GF/F glassfiber filters (nominal pore size 0.7 m) using low vacuum for pigment analysis. Filters were placed in liquid nitrogen until measurements were conducted.
Analytical methods
Salinity was measured using a salt analyzer (SAT-210; Toa Electronics Ltd, Japan). A standard deviation for salinity calculated from 15 subsamples taken from a reference water sample (S= 10.00) was 0.03. δ18O was determined with a mass spectrometer (DELTA plus; Finnigan MAT, USA). Results are reported as δ18O (‰), defined as the deviation of H2 18O/H2 16O ratio of the measured sample to that of Vienna Standard Mean Ocean Water (VSMOW). The precision of δ18O analysis from duplicate determinations is ±0.02% (Reference Toyota, Takatsuji, Tateyama, Naoki and OhshimaToyota and others, 2007). Chlorophyll a (Chl a) was determined with high-performance liquid chromatography (HPLC; Shimadzu SPD-M10AVvp with Class-VP Multi Workstation) following the methods of Reference Furuya, Hayashi and YabushitaFuruya and others (1998).
CDOM spectral absorbance (A(λ)) was measured using a Shimadzu UV-PC2501 spectrophotometer with a 10 cm quartz cell, from 190 to 800 nm, with 2 nm slits and at 1 nm intervals, after samples had been allowed to warm to room temperature, with ultrapure water (Milli-Q) as reference. A baseline correction was carried out, with average absorbance at 690-710 nm subtracted from the spectra. Absorbance was converted to absorption coefficients (a CDOM(λ) (m-1) = [2.303∆(λ)]/0.1 m). CDOM samples were measured within 2-3 months after collection. The spectral slope coefficient S at 250-400 nm range was calculated using a linear fit to log-transformed absorption data.
The CDOM fluorescence was measured using a Varian Cary spectrofluorometer with excitation at 240-450 nm (every 5 nm), and the resulting emission was measured from 300 to 600 nm (every 2 nm). The resulting excitation-emission matrices (EEMs) were corrected for instrument-specific spectral corrections, inner filter effects (based on the absorption of the sample) and Raman and Rayleigh scattering, and finally converted to Raman units (RU) based on the Raman signal from ultrapure water (ultrapure water was run as a sample for each batch of samples measured to account for possible instrument drift). Corrected fluorescence data (i.e. EEMs) were analyzed using the DOMFluor Toolbox v1.7 (Reference Stedmon and BroStedmon and Bro, 2008).
PARAFAC modeling
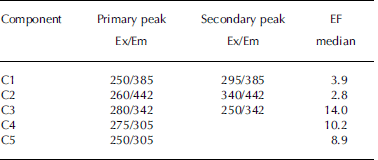
A total of 65 EEMs were collected and modeled in this study, which included 14 sea-water samples and 51 samples from sea-ice cores. However, the dataset was very diverse; it was not collected along a mixing gradient or time series, but contained extreme samples. Therefore it is not ideal for parallel factor analysis (PARAFAC) modeling (see Reference Stedmon and BroStedmon and Bro, 2008). Thus weighing was carried out in accordance with Reference Kowalczuk, Durako, Young, Kahn, Cooper and GonsiorKowalczuk and others (2009), who also had a diverse EEM dataset. Samples with extremely high fluorescence were diluted, such that all samples were within a maximum intensity range of 0-0.5 RU. This weighed dataset was split-half validated for a five-component PARAFAC model, and a residual analysis also confirmed that a five-component model was appropriate (Reference Stedmon and BroStedmon and Bro, 2008). Table 2 lists the excitation and emission peaks of the components derived for the validated PARAFAC model. The fluorescence intensities in the subsequent data analysis refer to the maximum fluorescence signals of each component.
Table 2. Primary and secondary excitation–emission (Ex/Em) peaks for PARAFAC model components on Okhotsk sea ice and surface waters. Median EFs for components in sea ice relative to underlying sea water
Results and Discussion
Sea-water and sea-ice characteristics
In 2008 the ice sampled was first-year ice (˃0.3 m thick), while in 2009 the majority of the ice sampled in the Sea of Okhotsk was young ice, pancake ice or dark nilas (˂0.3 m thick). This is reflected in the higher salinity of the younger ice (Table 1). From previous studies it is known that snow can contribute significantly to sea-ice mass both in the southern Sea of Okhotsk (Reference Ukita, Kawamura, Tanaka, Toyota and WakatsuchiUkita and others, 2000; Reference Toyota, Takatsuji, Tateyama, Naoki and OhshimaToyota and others, 2007; Reference NomuraNomura and others, 2010) and in Saroma (Reference Shirasawa, Leppäranta, Saloranta, Kawamura, Polomoshnov and SurkovShirasawa and others, 2005; Reference Nomura, McMinn, Hattori, Aoki and FukuchiNomura and others, 2011), although considerably more so in the latter. Our observations concurred with earlier observations: in 2008 -10% of the total length of the Okhotsk ice samples was snow-ice, while in Saroma snow-ice contributed 30-40% of the total ice thickness based on δ18O composition. Dynamic thickening and rafting of young floes contributed significantly in the Sea of Okhotsk in 2008. In 2009, as mainly new or young ice was sampled, the snow contribution to ice mass was negligible in the Okhotsk samples. In Saroma, snow-ice again contributed significantly, although less than in 2008.
Reference Uusikivi, Vähätalo, Granskog and SommarugaYamamoto and others (2001, 2002) proposed that deviations in sea-water composition (salinity and δ18O) from the western sub-arctic Pacific water (WSAP) line (δ18O = 0.3915x salinity-13.561, R 2 = 0.99, n = 59; Reference Yamamoto, Tanaka and TsunogaiYamamoto and others, 2001) can be used to explain the mechanism behind the sea-water composition. Our data are mainly displaced in a manner consistent with the addition of meteoric water (lighter δ18O) and brine from sea-ice formation (higher salinity), relative to the WSAP line (not shown) in both Saroma and Okhotsk samples. Samples collected 3 m below the ice bottom (2008) might not capture the shallow fresher under-ice layer in Saroma (see Reference Nomura, Takatsuka, Ishikawa, Kawamura, Shirasawa and Yoshikawa-InoueNomura and others, 2009), which is reflected in the lower salinity (Table 1) and light isotope values for under-ice water collected 1 m below the ice bottom in Saroma in 2009 (not shown).
Algal biomass
The algal biomass in sea ice in Saroma ranged from 2.0 to 9.0mgChl am–2 in 2008 and 0.3 to 10.6mgChl am–2 in 2009. This contrasts with previous observations which include 272.8±20.2 mgChl am–2 (Reference McMinn and HattoriMcMinn and Hattori, 2006), 2-119 mg Chl am–2 (Reference Robineau, Legendre, Kishino and KudohRobineau and others, 1997) and 38.25 mgChl am–2 (Reference Kudoh, Robineau, Suzuki, Fujiyoshi and TakahashiKudoh and others, 1997). Although biomass in Saroma was thus lower than in earlier studies, biomass in Okhotsk ice was even lower than in Saroma, where it ranged from 0.2 to 1.6 mgChlam–2 in 2008 and from 0.0 to 3.5 mg Chl a m–2 in 2009. The ice algal biomass distribution was typically accumulated in the bottom 5-10 cm. In only one core (2008) from Saroma was algal biomass distributed evenly throughout the length of the core (Saroma R station). Chl a levels were up to an order of magnitude lower in under-ice water than in ice, both in Okhotsk and Saroma. The standing stock in the ice cores corresponded to the total algal biomass in 5-15 m of the under-ice water column at the time of sampling.
CDOM absorption
Measurements of CDOM absorption in sea water showed the typical featureless exponential decay (e.g. Reference Bricaud, Morel and PrieurBricaud and others, 1981) with increasing wavelength, as shown in Figure 2a and b. Values of absorption at 350 or 375 nm (a350 and a375) were higher in Saroma than in Okhotsk waters (Table 3). The spectral slope coefficient S had values between 0.0197 and 0.0280 in Okhotsk and 0.0183 and 0.0202 in Saroma (Table 3). The lower S values in Saroma imply a greater influence from rivers (e.g. Blough and Del Vecchio, 2002), and the lower values in 2009 vs 2008 also exemplify the less saline water sampled in 2009 belonging to a well-developed under-ice plume (Reference Nomura, Takatsuka, Ishikawa, Kawamura, Shirasawa and Yoshikawa-InoueNomura and others, 2009). The majority of the sea-ice samples had local absorption peaks (Fig. 2c and d), which made it impossible to compute S values as the fit was very poor. A distinct peak at -325 nm was present in the majority of sea-ice samples, while a secondary peak was visible in some sea-ice samples at -275 nm. Peaks in CDOM absorbance spectra similar to those observed here were reported by Reference Belzile, Johannessen, Gosselin, Demers and MillerBelzile and others (2000) in ice-core samples from Baffin Bay, Canadian Arctic, and similar peaks have recently been found to be a ubiquitous feature of CDOM in Antarctic sea ice (Reference NormanNorman and others, 2011). Patasayeva and others (2004) observed absorption peaks between 270 and 300 nm in sea-ice brine samples generated in an ice tank experiment. Similar absorbance spectra have also been found in the CDOM produced by cyanobacteria in culture (Reference Steinberg, Nelson and PrusakSteinberg and others, 2004). The exact source of these peaks is unknown; however, they seem to represent an ice-specific CDOM fraction, i.e. related to autochthonous production, and are suggested to be related to the production of ultraviolet-absorbing compounds for photoprotection (e.g. Reference Belzile, Johannessen, Gosselin, Demers and MillerBelzile and others, 2000). It is known that mycosporine-like amino acids (MAAs) absorb strongly at -330nm, while at 275-280nm aromatic amino acids (precursors to MAAs) and some nucleic acids absorb strongly (e.g. Reference Woźniak and DeraWoźniak and Dera, 2007). All these compounds are produced by marine organisms. MAAs were almost completely absent at the bottom of thick Antarctic sea ice (Reference Ryan, McMinn, Mitchell and TrenerryRyan and others, 2002), likely due to low exposure to ultraviolet light. In thin (snow-free) seasonal sea ice in the Baltic Sea, more representative of the conditions in the Sea of Okhotsk or Saroma, concentrations of MAAs were exceptionally high (with MAA to Chl a ratios up to 0.65) and could be detected as high particulate absorption peaks at -330 nm (Reference Uusikivi, Vähätalo, Granskog and SommarugaUusikivi and others, 2010).
Table 3. Optical properties of CDOM in sea water and sea ice. Mean and standard deviation (maximum) are given for a350, a375 and aR (ratio a330: a 300). Range is given for S. S values for sea ice are not given as they could not be computed reliably
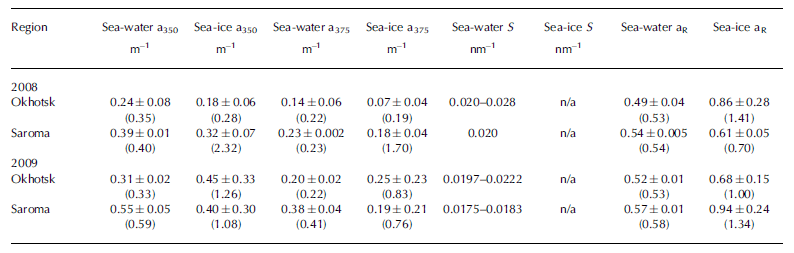
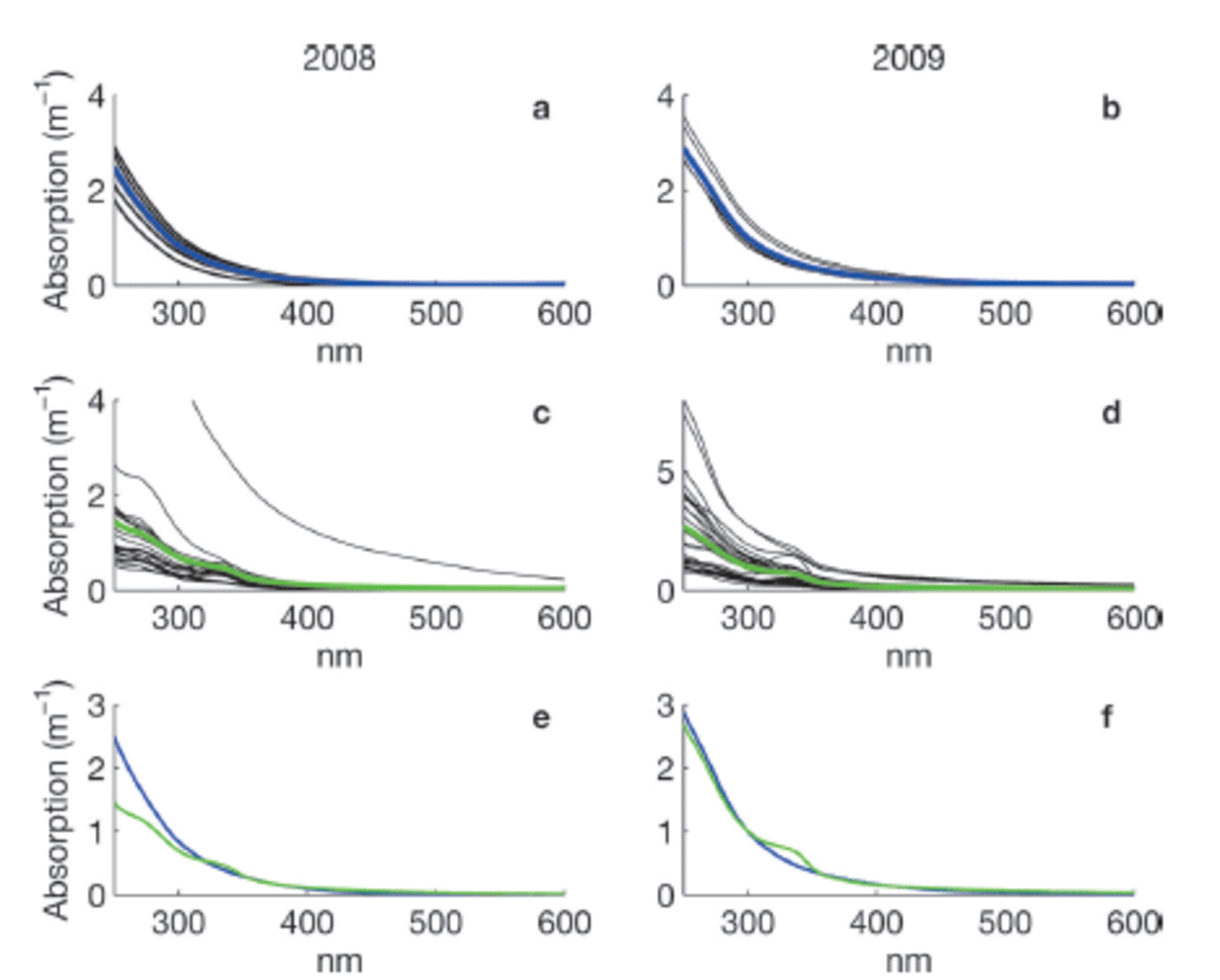
Fig. 2. Absorption spectra for sea-water (a, b) and sea-ice (c, d) samples collected in 2008 and 2009, respectively. Blue and green lines in (a–d) show the mean. The means in 2008 (e) and 2009 (f) for sea water (blue) and sea ice (green) are compared. Note the variable y-axis scale.
From Figure 2c and d it is apparent that many of the absorption spectra in sea ice are often very flat below 350 nm and do not have the characteristic strong exponential increase in CDOM absorption at shorter wavelengths (e.g. Blough and Del Vecchio, 2002). The samples with these flat absorbance spectra are predominantly from older first-year sea ice, while the younger ice tended to have absorbance spectra similar to that of sea water (besides the distinct absorption peaks also present in some of these samples). This could be partly due to the age of the ice, since either mature sea-ice communities produced this material or sunlight photobleached dissolved material, thereby flattening the absorption spectra. Flatter CDOM absorption spectra at shorter wavelengths have been observed in surface ice layers and for older ice in the Baltic Sea (Reference Uusikivi, Vähätalo, Granskog and SommarugaUusikivi and others, 2010; Muller and others, 2011).
Our observations indicate that sea ice can support concentrated production of specific light-absorbing compounds, presently of unknown origin or composition, which can be released during ice melt. Values in sea ice are considerably higher than expected from conservative incorporation relative to salinity. The absorption peak at -330 nm was particularly distinct in the majority of the sea-ice samples, while the absorption at -300nm has values expected from simple exponential decay with increasing wavelength. Hence, the ratio of absorption at 330 nm to that at 300 nm (aR = a330: a300), which was considerably higher in sea ice than in sea water (Table 3), can be used to show that CDOM released from sea ice appears to differ substantially from that of underlying sea water, although the concentration appears similar to sea water. Thus, sea ice does not provide an excess amount of CDOM during melt, as reported for Baffin Bay (Reference Nakatsuka, Toda, Kawamura and WakatsuchiScully and Miller, 2000), but rather is a source of CDOM of different quality (Reference NormanNorman and others, 2011).
CDOM fluorescence
Characteristic fluorescence excitation–emission
The majority of sea-water samples were dominated by a terrestrial humic-like fluorescence (Fig. 3) with excitation at 250 nm and maximum emission at ˃400nm (Reference CobleCoble, 1996). Some sea-water samples had another fluorescent compound with excitation/emission maximum at -275/305 nm. The EEMs in sea ice differed considerably from those in (parent) sea water (Fig. 3), and are dominated by emission below 400nm, similar to observations in Baltic Sea ice (Reference Stedmon, Thomas, Granskog, Kaartokallio, Papadimitriou and KuosaStedmon and others, 2007).

Fig. 3. Typical EEMs in the study region. EEMs shown are means for sea water in Saroma (a), sea water in the Sea of Okhotsk (b), sea ice in Saroma (c) and sea ice in the Sea of Okhotsk (d). Note the difference in intensity scale between sea-water (a, b) and sea-ice samples (c, d).
PARAFAC components
Components in the validated PARAFAC model (Table 2) overlap with results from earlier studies. Two of the identified components (C1 and C2) had properties that are often referred to as humic-like (Reference CobleCoble, 1996), with broader excitation spectra and emission maxima ˃380 nm. The other three components (C3-C5; Table 2) resemble protein-like material, with emission at ˂360nm (Reference CobleCoble, 1996), overlapping with findings from other studies. For example, C3 resembles tryptophan-like (free or bound to protein) fluorescence in Antarctic sea-ice brines (Reference Stedmon, Thomas and PapadimitriouStedmon and others, 2011), and C4 and C5 have been referred to as tyrosine-like fluorescence (Reference CobleCoble, 1996; Patasayeva and others, 2004). These are likely not pure compounds but bound to other compounds/molecules, which is why their fluorescence characteristics differ from those of pure amino acids.
Sea-water fluorescence in both Saroma and Okhotsk was dominated by the humic-like component C2, with a significant negative correlation with salinity and δ18O in sea water (r˂-0.76, p˂0.01), implying a terrigenous source. This relationship could not be found in sea ice. In contrast, sea-ice DOM fluorescence was generally dominated by varying degrees of the protein-like components. To examine whether these compounds are found in excess in sea ice relative to parent sea water, enrichment factors (EFs) were calculated as in Reference Müller, Vähätalo, Stedmon, Granskog and NormanMüller and others (2013), with values ˃1 indicative of an excess in sea ice that cannot be explained by conservative incorporation relative to salinity during ice formation (Table 2). The EFs for protein-like components are much higher (median values 8.9–14.0; Table 2) than observed during initial ice formation (EFs˂ 3; Reference Müller, Vähätalo, Stedmon, Granskog and NormanMüller and others, 2013, Fig. 11). EFs for humic-like material (EF ˂ 4) were considerably lower than for protein-like material, similar to what is found during initial ice formation (Reference Müller, Vähätalo, Stedmon, Granskog and NormanMüller and others, 2013). The considerable protein-like accumulation thus points towards biological activity in sea ice during ageing (cf. Reference Stedmon, Thomas, Granskog, Kaartokallio, Papadimitriou and KuosaStedmon and others, 2007, 2011).
The much higher biomass in sea ice relative to sea water points towards possible accumulation in sea ice as a result of sympagic production. CDOM released during sea-ice melt is therefore compositionally different than that present in parent sea water, and sea ice might therefore be a source of fresh and potentially more labile CDOM (Reference NormanNorman and others, 2011). However, more detailed studies during ice melt are needed to validate this assumption. Reference NormanNorman and others (2011) showed that the material released from sea ice is susceptible to exposure to sunlight and is easily photobleached; therefore, its lifetime could be transient.
Conclusions
We have examined the composition of CDOM in sea ice and surface waters in the southern Sea of Okhotsk and the adjacent semi-enclosed Saroma lagoon by resolving the absorbance and fluorescent properties of CDOM. Sea-water CDOM shows the typical exponentially decaying absorption spectra and is composed mainly of humic-like material. In sea ice, the absorption spectra deviated significantly from the classical exponential decay spectra (e.g. Reference Bricaud, Morel and PrieurBricaud and others, 1981), with distinct absorption peaks in many samples and flatter absorption spectra in the ultraviolet range in older first-year sea ice (Fig. 2). Flat spectra are perhaps a response to exposure to light, as the relatively low latitudes in the study area result in high insolation. This is also supported by recent observations of sea ice in the Baltic Sea (Reference Uusikivi, Vähätalo, Granskog and SommarugaUusikivi and others, 2010) and the Antarctic (Reference NormanNorman and others, 2011). The analysis of CDOM in sea ice therefore shows evidence for the production of compounds with strong ultraviolet absorption at certain wavelengths, suggestive of production of aromatic and mycosporine-like amino acids within sea ice (cf. Reference Uusikivi, Vähätalo, Granskog and SommarugaUusikivi and others, 2010). Support for this was found by the fluorescence of CDOM, which showed that protein-like compounds dominated in sea ice and were found in significant excess compared with underlying sea water, as has been found recently in Antarctic sea ice (Reference Stedmon, Thomas and PapadimitriouStedmon and others, 2011). The enrichment of these compounds in sea ice is much higher than can be explained by incorporation during ice formation (Reference Müller, Vähätalo, Stedmon, Granskog and NormanMüller and others, 2013). This suggests production and accumulation of specific protein-like compounds in sea ice (cf. Reference Stedmon, Thomas, Granskog, Kaartokallio, Papadimitriou and KuosaStedmon and others, 2007, 2011). Thus the material released from melting sea ice is apparently composed of labile material (cf. Reference NormanNorman and others, 2011). Further studies are needed, however, to better understand the role of sea-ice-derived DOM, especially in the highly productive ice edge or marginal ice zones.
Acknowledgements
This work was supported by a bilateral research mobility grant from the Academy of Finland and the Japan Society for Promotion of Science (JSPS) to M.A.G., and the Norwegian Polar Institute. We thank the reviewers and the Scientific Editor Gerhard Dieckmann for constructive comments.






