


INTRODUCTION
Antiviral drugs are a crucial method of defence against novel emerging strains of influenza for which there are no effective vaccines [Reference Moss1]. Understanding the impact of widespread antiviral usage on population immunity and long-term strain dynamics is key for controlling multiple waves of a pandemic. Antivirals can decrease household transmission by 42–80% [Reference Goldstein2, Reference Halloran3], decrease disease severity by 38% [Reference Treanor4] and decrease disease duration by 30–50% [Reference Nicholson5, Reference Ng6]. Due to decreased transmission and better clinical outcome, there is interest in evaluating cost-efficacy and implementation strategies of antiviral drugs relative to other methods of prophylaxis in the event of the emergence of a moderate or severe novel human pandemic strain of influenza [Reference Dimitrov7–Reference Lugner, Mylius and Wallinga12]. Oseltamivir is currently believed to be the most effective antiviral drug against influenza and was used in up to 40% of patients in some areas during the early emergence of pandemic influenza A (pH1N1) in 2009. The potential widespread administration of these treatments motivates consideration of the longer-term population-level effects.
A second potentially important ramification of widespread drug usage is decreased population immunity resulting from lowered immune responses within individuals and from averted infections. Understanding drug effects on population immunity is particularly important for pandemic control of partially immunizing, seasonal viruses such as influenza because hosts can be re-infected, contributing to multiple epidemic waves. Despite the important role of host-population immunity in seasonal diseases, the effect of antiviral regimens on host-population immunity and multi-season dynamics has rarely been explored [Reference Wessel13]. Mathematical models can help elucidate whether or how these effects should be considered in pandemic response planning by considering ‘what-if’ scenarios and evaluating the impact of various response strategies. For example, recent mathematical models have shown the impact of drug resistance on outbreak severity, thus guiding plans to minimize transmission of drug-resistant strains [Reference Alexander, Bowman and Feng14–Reference Debarre, Bonhoeffer and Regoes18].
The emergence of novel strains occurs during ongoing circulation of seasonal variants, as was the case with pH1N1 emergence in 2009 (e.g. [Reference Lucas19]). Cross-reactivity of pandemic strains with seasonal strains can impact the composition of host-population immunity making it difficult to predict and interpret dynamics of the emerging strain [Reference Viboud and Simonsen20, Reference Labrosse21] due to increased heterogeneity in immunity. Similarly, antiviral drugs such as oseltamivir can increase heterogeneity in host-population immunity because they impact within-host dynamics by reducing viral load [Reference Li22, Reference Yu23], reducing duration of viral shedding [Reference Na24] and reducing the strength of adaptive immunity [Reference Takahashi25] in a manner that depends on the time at which drugs are taken relative to the onset of infection. Furthermore, there have been reports of higher re-infection rates in patients that have a history of oseltamivir usage relative to those that did not take the drug during a previous infection [Reference Perez, Ferres and Labarca26, Reference Wu27]. Thus, in addition to evaluating the efficacy of antivirals in reducing transmission during a single outbreak, it is important to understand the potential implications of drug interventions on host-population immunity at an inter-season scale in order to develop appropriate public health policies for drug implementation and to plan for the ramifications of drug usage.
Heterogeneities in host-population immunity due to infection history occur at two scales: the magnitude of coverage in the population and the strength of immunity within individual hosts carrying some level of immunity. Although there is still much to be learned before individual-level immunity levels are predictable [Reference Handel, Longini and Antia9, Reference Handel, Longini and Antia28, Reference Mak29], viral and immune dynamics during previous infections clearly play a role in determining the strength and specificity of immune memory that is available to fight subsequent infections [Reference Mak29–Reference Hung31]. Thus, in order to evaluate effects of antivirals across multiple epidemic waves, we present a two-strain individual-based epidemiological model that explicitly accounts for viral and immune levels changing over time within individual hosts throughout the course of an epidemic; and uses the distribution of immunity from viral dynamics in the previous outbreak to initialize subsequent outbreaks. The population follows Susceptible–Infectious–Removed (SIR)-type dynamics [Reference Kermack and McKendrick32] with transmission being dependent on the infectious load of hosts at contact. We explored effects of the timing of drug treatments, R 0 (the average number of transmissions made by an infected host in a completely susceptible population; a transmission rate) and drug coverage, on multiple epidemic waves of a novel strain (invader) that cross-reacts with a resident strain. Specifically, we examined the case where a seasonal strain was introduced to a naive population followed by later introduction of a novel strain into the same population. We seeded the multiple waves with this initial situation (as opposed to the situation where the population had immunity to the seasonal strain) because we were interested in the case where the seasonal strain had experienced antigenic drift prior to the seasonal epidemic. The second strain, which could have arisen by mutation or reassortment, was assumed to be substantially antigenically different to the wild-type strain.
METHODS
Within-host model
In previous work we studied a Lotka–Volterra-type model [Reference Volterra and Chapman33, Reference Lotka34] of the within-host dynamics of two strains that interact with two types of immune effectors through primary and secondary interactions [Reference Pepin35]. Here we extended the model to include effects from antiviral drugs [equations (1)–(4)].
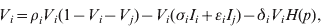 $$V_{i} \equals \rho _{i} V_{i} \lpar 1 \minus V_{i} \minus V_{j} \rpar \minus V_{i} \lpar \sigma _{i} I_{i} \plus \epsiv _{i} I_{j} \rpar \minus \delta _{i} V_{i} H\lpar p\rpar \comma \hfill$$
$$V_{i} \equals \rho _{i} V_{i} \lpar 1 \minus V_{i} \minus V_{j} \rpar \minus V_{i} \lpar \sigma _{i} I_{i} \plus \epsiv _{i} I_{j} \rpar \minus \delta _{i} V_{i} H\lpar p\rpar \comma \hfill$$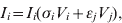 $$I_{i} \equals I_{i} \lpar \sigma _{i} V_{i} \plus \epsiv _{j} V_{j} \rpar \comma \hfill$$
$$I_{i} \equals I_{i} \lpar \sigma _{i} V_{i} \plus \epsiv _{j} V_{j} \rpar \comma \hfill$$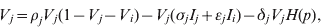 $$V_{j} \equals \rho _{j} V_{j} \lpar 1 \minus V_{j} \minus V_{i} \rpar \minus V_{j} \lpar \sigma _{j} I_{j} \plus \epsiv _{j} I_{i} \rpar \minus \delta _{j} V_{j} H\lpar p\rpar \comma \hfill$$
$$V_{j} \equals \rho _{j} V_{j} \lpar 1 \minus V_{j} \minus V_{i} \rpar \minus V_{j} \lpar \sigma _{j} I_{j} \plus \epsiv _{j} I_{i} \rpar \minus \delta _{j} V_{j} H\lpar p\rpar \comma \hfill$$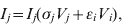 $$I_{j} \equals I_{j} \lpar \sigma _{j} V_{j} \plus \epsiv _{i} V_{i} \rpar \comma \hfill$$
$$I_{j} \equals I_{j} \lpar \sigma _{j} V_{j} \plus \epsiv _{i} V_{i} \rpar \comma \hfill$$Here, V is viral load, I is immunity level, i is strain 1 and j is strain 2. ρ, σ, ε and δ are constant parameters for replication rate, interaction with primary effectors, cross-interactions and drug effects, respectively (Table 1a). H(p) is a step function such that H(p) = 0 for t < p and H(p) = 1 for t ⩾ p, to allow a delay between the time of infection and the time at which drugs were initiated. For simplicity, variables were re-scaled from the original form in the logistic population growth model (rV i(1 – (V i+V j)/K)) to units of the carrying capacity, K, which is the maximum viral load due to cell depletion, and time was re-scaled by the inverse of the carrying capacity, resulting in an effective carrying capacity of 1. Immunity levels had an upper bound determined by total viral load, and a minimum of 10−3 (i.e. naive hosts; value chosen via sensitivity analyses in Pepin et al. [Reference Pepin35]). We assumed that two types of cross-reactivity were possible: (i) cross-immunity where strains are suppressed by primary antibodies of the other strain [ε in equations (1) and (3)], and (ii) cross-stimulation where strains stimulate production of the alternate immune effectors [ε in equations (2) and (4)]. Each strain had more efficient interactions with one type of immune effector (primary effector) and weaker interactions with the alternate effector (cross-effector). We assumed that re-infections were similar to primary infections except that increased initial immunity levels caused decreased viral loads and infectious periods, and increased levels of adaptive immunity.
Table 1. Description of parameters used in main results

In all analyses, the intrinsic characteristics of the two strains were symmetric (the same values for replication rate and immunity interaction parameters) in order to focus on the effects of time-varying infection profiles and drug regimens (Table 1a). In rare cases where super-infection occurred, strains competed within hosts resulting in altered strain-specific viral load and immunity levels. For simplicity, we did not model the innate immune response explicitly, rather we assumed it was a constant and that differences in the ability of the immune system to control infection were due to differences in adaptive immune system responses. Target-cell depletion was also modelled implicitly as a constant through the carrying capacity term. Thus, we assumed that the viral load dynamics of a particular strain were impacted entirely by: adaptive immunity, the presence of another strain, an upper limit on viral load, and drugs.
Detailed behaviour of the within-host model with drug effects is shown in Supplementary Figures S1 & S2 (see also [Reference Pepin35] for a version of the model with no drug effects). Briefly, viral population size increases sharply to peak load followed by a slower rate of decrease to complete clearance. In the absence of drugs, total viral load, final immunity and infectious period do not depend on initial viral dose unless the strains co-infect. Differences in population growth rate (ρ) and cross-interactions with immunity (ε, δ) cause complex behaviour in relative strain dynamics, especially when the parameter values are asymmetric between strains. Here, we exclude these effects by considering strains with the same values of population growth and cross-interaction.
Transmission model
Multiple factors influence transmission – from host contact structure to within-host viral abundance [Reference Bansal36–Reference Miller38]. In order to isolate effects of viral infection profiles on transmission and immunity, we compared two types of simple population-patch structure: random global mixing and a 10-patch metapopulation with random mixing within and 0·25% movement between patches per time step. The scenarios had equal R 0 values. We used an agent-based simulation approach to model transmission in discrete time such that infectiousness and infectious doses depended on viral abundance in donor hosts at the time of contact. Simulations were conducted in Matlab (R2009b, MathWorks Inc., USA). Transmission from an infected host occurred with a fixed transmission probability given that the donor had a total viral abundance above the threshold of infectiousness (⩾102) and the recipient candidate was susceptible. Infection in the recipient was established with a fixed transmission probability if the amount of virus, after sampling by a fixed bottleneck factor (10−3), was above the threshold for establishment (⩾10−6, arbitrarily chosen to reflect 1 viral particle assuming that a host expels up to 107 particles per transmission event). The bottleneck parameter was chosen under the assumption that the number of particles transmitted is 1/1000 times more dilute than that in the infected host, and sensitivity analyses showed that bottleneck factors ⩾10−4 did not increase stochastic extinction of either strain. Super-infections, although rare, could occur when multiple infected hosts attempted transmission to the same susceptible host during the same time step (if both were successful according to the random transmission probability then the recipient was infected with the sum of the donor virus). Host transition from infected class to recovered class depended on infection period (the time from infection start to clearance which includes infectious and non-infectious time).
Simulations were mainly conducted under R 0 = 1·5 (mean time between serial infections 2–3 days) for the case without drugs to mimic conditions that are typical for influenza [Reference Ferguson8, Reference Fraser39, Reference White40], but effects of R 0 between 2 and 3 were also explored since they have been reported for a pandemic strain [Reference Mills, Robins and Lipsitch41, Reference Boelle42]. For analyses of drug effects, 40% coverage was used in the main text since this is a realistic level of drug stockpiling for pandemic preparedness (e.g. http://www.chp.gov.hk/en/guideline1/29.html). Hosts that took antivirals were chosen at random (implemented by assigning a 40% chance of taking antivirals to each host that became infected). Although we only investigated effects of post-infection drug treatment, simulations where antivirals were taken on the first day are similar to prophylactic drug use. The effects of 100% drug coverage were simulated for comparison and to show that the model produces realistic decreases in transmission due to drug treatment under the fixed parameters (38–65%, Fig. S2; similar to a decrease in household transmission of 42–80% [Reference Goldstein2, Reference Halloran3]). In order to focus on the epidemic dynamics rather than stochastic extinction, all simulations were begun with 10 infected hosts that were randomly selected from the population. The invader was introduced in 10 other hosts when 5% of hosts had been infected with the resident strain, which was approximately the midpoint of the epidemic growth curve under R 0 = 1·5. A sensitivity analysis on invasion time is presented in Figure S3.
Transmission across multiple waves
Each wave was initiated by introducing 10 infected hosts into a population consisting of individuals whose immunity levels were determined from the last wave. We assumed that hosts could only be re-infected between seasons since re-infection with a completely homologous strain within the same season does not cause productive infection [Reference Carroll43]. Thus, each wave faded out when contact rates with susceptible hosts (i.e. hosts that had not yet been infected within the season) decreased below the infectious period of infected hosts. When we re-introduced the virus in the next season, we assumed that previously infected hosts could again be infected since each the resident strain and invader would have experienced a small amount of antigenic drift, which is typical for influenza viruses. However, this assumption was only included implicitly rather than explicitly to reduce model complexity and uncertainty (i.e. decrease the number of parameters for which there are few empirical data). Thus, in our model, the small amount of antigenic drift enabled re-infection of hosts that had previously been infected with a similar strain and the infection profiles generated by these previously infected hosts correlated with immunity generated during prior infection [Reference Carroll43, Reference Greenberg, Couch and Kasel44]. Specifically, immunity was not prohibitive to infection if the viral dose was high enough to generate an infectious period despite a given immunity level or if the use of drugs in season 1 caused decreased immunity.
RESULTS
Within-host dynamics
The strength of adaptive immunity that arose in an individual following recovery from infection was influenced by two factors in our model. The first was whether and when an antiviral drug was taken during infection (Fig. 1). When an antiviral was not taken, peak viral load and final immunity were similar regardless of the seeding dose. However, antivirals induced dose-dependent final immunity and infectious periods. Immunity strength was lower and infectious period was shorter when drugs were taken earlier during the infection time-course (Fig. 1, Fig. S1). The size of seeding dose enhanced these drug-induced effects – lower seeding dose led to even lower final immunity and infectious periods. The effects of resident strain initial immunity on re-infection of the resident and cross-infection of the invader are depicted in Figure S2.

Fig. 1. Infection time-course for different initial doses (indicated in right-hand panel) and drug regimens (t): the time at which drugs were begun during the infection time-course. Viral load (solid lines, y axis, left) over time is plotted on a log10 scale. Immunity level (dotted lines, y axis, right) is on a natural log scale. Parameters are given in Table 1.
Infection and immunity dynamics in the host population
In order to understand the potential effects of within-host immunity suppression at the population level, we simulated transmission of the two strains in populations with different drug therapy regimens at the individual level (early vs. late during infection). For all simulations we assumed that the invading strain was seeded at a lower dose (i.e. invasion of a rare strain) and arrived during an outbreak of the resident strain at the midpoint of epidemic growth. We also assumed that stockpiles and human behaviour allowed for 40% drug coverage in infected hosts. Even when only 40% of hosts took drugs, transmission was significantly decreased (cf. Fig. 2a with Fig. 2b, c) and variability in mean population immunity increased (cf. Fig. 2d with Fig. 2e, f). Taking drugs early resulted in stronger population immunity for the invader, relative to taking drugs late or not taking drugs at all (cf. Fig. 2e with Fig. 2d, f).

Fig. 2. [colour online]. Infection and immunity dynamics. Top row shows disease states of hosts over time in populations where all hosts: (a) do not take drugs, (b) take drugs early during their infectious period, (c) take drugs late during their infectious period. Bottom row shows the mean final immunity for hosts that recover during each time step. Panels (d–f) correspond to drug conditions in panels (a–c). The initiation time of drug treatment was fixed at 6 (early) or 9 (late). Bottleneck at transmission = 10−3. The host population mixed globally at random. R 0 in panel (a) (no drug case) was 1·5. Only 40% of hosts took drugs. The resident strain was seeded in 10 hosts at the outset; the invader strain was seeded in 10 hosts after 5% of hosts had been exposed to the resident strain. Thin lines (top) represent 10 individual realizations of stochastic transmission and the thick line is a mean of these 10 runs (top and bottom). Within-host parameters are as in Table 1.
Effects of drug timing during the first wave
Drug timing affected two epidemic components: transmission in hosts and immunity levels in the host population. Effects on transmission were assessed by the proportion of cases averted relative to epidemics where no drugs were taken. Population immunity is presented on a log2 scale to be easily comparable with typical output from serological assays. Thus, a twofold reduction in population immunity reflects an average twofold reduction in the strength of immunity in recovered hosts, which has been shown to cause significant increases in infectivity and peak viral load upon re-challenge [Reference Carroll43, Reference Davies and Grilli45, Reference Delem and Jovanovic46]. Under realistic conditions for an influenza outbreak (R 0 = 1·5 and 40% drug coverage), taking drugs earlier during infection led to a significantly greater decrease in transmission and mean population immunity relative to taking drugs late (Fig. 3a, 30% vs. 16%). However, beyond a threshold of eight infection time steps (4 days post-infection), the reduction in transmission was constant at 16%. Both early- and late-administered drugs also caused a 2·9- and 1·4-fold decrease in immunity in recovered individuals, respectively (Fig. 3b), which translated to a 3- and 1·7-fold decrease in the full population (i.e. accounting for both averted cases and drug coverage, data not shown). Effects on population immunity did not plateau with later drug timing; instead, there was a positive linear relationship between drug timing and reduction in immunity (after 1·5 days post-infection). For comparison with the 40% case, Figure S4 shows the results when 100% of hosts take drugs.

Fig. 3. Effects of drug timing. (a) Total incidence following an epidemic with two strains in a completely naive population when 40% of hosts take drugs. Percent reductions in incidence for the ‘early’ and ‘late’ drug treatments are indicated. (b) Average immunity to each strain (resident, ▴; invader, ![]() ) for all hosts that were infected during the first wave. Numbers in panel (b) indicate the log2-fold reduction in immunity of infected hosts for the early and late treatments. Error bars represent 2 standard errors of the means of 100 replicate simulations. Only epidemics that reached 25% of the maximum (out of 100) were included in the averages such that stochastic loss at the beginning was excluded from the mean behaviour. Parameters are as in Figure 2. R 0 for the no drug case was 1·5.
) for all hosts that were infected during the first wave. Numbers in panel (b) indicate the log2-fold reduction in immunity of infected hosts for the early and late treatments. Error bars represent 2 standard errors of the means of 100 replicate simulations. Only epidemics that reached 25% of the maximum (out of 100) were included in the averages such that stochastic loss at the beginning was excluded from the mean behaviour. Parameters are as in Figure 2. R 0 for the no drug case was 1·5.
Effects of drugs across sequential waves
Next we examined the effects of decreased population immunity on subsequent epidemic waves by running sequential epidemics using the population immunity from the previous epidemic (i.e. at roughly 2–4 months previously). The total number of cases during all waves was higher when drugs were not taken (Fig. 4). At R 0 < 1·5 (where R 0 is measured from the first wave when the population is completely naive), the strain dynamics were qualitatively similar regardless of whether or not drugs were taken. However, at R 0 ⩾ 1·5, the relative frequency of each strain depended on drug usage. There was an increase in the ratio of resident:invading strain infections in subsequent waves when drugs were taken compared to when they were not taken (Fig. 4c, d). More specifically, at R 0 = 1·5, the resident strain caused 17% more infections relative to the invader in subsequent waves when drugs were taken, but 27% fewer infections when drugs were not taken. At R 0 = 2, both strains caused a similar proportion of infections in later waves when drugs were taken, whereas the invader strain caused 56% more infections in later waves when drugs were not taken. Thus, even under only 40% drug coverage, more cases were averted across multiple waves when drugs were taken compared to when they were not taken, but the relative frequency of different strains changed. The invader strain was more successful at re-invasion when no drugs were taken, while taking drugs facilitated persistence of the resident strain.

Fig. 4. Effects of drugs across sequential epidemic waves. (a, b). Total cases (thin grey line) of the resident (▴) and invader (![]() ) strains after the first wave in a completely naive host population. (c, d) The sum of cases during three subsequent waves in the populations in panels (a) and (b). Each wave was seeded with the immunity profile from the previous wave. Only 40% of hosts took drugs in the drug treatment and, when taken, drugs were started at time step 6 after infection. The invader strain arrived at approximately the midpoint of the epidemic growth curve. Error bars are 2 standard errors of the mean of 100 replicate simulations. All other parameters are as in Figure 2.
) strains after the first wave in a completely naive host population. (c, d) The sum of cases during three subsequent waves in the populations in panels (a) and (b). Each wave was seeded with the immunity profile from the previous wave. Only 40% of hosts took drugs in the drug treatment and, when taken, drugs were started at time step 6 after infection. The invader strain arrived at approximately the midpoint of the epidemic growth curve. Error bars are 2 standard errors of the mean of 100 replicate simulations. All other parameters are as in Figure 2.
Population structure also impacted the multi-wave dynamics by decreasing the relative difference between strains in all waves (Fig. S5). Proportionately more invader cases occurred in the first wave in the structured population relative to the population that mixed homogenously (Fig. 4a, b vs. Fig. S5 a, b). This decreased the number of invader cases in subsequent waves (relative to the homogenously mixed population), especially at R 0 ⩾ 2, regardless of whether drugs were taken. Thus, population structure allowed higher invasion during the first wave, more equal strain frequencies in subsequent waves and less impact of drugs on strain dynamics in subsequent waves. Effects of the interaction of population structure, drug coverage, drug resistance and drug timing are presented in the Supplementary Text and Figures S6 and S7. Most importantly, there was a non-trivial interaction between drug timing and coverage such that intermediate levels of drug coverage minimized cases in subsequent seasons in homogenously mixing populations; 40% coverage minimized cases during early drug treatment, 60% during late drug treatment. However, interestingly, in structured populations, any level of drug coverage beyond 40–60% minimized the number of cases in subsequent seasons. In the randomly mixing population, the invader strain caused more cases than the resident strain in later waves at drug coverages below 20% (under both early and late drug treatments), whereas strain dynamics were qualitatively similar across all drug coverages in the structured population.
DISCUSSION
We used a two-strain model of within-host dynamics embedded in a transmission model to examine mechanisms by which antiviral drugs impact recurrent epidemics. Our model showed that small changes in drug timing at the individual level can cause large differences in the number of cases averted and the level of population immunity in recovered hosts. Drug regimens also altered strain dynamics such that earlier drug usage decreased the incidence of the novel strain and increased the incidence of the resident strain in later epidemic waves. Our results suggest that preparedness plans for moderate and severe pandemics should consider sequential seasons, especially in relation to the knock-on effects of large-scale drug use during early waves.
Administration of drugs and timing of drug treatment are important considerations for controlling the emergence of a novel strain and predicting multi-strain dynamics across epidemic waves. Transmission and population immunity were reduced ∼50% more when hosts took drugs early compared to when they took drugs late. Furthermore, earlier drug usage led to proportionately higher levels of immunity to the invader strain during the first season relative to late drug usage, which translated to lower incidence of the invader relative to the resident strain in later waves. Thus, the timing of drug treatment not only impacted incidence in later waves, but also determined success of a novel invader strain. These results emphasize that even when the invading strain has the same inherent fitness as a resident strain, the interaction of antiviral drugs and population immunity alter the relative fitness between strains, and hence prediction of the levels of strain recurrence. Population structure was another factor that affected relative strain dynamics. When the population did not mix randomly, there was less difference in primary immunity to each strain, which led to levels of incidence between strains across seasons that were more similar. Thus, when there is increased population structure or partial coverage of anitivirals that are administered early, it is important to plan for vaccines against both strains (if there is only partial cross-reactivity, as is the case here).
It is extremely difficult to achieve high levels of drug coverage (discussed thus far) due to limited resources and human behaviour, but up to 40% coverage has been stockpiled as part of pandemic preparedness plans (http://www.chp.gov.hk/en/guideline1/29.html). When 40% of hosts took drugs early during infection, transmission was decreased by roughly one third in later waves, even despite decreased population immunity in the first wave. This was because the decrease in population immunity was not large enough to overcome the decreased effective reproductive number in later seasons (R e) from the combined effects of antivirals and the presence of some level of immunity. However, under higher levels of drug coverage and R 0 ⩾ 2, antiviral usage led to higher incidence in subsequent epidemic waves, since population immunity was further depressed during the first wave and inherent transmissibility was high enough to overcome suppression by drugs. Moreover, the level of drug coverage that minimized cases in subsequent waves depended on the time at which drugs were initiated during the infection time-course. These results highlight that the potential perverse effects of large-scale drug usage could lead to increased persistence of a novel pandemic strain and that the interaction effects of drug coverage and timing merit attention in disease control planning. Similarly, the interaction effects of drug coverage and R 0 of the resident strain suggest that drug-treatment policies should consider conditions of individual emergence events (e.g. fitness of the dominant strain and tendencies for cross-reactivity) for determining which antiviral strategy to implement during a given event rather than aiming to develop an ultimate strategy.
As with all mathematical models our results are subject to the assumptions of our model. We assumed that selection was limited to two genotypes (i.e. no antigenic drift across waves) which excludes adaptations that could cause increased fitness in either strain or additional changes in the structure of host-population immunity. Our analysis assumed that patients undergoing treatment follow their treatment to the end. In reality, the side-effects of drugs such as oseltamivir can be severe so that patients do not follow the treatment plan [Reference Strong47, Reference Wallensten48], which would lead to more heterogeneity in immunity in the host population and possibly overestimation of decreased population immunity due to 40% drug coverage. Next, in order to isolate the effects of drug resistance and drug usage on population immunity, we assumed that the novel strain (sensitive or drug-resistant) had the same inherent fitness as the resident strain. Although this is unlikely to be true, it is thought that drug-resistant strains of the 2009 pH1N1 showed similar fitness to the sensitive strains [Reference Hamelin49]. Moreover, the simplification allowed us to dissect effects of drugs from those of inherent viral fitness and reflects two-strain dynamics between strains with similar fitness levels. We also assumed that strains had 50% cross-reactivity. Higher or lower levels of cross-reactivity would alter the relative strain dynamics in later waves and these effects should be investigated in future work. There are also some population caveats, including variable seasonality trends and age heterogeneities [Reference Bansal36, Reference Tamerius50], which are discussed in the Supplementary text.
Finally, we used a simple model of within-host dynamics that excluded effects from innate immunity and approximated effects from target-cell depletion implicitly through a fixed carrying capacity. Our model had the advantage of reducing the number of free parameters for which there are few empirical data, and of reducing complexity so that the effects of drugs on within-host dynamics could be investigated at an epidemic scale. However, we emphasize that our model is not meant to make quantitative predictions of within-host dynamics. Quantitative models of antiviral effects on influenza within-host dynamics do exist [Reference Beauchemin51, Reference Dobrovolny52]; but are too parameter rich for extension to populations and recurring outbreaks in an agent-based framework (but see [Reference Handel, Longini and Antia9] for an elegant example of extension of a moderately complex within-host model to the single outbreak scale). Nonetheless, our model produces behaviour that is qualitatively similar to more complex models of influenza dynamics within hosts [Reference Handel, Longini and Antia28, Reference Beauchemin51–Reference Saenz55] and to empirical patterns of within-host dynamics under different drug treatments [Reference Li22, Reference Takahashi25, Reference Aoki56]. Thus, our simplified within-host model was effective for exploring how antiviral regimens impact epidemic dynamics during invasion of a cross-reactive novel strain.
In conclusion, we showed how drug regimens could alter the profile of population immunity through effects on within-host viral dynamics. It is critical to identify factors that shape population immunity since it is the main fabric of selection on co-circulating strains. Large advances have been made in understanding immunity mechanisms to influenza, but little is known about how drugs impact population immunity, how prior immunity impacts within-host dynamics in subsequent infections and how strain co-circulation alters the distribution of population immunity through within- and between-host interactions. Our results emphasize that these data gaps should be addressed since drug interventions, strain interactions and population-immunity structure all determine viral fitness. Once addressed, these data can be incorporated into quantitative epidemiological models that can be used for anticipating outbreak severity and strain persistence from early stage novel-strain emergence data. In the meantime, groundwork for practical models that will be used for pandemic preparedness should be developed with a structure that accommodates strain interactions and disease recurrence.
SUPPLEMENTARY MATERIAL
For supplementary material accompanying this paper, visit: http://dx.doi.org/10.1017/S0950268812000477.
ACKNOWLEDGEMENTS
Thanks to Colleen Webb for helpful comments on the manuscript. This work was supported by the National Science Foundation grant 0742373. K.M.P., S.R. and B.T.G. were also supported by the RAPIDD program of the Science and Technology Directorate, U.S. Department of Homeland Security, and the Fogarty International Center, NIH.
DECLARATION OF INTEREST
None.







