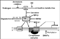
Introduction
Estrogen receptor-alpha (ER-α) is a member of the nuclear receptor (NR) family of ligand-activatable transcription factors. It is classified as a Type I NR, along with other steroid hormone receptors, including the progesterone receptor (PR), and androgen receptor (AR). When activated by their specific ligands, Type I receptors bind as homodimers to specific hormone response elements that are inverted half-sites: estrogen response elements (EREs), progesterone response elements (PREs), and androgen response elements (AREs). Type II receptors (e.g. thyroid receptor and retinoic acid receptors (RARs)) are usually localized in the nucleus even in the absence of ligands; and following ligation, they bind to their specific response elements as heterodimers with the retinoid X receptor (RXR, the 9-cis-RAR). Type III receptors (also called orphan NRs) have domain structures similar to other NRs but have no known ligands.
A second receptor with a high degree of homology to ER-α was discovered nearly a decade ago [1]. This receptor, called ER-β, has an overlapping but distinct set of functions and a different tissue distribution. A set of estrogen-related receptors (ERR-α, ERR-β, and ERR-γ) has also been identified [2]. The ERRs (also called ESRRA, ESRRB, and ESRRG, respectively with the above) are considered to be orphan NRs since high-affinity ligands have not been identified. Their physiologic functions are unclear, although they may play roles in normal development. This review will focus on ER-α, which is the major estrogen receptor (ER) in mammary tissue and which has been linked to the development and progression of human breast cancer. To understand how ER-α signaling can be inhibited, it is first necessary to understand a little about its basic structure and mechanism of activation.
Like other NRs, ER-α has a modular structure, consisting of six domains designated alphabetically (A–F). The A and B domains located at the aminoterminus of the ER-α protein contain a transcriptional activation function (AF-1) that does not require a ligand for activity and is constitutively active when linked to a suitable DNA-binding domain (DBD) [3]. The C domain contains the DBD, which contains two zinc fingers, structures that specifically recognize the ERE [4]. The D domain contains a flexible hinge domain; while the E domain contains the ligand-binding domain (LBD) and a second transcriptional activation domain that requires ligand for activation (AF-2). The carboxylterminal F domain contributes to the ligand-binding pocket when the receptor is activated and also to the binding of coactivators such as steroid receptor coactivator-1 (SRC-1) [5]. Maximal activation of ER-α requires an interaction between the two activation domains AF-1 and AF-2, which occurs when the ligand and coactivator proteins are present [6].
A classical scheme for ER-α activation involves a series of events. In this scheme, ligation of cytoplasmic ER-α with 17β-estradiol (E2) induces a conformational change in the receptor that causes:
- release of heat shock proteins (HSPs, chaperones that maintain ER-α in an inactive but activatable configuration);
- translocation of ER-α to the nucleus;
- homodimerization;
- binding of the dimer to two ERE half-sites within the regulatory regions of target genes;
- formation of a coactivator complex;
- recruitment of the basal transcription factor (BTF).
(Note: Not necessarily in that exact order.) ER-α mediated stimulation of expression of expression of target genes is a complex process that also involves:
- post-translational modification of histones (methylation and acetylation) to convert the local chromatin structure to a permissive state for transcription;
- ATP-dependent remodeling of chromatin by the SWI/SNF chromatin remodeling complex;
- recruitment of a ‘mediator’ (TRAP/DRIP) complex that makes stimulatory contacts with the BTF.
The main topic of this review is the mechanisms by which ER-α signaling may be inhibited, with particular emphasis on mechanisms that may be relevant to the etiology of breast cancer, its prevention, and its treatment. We include here a discussion of corepressors (coregulatory proteins that bind and inhibit ER-α and other NRs); the tumor suppressor BRCA1; anti-estrogens (selective estrogen receptor modulators (SERMs) that have found usage in breast cancer treatment and prevention); modification of estrogen production (by aromatase inhibitors); ER-β (which can oppose the action of ER-α); epigenetic silencing; and dietary factors that can inhibit ER-α by different mechanisms. This list is not complete. For example, ER-α signaling can be blocked by downregulation of its expression or its targeting for destruction in proteasomes [7]. However, it includes the mechanisms generally considered to be the most relevant to breast cancer treatment and prevention.
Inhibition of ER-α signaling
Corepressors
Transcriptional inhibition is now known to be an active process, mediated by high molecular weight multiprotein complexes that include transcriptional repressors, corepressors, histone deacetylases (HDACs) and a variety of auxiliary proteins (e.g. the Mi-2/NuRD complex, mSin3/HDAC1/2- and NCoR (nuclear receptor corepressor)/SMRT (silencing mediator of retinoid and thyroid receptors)/HDAC3-containing complexes) [8]. The role of corepressor proteins in negatively regulating ER-α signaling has been reviewed elsewhere in depth [9]. Here, we will highlight a few mechanisms through which different corepressors may block ER-α signaling. Transcriptional coregulators are auxiliary proteins that can either increase (coactivators) or decrease (corepressors) the rate of transcription of target genes for NRs such as ER-α. In some cases, the same coregulator can stimulate or inhibit transcription in different contexts.
The idea of ER-α corepressors has been somewhat controversial until recently, because it is in conflict with the classical scheme for ER-α activation described above, in which the unliganded ER-α is held in an inactive configuration in the cytoplasm complexes with chaperones (e.g. HSP90α and cochaperone p23) [10]. In the corepressor model, ER-α is localized in the nucleus, bound to DNA of target genes. However, it is transcriptionally inactive because it is associated with a corepressor complex, which converts the local chromatin structure into a repressive state. Over 20 different corepressors for ER-α have been identified [9]. These proteins bind to different portions of ER-α (e.g. AF-1, the DBD, and the AF-2/LBD region) and mediate repression by different mechanisms. For example, some corepressors act by recruiting HDACs (e.g. NCoR and SMRT); while other mediate HDAC-independent repression (e.g. RTA (repressor of Tamoxifen transcriptional activity) and LCoR (ligand-dependent corepressor)). Others act by competition with ER-α coactivators (e.g. REA (repressor of estrogen receptor activity) and SHARP (SMART/HDAC1-associated repressor protein)), interference with ER-α DNA binding (e.g. SHP (small heterodimer partner) and COUP-TF (chicken ovalbumin upstream promoter–transcription factor)), inhibition of ER-α dimerization (DAX-1 (DSS-AHC critical region on the X chromosome, gene 1) and TR2 (testis-derived orphan NR)), and/or sequestering ER-α in the cytoplasm (MTA1s, metastatic tumor-associated 1). A number of these corepressors work through more than one mechanism. Several of the above ‘corepressors’ are actually Type III (orphan) NRs, a number of which function, in part, to regulate the activity of Type I and Type II NRs (SHP, COUP-TF, DAX-1, and TR2).
An advance in the understanding of how NR coregulators work was the identification of helical motifs that mediate stimulator interactions with NRs (the ‘LxxLL’ motif (NR box)) or that mediate inhibitory interactions (the ‘(L/I)XX(I/V)I’ motif (CoRNR box) or the ‘Lxx(I/H)Ixxx(I/L)’ motif (extended helix motif)) [11–13]. Many coactivators contain one or more NR boxes (e.g. SRC-1, GRIP1, p300/CBP); while many corepressors contain CoRNR or extended helix motifs (e.g. NCoR and SMRT). The stimulatory and inhibitory motifs yield different helical configurations when they interact with NRs, such as ER-α. As noted below, the binding of some ligands (e.g. Tamoxifen) confers a three-dimensional conformation of ER-α that promotes the binding of corepressors rather than coactivators. Although many different corepressors clearly inhibit ER-α signaling in cell culture experiments, a lot less is known about the function of these molecules in vivo and, in particular, their roles in breast cancer development and progression.
BRCA1
The breast cancer susceptibility gene 1 (BRCA1) is a tumor suppressor gene, mutations of which are linked to hereditary early-onset breast cancer and breast/ovarian cancer syndromes [14]. A body of evidence suggests that BRCA1 may function, in part, as a caretaker gene to protect the integrity of the genome [15]. However, this generic function does not explain why BRCA1 mutations are linked to development of specific cancer types, and particularly to estrogen-responsive cancers (breast, endometrial, and cervical cancers) [16]. A molecular linkage between BRCA1 and ER-α was demonstrated by the finding that BRCA1 over-expression inhibits signaling by the liganded ER-α through the classical ERE in cultured cells [17]. This and subsequent studies revealed that BRCA1 blocks the activity of the intact ER-α and the E2-activated AF-2/LBD but has no effect on the activity of the E2-independent AF-1 domain [18]. BRCA1 also blocked the E2-stimulated expression of two estrogen-responsive genes, pS2 and cathepsin D [19].
Several potential mechanisms for BRCA1 inhibition of ER-α have been identified: (1) a physical interaction between the BRCA1 and ER-α proteins, which occurs independently of E2; and 2) downregulation of expression of p300, an NR coactivator [18,19]. The BRCA1:ER-α interaction was mapped to the aminoterminus of BRCA1 and a helical region within the AF-2 domain of ER-α [20]. The relevance of this mechanism to breast cancer was suggested by the finding that various cancer-associated BRCA1 mutant proteins failed to or showed reduced ability to inhibit ER-α activity [19,20]. Over-expression of p300 or its functional homolog CBP (the CREB-binding protein) rescued the BRCA1 inhibition of ER-α [18]. Interestingly, the ability of p300/CBP to rescue the inhibition of ER-α mapped to a conserved cysteine–histidine rich region (CH3), that was necessary and sufficient for rescue [18]. This finding suggests that the rescue function is distinct from the ability of p300/CBP to coactivate ER-α, since the histone acetyltransferase (HAT) and SRC-1-binding domains were dispensable for rescue. A further indication that coactivation and derepression are distinct mechanisms is the finding that GRIP1 (glucocorticoid receptor interacting protein-1) and PCAF (p300/CBP-associated factor) failed to rescue the BRCA1 inhibition of ER-α.
A higher resolution analysis of the BRCA1:ER-α interaction revealed two potential contact sites for BRCA1 on ER-α (the major site in a helical region within amino acids 338–379) and two contact sites for ER-α on BRCA1 (the major site containing a conserved helical motif (amino acids 86–95) resembling a previously identified nuclear corepressor motif (Lxx(I/H)Ixxx(I/L), where x = any amino acid) [20]. Based on a computer-generated model, BRCA1 heterodimerizes with ER-α via the anti-parallel α-helix domain, mainly using the third helix (amino acids 80–96) of BRCA1. The ER-α side of the interface is an α-helix of ER-α (amino acids 338–379), which is at the opposite side of the ER-α homodimerization interface. Two tumor-associated BRCA1 mutations at the BRCA1: ER-α interface (L63F and I89T) impaired the ability of BRCA1 to inhibit ER-α activity [20].
Several studies suggest that in addition to inhibiting E2-stimulated ER-α activity, BRCA1 also mediates ligand-independent repression of ER-α activity. Thus, ER-α was found to be constitutively active in the absence of ligand in cells harboring a homozygous null mutation of BRCA1; and BRCA1 was present at the ERE site on the promoter of the E2-responsive gene cathepsin D in MCF-7 human breast cancer cells before but not after stimulation with E2 [21]. Consistent with these findings, knockdown of endogenous BRCA1 by RNA interference conferred ligand-independent activation of ER-α in MCF-7 cells [21]. Furthermore, BRCA1 knockdown enhanced the degree of E2-stimulated ER-α activity, with a higher fold-stimulation of ER-α activity at lower doses of E2. Thus, the absence or inactivation of BRCA1 may promote ER-α activity under physiologic conditions of low or zero E2.
Recent studies have identified a pool of ER-α at the plasma membrane [22]. The membrane-localized ER-α is G-protein coupled and mediates signaling through cross-talk with the epidermal growth factor receptor (EGFR) or insulin-like growth factor-1 receptor (IGF1R) [22]. In several E2-responsive breast cancer cell lines, E2 caused activation of extracellular signal-related kinase (ERK) signaling that was blocked by wild type but not mutant BRCA1 [23]. BRCA1-blocked EGF-induced ERK activation and cell proliferation through a mechanism involving an ERK phosphatase [23]. Thus, BRCA1 may inhibit E2-stimulated cell proliferation by blocking cross-talk with the EGFR.
SERMs
Anti-estrogen therapy has been a mainstay in the treatment of hormone-responsive breast for over 20 years [24]. Tamoxifen, a SERM, has been the agent of choice for hormone-responsive cancers in post-menopausal women. SERMs have mixed ER-α antagonist/agonist activity in different tissues. Thus, Tamoxifen acts as an ER-α antagonist in the breast (hence its use in breast cancer) but functions as an ER-α agonist in bone, the cardiovascular system, and the uterus [25]. Manifestations of its agonistic activity include beneficial actions (potential reduction in osteoporosis and fractures) and harmful actions (increase in the incidence of endometrial carcinoma). Tamoxifen efficacy has been demonstrated as an adjuvant treatment for post-menopausal hormone receptor-positive breast cancer and, more recently, as a chemoprevention agent for younger women at high risk for the development of breast cancer [26]. Recent studies indicate that Tamoxifen binding to ER-α causes it to adopt a configuration that is different from that induced by E2 [27]. This configurational change not only inhibits the ability of ER-α to recruit coactivators but also enhances the recruitment of corepressors. Thus, the presence of Tamoxifen enhances the binding of NCoR, SMRT, REA, RTA, and other corepressors to ER-α; and over-expression of some corepressors enhance the antagonist activity of Tamoxifen, while under-expression of corepressors stimulate Tamoxifen's agonist activity [28].
Thus, one explanation for the ability of Tamoxifen to act as an antagonist vs. agonist in different tissues is tissue-specific differences in the balance of corepressors vs. coactivators. Because AF-1 can function autonomously as a ligand-independent transcriptional activation domain, it has been suggested that conditions that enhance AF-1 activity promote Tamoxifen's agonist activity, as observed in endometrial cells [29–31]. Various factors that may contribute to increased AF-1 activity have been identified, including [31–38]:
- differential effects of different anti-estrogens upon AF-1 activation (Tamoxifen allows greater AF-1 activation than Raloxifene, a SERM that does not increase the risk of endometrial carcinoma);
- differential effects of anti-estrogens in allowing the recruitment of corepressors and HDACs;
- signal transduction pathways that promote activation of AF-1 (e.g. MAP kinase mediated phosphorylation of serine-118 and c-Akt mediated phosphorylation of serine-167, both within the AF-1 domain of ER-α);
- increased ER-α expression and promoter occupancy.
These mechanisms may contribute to both the agonist activity of Tamoxifen in the uterus and the development of Tamoxifen resistance in breast cancer.
Other anti-estrogens include Raloxifene (Evista, a second generation SERM) and ICI 182,780 (Faslodex). Raloxifene exerts anti-estrogenic activity in the breast and uterus and pro-estrogenic activity in bone and in the cardiovascular system [39]. The Multiple Outcomes of Raloxifene Evaluation (MORE) trial was originally designed to test the ability of Raloxifene to reduce the incidence of osteoporosis and fractures in older women. Although the women in this trial were not specifically at increased risk for breast cancer, this trial showed a significant risk reduction for breast cancer, without a concomitant increase in the risk for endometrial cancer [40–42]. In both the MORE trial and the NSABP-P1 breast cancer prevention [26], Raloxifene and Tamoxifen, respectively, effectively prevented the development of ER-positive breast cancers but not ER-negative cancers. These findings suggest a difference in the cell of origin for ER-α (+) vs. ER-α (−) cancers or the conversion of ER-α (+) to ER-α (−) status during the pathogenesis of Tamoxifen-resistant ER-α (−) cancers. Faslodex (also called Fulvestrant) is a pure anti-estrogen that inhibits both AF-1 and AF-2 activity, promotes the degradation of ER-α, and, as far as we know, acts as an ER-α antagonist in all estrogen-responsive tissues (e.g. breast, bone, cardiovascular, and uterus) [43–45]. Its major use has been in treatment of advanced ER-α (+) breast cancers, particularly in the treatment of Tamoxifen-resistant cancers [45,46]. Interestingly, ICI 182,780 requires a specific ER-α corepressor (NEDD8, neural precursor cell expressed developmental downregulated 8) to exert its anti-estrogenic activity. Thus, inhibition of the NEDD8 pathway conferred cellular resistance to ICI 182,780 [44].
Modification of estrogen production
A recent advance in the treatment of estrogen-responsive breast cancer is the development of aromatase inhibitors. The clinical aspects of aromatase inhibitors have been reviewed elsewhere [47–49]. The enzyme aromatase (also called CYP19 or CYP19A1) is a member of the cytochrome P450 superfamily of microsomal monoxygenases that mediate the metabolism of drugs and xenobiotics, synthesis of cholesterol, and synthesis of steroids and other lipids [50]. Aromatase is localized in the endoplasmic reticulum and mediates the conversion of androgens into estrogen. In post-menopausal women, the major source of estrogen is the peripheral conversion of adrenal androgens into estrogen, which occurs in fat and other tissues [51]. Third generation aromatase inhibitors that have been tested in clinical trials include anastrazole (Arimidex), letrazole (Femara), and exemestane (Aromasin). These trials suggest that aromatase inhibitors are at least as effective as or more effective than Tamoxifen in reducing the risk of recurrence or new breast cancers in post-menopausal women with ER-positive tumors when used in an adjuvant setting [47]. A potential advantage of aromatase inhibitors is that unlike Tamoxifen, which acts as ER agonist in the uterus, aromatase inhibitors do not have pro-estrogenic effects and do not appear to increase the risk of endometrial cancer. A disadvantage is that aromatase inhibitors may increase the risk of osteoporosis and fractures because of the reduction of circulating estrogens. In contrast, Tamoxifen acts as an ER agonist in bone and does not increase the risk of fractures. Anastrazole has been approved by the Food and Drug Administration (FDA) for use as adjuvant treatment for breast cancer in post-menopausal women and appears to be a reasonable alternative to Tamoxifen.
Estrogen receptor-β
ER-β (also called ESR2, or estrogen receptor 2) is structurally similar to ER-β, particularly within the DBD, but exhibits a different tissue distribution and overlapping but distinct functional properties with respect to ligand selectivity, binding affinity, and transcriptional activation [1]. ER-β is highly expressed in the ovary, testis, prostate spleen, and thymus; but it is also found in other tissues, including the uterus and the breast [1,52]. In ER-β knockout (BERKO) mice, E2 caused an exaggerated proliferative and biochemical response in the uterus, suggesting that ER-β negatively regulates ER-α signaling [53]. In response to E2, ER-β may form heterodimers with ER-α, suggesting that heterodimerization with ER-β may modulate the function of ER-α [54,55]. In a study of ER-α or ER-β mediated signaling, co-expression of ER-α and ER-β caused reduced E2-stimulated transcription at low concentrations of E2 [55]. Similar findings were reported in an earlier study in which it was also observed that in contrast to ER-α, the ER-β AF-1 was a weak activator domain that actually reduced the transcriptional activity of the intact ER-β [55]. Co-expression of ER-α and ER-β not only resulted in a reduced sensitivity to E2, but also caused a reduction of the agonist activity and an increase in the antagonist activity of Tamoxifen [55–57]. These findings further support the idea that AF-1 may mediate the agonistic activity of Tamoxifen.
The vascular endothelial growth factor (VEGF) gene promoter contains an ERE and is E2 responsive. Interestingly, in human breast cancer cells expressing ER-α or ER-β alone, E2-stimulated VEGF transcription, while E2 inhibited VEGF transcription in cells expressing both ERs, suggesting that the heterodimer mediates an inhibitory signal [56]. While most studies suggest an antagonistic relationship between ER-α and ER-β when both are present in the form of a heterodimer, one recent study has challenged this view [58]. Here, it was reported that a genetic fusion of ER-α, with ER-β to form a single-chain ER, the heterofusion ER mimicked the properties of ER-α rather than ER-β, as suggested by the co-expression studies. The story is even further complicated by the finding that both ER-α and ER-β have naturally occurring splice variants that can modulate the activity of the full-length receptors [59]. Further studies will be required to determine the physiologic significance of ER-α/ER-β interactions in vivo and their relevance to breast cancer.
Epigenetic silencing of ER-α
Silencing of expression of subsets of genes occurs as a consequence of normal development (e.g. imprinting), aging and neoplasia and may contribute to these processes [60–62]. Expression of DNA (cytosine-5)-methyltransferase 1 (DNMT1) is called a ‘maintenance DNA methyltransferase’, because it methylates newly replicated DNA that is hemimethylated, thus maintaining the pre-replication methylation status [63]. DNMT3a and DNMT3b are classified as de novo methyltransferases, because they can methylate previously unmethylated DNA [64]. DNA methyltransferases may work in conjunction with HDACs and other repressors to maintain chromatin in a transcriptionally silent state. This mechanism is suggested by the observation that the methyl CpG-binding protein MeCP2 can recruit a multiprotein repressor complex containing the transcriptional repressor mSin3A, HDAC1, and HDAC2 [65]. DNA methyltransferases have also been shown to be present in repressor complexes containing HDACs and non-HDAC transcriptional repressor proteins [66,67]. DNA methyltransferase activity appears to be low in normal human cells, but DNMT1, DNMT3a, and DNMT3b may be over-expressed, with concomitant increases in DNMT activity in a variety of cancer types [68]. This correlates with the observation that the patterns of DNA methylation are consistently abnormal in human cancers; and the expression of tumor suppressor genes (e.g. the cell cycle inhibitor p16) may be reduced due to promoter methylation in some cancers [68,69]. Hypermethylation of CpG dinucleotide islands within gene promoters correlates strongly with transcriptional silencing of gene expression [69].
Epigenetic silencing of gene expression may contribute to the development of breast cancer. Thus, BRCA1 expression is frequently decreased or absent in sporadic breast and ovarian cancers [70–72]; and a subset of these cancers exhibit hypermethylation of the BRCA1 gene promoter on CpG islands [73,74]. In addition, some hormone receptor-negative breast cancers exhibit hypermethylation of the ER-α and/or PR promoters. Thus, about 25% of primary ER-α-negative breast cancers and about 40% of primary PR-negative cancers exhibit hypermethylation of the ER-α and PR promoters, respectively; whereas very few ER-α/PR-positive cancers showed methylation of these sites [75,76]. About 80% of BRCA1 mutant breast cancers are ER-α and PR negative; and a subset of these cancers exhibit hypermethylation of the ER-α promoter [77]. In this regard, dysregulation of expression of DNMT1 has been linked to loss of ER-α expression in ER-α-negative breast cancer cells [78]. Interestingly, a recent study indicates that antisense inhibition of DNMT1 causes conversion of ER-α-negative human breast cancer cells to ER-α positivity [79]. On the basis of these considerations, it has been suggested that inhibition of DNMT1 and HDAC inhibitors may be a useful therapeutic strategy for breast cancer [79]. It is, as yet, unclear if hypermethylation of the ER-α promoter is a primary event in the development of ER-α-negative breast cancer or if hypermethylation occurs following other mechanisms of transcriptional inhibition.
Methylation of the ER-α gene has been observed in a variety of other cancer types [68]. In an interesting study of colon cancer, hypermethylation of the ER-α promoter was observed in colon carcinoma cells, precancerous adenomatous polyps, and the normal colonic mucosa [80]. The incidence of ER-α promoter methylation appeared to be age related; and it increased sharply after age 50. In the same study, expression of exogenous ER-α inhibited the growth of colon cancer cell lines [80]. The authors proposed that hypermethylation of the ER-α gene is an age-related phenomenon; and colon cancer growth selects for hypermethylation because ER-α inhibits cancer growth.
As noted above, methylation of the PR promoter is also a common event in human breast cancers. The PR is a major transcriptional target of ER-α. Treatment of ER-α/PR-negative MDA-MB-231 breast cancer cells with a methylation inhibitor (5-azacytidine) lead to the re-expression of both ER-α and PR [81]. In this study, an anti-estrogen-blocked PR expression, while PR expression could be induced by the estrogen-activated ER-α without demethylation, suggesting that ER-α could overcome the repression of PR expression even in the presence of a methylated PR gene promoter. A final point of interest is the observation that chronic exposure of ER-α breast cancer cells to adriamycin (doxorubicin) caused their conversion to ER-α-negative status; and those ER-α-negative cells exhibited hypermethylation of the ER-α promoter [82]. These findings raise the possibility that adriamycin might promote hormone insensitivity by inducing the loss of ER-α expression.
Dietary factors
This section will not attempt to be comprehensive but will review a few of the dietary factors that can influence ER-α signaling. The relationship between obesity and breast cancer has been researched extensively and is reviewed elsewhere [83]. Obesity at menarche and during the post-menopausal years, as quantified by an increased body mass index (BMI), has been shown to increase the risk for breast cancer in the post-menopausal years. While increased endogenous estrogens derived from adipose tissue may be a contributory factor, the mechanism(s) underlying the increased breast cancer risk are probably considerably more complex [83].
Epidemiologic studies suggest that while a diet rich in total vegetables does not affect breast cancer risk, a diet rich in cruciferous vegetables (e.g. cabbage, cauliflower, and broccoli) confers a reduced risk for breast cancer [84,85]. Indole-3-carbinol (I3C), a micronutrient derived from glucobrassein, a component of cruciferous vegetables, has been found to exhibit anti-estrogenic properties and to prevent mammary carcinogenesis and other estrogen-responsive cancer types in experimental mouse and rat models [86–88]. One potential mechanism of action of I3C is through an alteration in hepatic microsomal metabolism of estrogen [86,89]. Thus, I3C stimulates 2-hydroxylation of estrone (E1) at the expense of 16α-hydroxylation, resulting in increased production of 2-OH-E1 and decreased production of 16-OH-E1, a potential carcinogen. I3C causes an increase in the ratio of urinary 2-OH-E1 to 16-OH-E1 in humans at well-tolerated doses [90–92], suggesting that it may be an attractive as a breast cancer chemoprevention agent. I3C exerts other actions that may contribute to its anti-estrogenic actions. Thus, I3C can bind to ER-α and inhibit E2-stimulated ER-α transcriptional activity [93,94]. The ability of I3C to inhibit E2-stimulated ER-α activity may be due, in part, to its ability to upregulate BRCA1 expression [94,95]. Finally, I3C and its major active metabolite diindolylmethane (DIM) also mediate E2-independent actions that may contribute to its anti-carcinogenic activity, including upregulation of expression of tumor suppressor genes ([BRCA1, BRCA, GADD45α (growth and DNA damage inducible 45α), E-cadherin, and PTEN (phosphatase and tensin homolog/mutated in multiple advanced cancers 1)) and decreased expression of cyclin-dependent kinase 6, among other activities [94–99].
Genistein is the major isoflavone derived from soy and is classified as a phytoestrogen. A high intake of soy contributes to the decreased incidence of breast cancer and prostate cancer in Asian women and men, respectively [100]. A recent meta-analysis suggests that increased intake of genistein contributes to a reduced breast cancer risk in pre-menopausal women [101]. One possible explanation is that genistein is a weak estrogen that stimulates ER-α activity in the absence of E2 but inhibits E2-stimulated ER-α activity [102]. Alternatively, genistein exhibits a higher affinity for ER-β than for ER-α [103], and its effect on breast cancer risk could be mediated through ER -β. In this regard, genistein mediates differential coactivator recruitment for ER-α vs. ER-β [104]. Genistein can also bind to and activate AR, suggesting it is also a phytoandrogen [105]. Genistein exhibits a variety of hormone-independent actions, including inhibition of cell proliferation, but these actions are usually observed at pharmacologic (>10 μM) rather than physiologic concentrations.
Lignans (enterodiol and enterolactone) are phytoestrogens derived from flaxseed through the action of bacteria in the colon on their precursor molecules. These compounds may protect against the development of pre-menopausal breast cancer [106,107]. Proposed mechanisms of action include inhibition of E2-stimulated ER-α mediated cell proliferation, stimulation of ER-β activity, induction of the expression of sex-hormone-binding globulin (SHBG), and an antioxidant activity [106]. Finally, alcohol (ethanol), while not a ‘dietary’ factor in the nutritional sense can modulate the risk for breast cancer. Thus, it has been established that moderate levels of alcohol consumption increase the risk for post-menopausal breast cancer, particular in collaboration with exogenous estrogens [108]. While the mechanism by which alcohol increases breast cancer risk are uncertain, a recent study indicates that ethanol increases ER-α expression and E2-stimulated ER-α transcriptional activity in cultured ER-α-positive human breast cancer cells [109]. The fold-stimulation of ER-α transcriptional activity was significantly greater than that of ER-α protein levels, suggesting that ethanol stimulates the intrinsic ER-α activity. This may be due in part to BRCA1, since ethanol caused corresponding decreases in the expression levels of BRCA1.
Conclusions
We have reviewed various mechanisms by which ER-α signaling can be inhibited, including transcriptional repression mediated by corepressors and BRCA1; inhibition of ER-α activity by SERMs; inhibition of ER-α production via aromatization; transcriptional silencing of the ER-α gene; heterodimerization with ER-β; and dietary agents that regulate ER-α signaling by various mechanisms (see Fig. 1). Where appropriate the clinical implications of these mechanisms have been discussed. None of the agents currently utilized to inhibit ER-α signaling are 100% effective. Thus, recurrences are observed after the use of Tamoxifen or aromatase inhibitors in the adjuvant setting for breast cancer; and neither Tamoxifen nor Raloxifene prevent the development of ER-α-negative breast cancers. And ER-α-positive cancers may develop resistance to hormonal manipulations or recur as ER-α-negative cancers [110]. Finally, existing therapeutic chemoprevention agents have side effects due to pro-estrogenic or anti-estrogenic activities in different tissues. These considerations indicate that although much progress has been made, further research is required to develop better strategies to inhibit ER-α signaling in breast cancer cells and avoid the adverse consequences of ER-α inhibition and/or stimulation in extra-mammary sites.

Figure 1. Schematic diagram illustrating some of the pathways of inhibition of ER-α signaling. See text for definition of abbreviations.
Acknowledgements
Dr. E. M. Rosen has been supported, in part, by grants from the United States Public Health Service (RO1-CA82599, RO1-CA80000, and RO1-ES09169) and by a grant from the Susan G. Komen Breast Cancer Foundation (BCTR0201295).