Introduction
Digenea Carus, 1963 and Aspidogastrea Faust and Tang, 1936 are two subclasses within the class Trematoda Rudolphi, 1808 (Cribb et al., Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a; Gibson et al., Reference Gibson, Jones and Bray2002; Kostadinova & Pérez-del Olmo, Reference Kostadinova, Pérez-del Olmo, Toldedo and Fried2014). Digeneans are the most diverse and numerically abundant group of parasitic metazoans, with approximately 18,000 nominal species included in the c. 2500–2700 nominal genera (see Cribb et al., Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a; Pérez-Ponce de León, Reference Pérez-Ponce de León2001; Gibson et al. Reference Gibson, Jones and Bray2002; Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003; Kostadinova & Pérez-del Olmo, Reference Kostadinova, Pérez-del Olmo, Toldedo and Fried2014; Littlewood et al., Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015). Several attempts have been made since the mid-19th century to achieve a stable classification scheme based on useful criteria (see Gibson, Reference Gibson, Gibson, Jones and Bray2002). Chronologically, the first attempts utilized the sucker arrangement. Later, other authors used cercarial morphology and life cycle patterns. More modern versions of the classification of the major groups of Digenea were proposed on cladistic analyses using morphological data from all life-history stages (Brooks et al., Reference Brooks, O'Grady and Glen1985; Gibson, Reference Gibson1987; Pearson, Reference Pearson1992). Then, a novel combination of morphological and molecular data was proposed (Cribb et al., Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a). Nowadays, the most accepted classification of the major groups is based on molecular data derived from phylogenetic assessments of two ribosomal genes (Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003; Littlewood et al., Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015).
Gibson & Bray (Reference Gibson and Bray1994) proposed a morphology-based classification of Digenea and subdivided the subclass into three orders, i.e. Strigeida, Echinostomida and Plagiorchiida; actually, these orders were used to organize three comprehensive volumes aimed at providing keys for the identification of the sexual adult trematodes (Gibson et al., Reference Gibson, Jones and Bray2002; Jones et al., Reference Jones, Bray and Gibson2005; Bray et al. Reference Bray, Gibson and Jones2008). Gibson & Bray (Reference Gibson and Bray1994) also predicted the increase in our understanding of the diversity, systematics and evolution of digeneans through molecular-based studies. Much progress has been gained in the last 25 years through continuous advancements of analytical and methodological tools to understand digenean evolution, and therefore, several phylogenetic classifications have been proposed. Brooks et al. (Reference Brooks, O'Grady and Glen1985, Reference Brooks, Bandoni, MacDonald and O'Grady1989) performed the first cladistic phylogenetic analyses of the subclass Digenea using morphological traits from all life-history stages. These studies represented a framework upon which the proposed phylogenetic classification and the character evolution of Digenea were further tested. The use of molecular tools in studies of intra-family and inter-family relationships of digeneans started in the 1990s (see Cribb et al., Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a and references therein). However, it was not until 2001 that Cribb et al. (Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a) used morphological and life-cycle characteristics, in combination with sequencing data from the 18S rRNA gene of 75 species of digeneans and three aspidogastreans as outgroups, to study the evolution of Digenea (also see Cribb et al. Reference Cribb, Bray and Littlewood2001b; Reference Cribb, Bray, Olson and Littlewood2003). The pioneer study by Cribb et al. (Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a) marked the starting point of what we currently know about the molecular phylogeny of Digenea. Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) provided a comprehensive molecular phylogenetic tree of Digenea by using the complete small subunit of the ribosomal RNA gene (ssrDNA = 18S rDNA) and partial (D1–D3) large subunit ribosomal gene (lsrDNA = 28S rDNA) of 163 digenean taxa representing 77 nominal families and seven aspidogastreans as outgroups; these nuclear genes were analysed independently and combined through parsimony and Bayesian inference (BI) methods. The analysis by Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) proposed a molecular phylogeny-based classification scheme for the subclass Digenea, and it has been widely used as a framework upon which modern studies have tackled questions regarding the evolution of digeneans (e.g. Fraija-Fernández et al., Reference Fraija-Fernández, Olson, Crespo, Raga, Aznar and Fernández2015).
The most recent molecular phylogenetic analysis of this group of parasitic platyhelminths was conducted by Littlewood et al. (Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015); these authors studied the diversification patterns of trematodes in reference to molecular phylogenies and definitive vertebrate host usage. A database was built by these authors with all the available data in GenBank (up to January 2013) for the 18S and 28S rRNA genes. Phylogenetic analyses were undertaken considering information for discrete species (Littlewood et al., Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015), including 202 and 556 sequences of 18S and 28S, respectively (Littlewood et al., Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015). Diversity and diversification patterns and processes were then described by reference to the phylogenetic analysis, where terminals were condensed to depict superfamilies (see figs 16.1 and 16.3 in Littlewood et al., Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015). Furthermore, the number of DNA sequences of nuclear and mitochondrial genes of digeneans has increased steadily in the last 15 years. Studies aimed at establishing more robust species limits and in detecting cryptic species increased the number of mitochondrial DNA sequences, particularly cytochrome c oxidase subunit 1 (cox1) (see Pérez-Ponce de León & Nadler, Reference Pérez-Ponce de León and Nadler2010); instead, studies of species diagnostics and phylogenetic assessments increased the taxon representation, at species, genus and family levels, for the 18S and 28S rRNA genes; still, these genes are the bedrock of digenean molecular systematics (see Lockyer et al., Reference Lockyer, Olson and Littlewood2003; Littlewood, Reference Littlewood2008).
Even though molecular data from nuclear rRNA genes have consistently resolved the inter-relationships among platyhelminths (see Littlewood, Reference Littlewood2008), the use of next-generation sequencing (NGS) methods in the last decade has increased our ability to readily sequence whole genomes leading to the possibility to explore new areas in the study of biodiversity and evolution (Olson et al., Reference Olson, Hughes and Cotton2016). As a result, the complete mitochondrial genome (mitogenome) of some digenean species is now available, and it has been used for phylogenetic analyses based on the premise that the number of sequenced base pairs, gene order and genome arrangement may provide evidence of a shared ancestry (see Littlewood et al., Reference Littlewood, Lockyer, Webster, Johnston and Le2006). As mentioned by Blasco-Costa et al. (Reference Blasco-Costa, Cutmore, Miller and Nolan2016): ‘sequencing whole mitochondrial genomes is possibly the next step beyond traditional DNA barcoding’. In this context, phylogenies of mitogenomes of closely related species encompassing c. 14,000 bp may provide better resolution of inter-relationships among digeneans and higher nodal support for phylogenetic relationship inference. NGS technologies are undoubtedly revolutionizing current taxonomic practices (see Olson et al., Reference Olson, Hughes and Cotton2016); in the case of digeneans, discrepancies in the higher-level classification system have been raised. Still, the results of phylogenetic analyses using the very few mitogenomes available in GenBank are questionable, their power to resolve deep levels of the tree such as superfamilies and suborders remain controversial and, additionally, taxon representation is still incomplete to generate a robust classification system. Considering the recent advancements of molecular techniques, the current review is an attempt to analyse once again the higher-level classification of the subclass Digenea using a more comprehensive taxon sampling of digeneans through conventional Sanger sequencing methods of nuclear rRNA genes. Here, we updated the database of the 28S and 18S rRNA genes of digeneans; a dataset of 1077 taxa representing 518 genera and 106 families, and 419 taxa representing 296 genera and 98 families, respectively (up until December 2017) was built. The new datasets were used to explore the robustness of the phylogenetic signal of ribosomal genes in resolving higher-level phylogenetic inter-relationships within Digenea; we discussed potential inconsistencies with previous molecular phylogenetic assessments in terms of the classification of digeneans at the superfamily and suborder levels, and evaluated the stability of the classification in comparison with the still incipient system derived from complete mitochondrial genomes and their performance to infer the higher-level inter-relationships among digeneans.
Gathering the data: sampling of sequences from GenBank
First, we analysed current trends in the use of molecular markers in the study of digenean taxonomy; we searched the GenBank database exhaustively for DNA sequences of species allocated in the subclass Digenea for different molecular markers for a period between January 1990 and December 2017. Only the most commonly used markers were considered for searching in the GenBank database, i.e. the large subunit of the ribosomal 28S rRNA gene (LSU); the small subunit of the ribosomal 18S rRNA gene (SSU); the internal transcribed spacers (ITS), including the ITS1, 5.8S and ITS2; the cytochrome c oxidase subunit I (cox1); and NADH1. The search strategy included seven combinations of terms to retrieve all the available information on trematode sequences:
(1) 28S: (‘trematoda’ [Organism] AND (‘28S’ OR ‘large subunit ribosomal’) AND ‘year’ [PDAT]) NOT ‘ITS’ NOT ‘Internal’ NOT ‘5.8S’ NOT ‘18S’ NOT ‘genome’.
(2) 18S: (‘trematoda’ [Organism] AND (‘18S’ OR ‘small subunit ribosomal’) AND ‘year’ [PDAT]) NOT ‘ITS’ NOT ‘Internal’ NOT ‘5.8S’ NOT ‘28S’ NOT ‘genome’.
(3) Only ITS1: (‘trematoda’ [Organism] AND ‘year’ [PDAT] AND (‘Internal transcribed spacer 1’ OR ‘ITS1’)) NOT ‘28S’ NOT ‘Internal transcribed spacer 2’ NOT ‘Internal transcribed spacer II’ NOT ‘ITS2’ NOT ‘genome’ NOT ‘NADH’ NOT ‘cytochrome’ NOT ‘NDH’.
(4) Only ITS2: (‘trematoda’ [Organism] AND ‘year’ [PDAT] AND (‘Internal transcribed spacer 2’ OR ‘ITS2’)) NOT ‘18S’ NOT ‘Internal transcribed spacer 1’ NOT ‘Internal transcribed spacer I’ NOT ‘ITS1’ NOT ‘genome’ NOT ‘NADH’ NOT ‘cytochrome’ NOT ‘NDH’.
(5) ITS1-5.8S-ITS2: (‘trematoda’ [Organism] AND ‘year’ [PDAT] AND (‘Internal transcribed spacer 1’ OR ‘Internal transcribed spacer I’ OR ‘ITS1’) AND (‘Internal transcribed spacer 2’ OR ‘Internal transcribed spacer II’ OR ‘ITS2’) AND ‘5.8S’) NOT ‘genome’ NOT ‘NADH’ NOT ‘cytochrome’ NOT ‘NDH’.
(6) cox1: (‘trematoda’ [Organism] AND ‘cytochrome’ AND ‘oxidase’ AND ‘subunit’ AND ‘year’ [PDAT]) NOT ‘ITS’ NOT ‘Internal’ NOT ‘5.8S’ NOT ‘28S’ NOT ‘genome’ NOT ‘18S’ NOT ‘subunit 3’ NOT ‘subunit III’ NOT ‘subunit 2’ NOT ‘subunit II’.
(7) NADH1: (‘trematoda’ [Organism] AND ‘dehydrogenase’ AND ‘subunit’ AND ‘year’ [PDAT]) NOT ‘genome’ NOT ‘subunit 3’ NOT ‘subunit III’ NOT ‘subunit 2’ NOT ‘subunit II’ NOT ‘subunit 4’ NOT ‘subunit IV’ NOT ‘subunit 4L’ NOT ‘subunit 5’ NOT ‘subunit V’ NOT ‘subunit 6’ NOT ‘subunit VI’.
For all the sequences retrieved for each molecular marker, we determined two parameters: the number of published sequences (individuals), and the number of species sequenced. Both parameters were organized by publication year as they appeared in GenBank, and accumulation curves were built to visualize the increase in both parameters over time (see fig. 1). The accumulation curve of the number of species per year only considered those identified up to the species level; instead, taxa identified up to the genus level and recorded in GenBank as ‘genus’ ‘sp.’ were excluded.
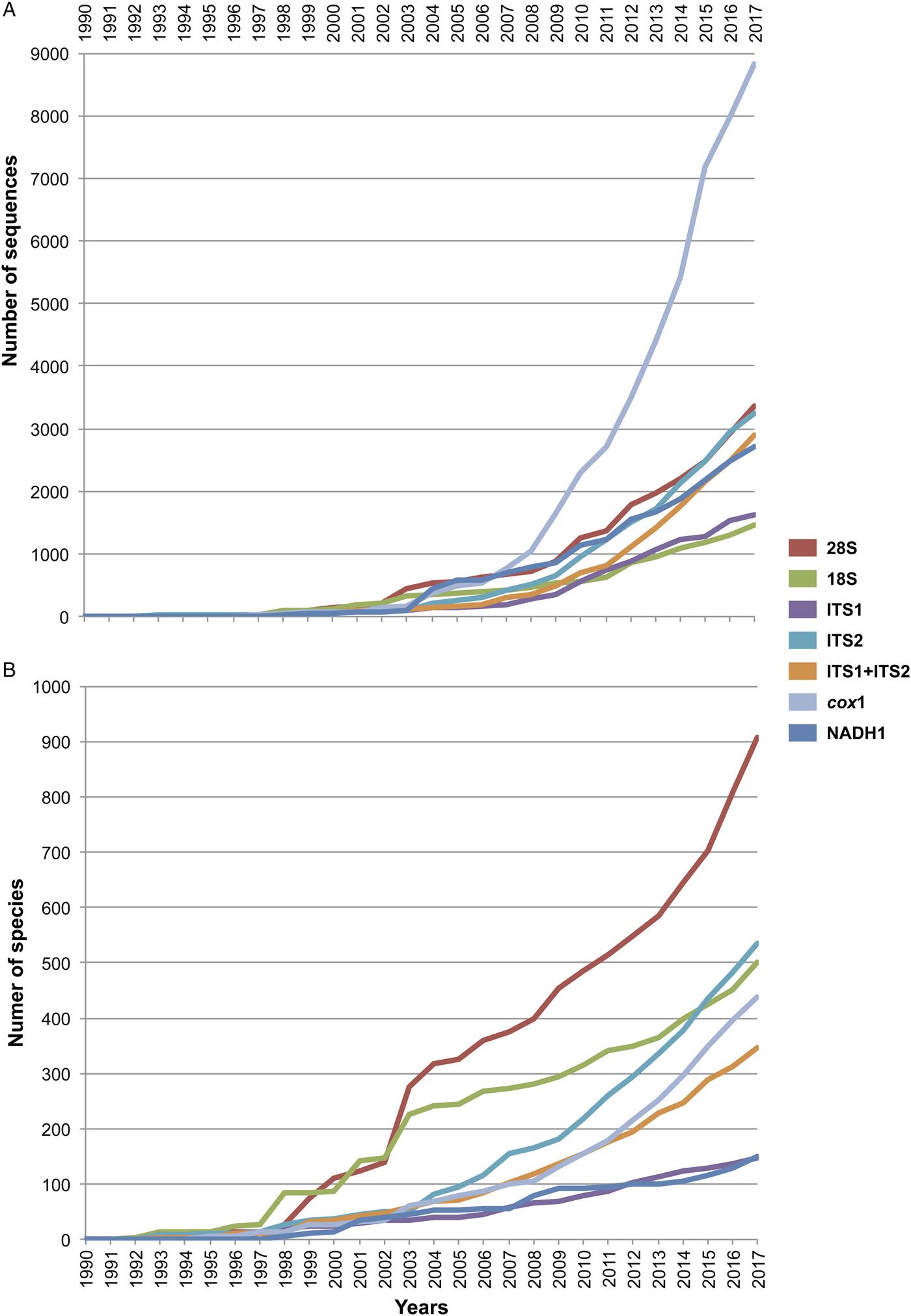
Fig. 1. Cumulative curve of the number of sequences (A) and number of sequenced species (B) of digeneans between 1990 and 2017 retrieved from GenBank for seven molecular markers. The internal transcribed spacers were analysed separately as ITS1 and ITS2, and together as ITS1-5.8S-ITS2.
Figure 1 depicts the accumulation curve of the number of sequences for each molecular marker, as well as the number of species of digeneans sequenced between 1990 and 2017, to determine the general trend in the generation of molecular data for this group of parasites. The number of sequences for individual digeneans has grown exponentially for the mitochondrial cox1 gene, probably due to its wide use in population genetics and phylogeographic studies, as well as in those related to DNA barcoding. Furthermore, the genetic library for the number of species of the 28S rRNA gene has grown at a faster rate (fig. 1); the number of genera and families to which these species belong has also increased, but at a slower rate. Figure 1 also shows that the number of sequenced species for the 18S ribosomal gene has not increased at the same rate in comparison to those of 28S, rendering the use of both markers in a concatenated analysis practically impossible for a comprehensive taxon sampling. Furthermore, the number of species of digeneans for which the internal transcribed spacer 2 (ITS2) has been sequenced increased at an even faster rate than the 18S gene. Thus far, the 28S rRNA ribosomal gene represents the largest dataset of digenean sequence data (fig. 1).
New molecular phylogenetic analyses: assembling newly generated sequences with data from GenBank
An exhaustive search was carried out in GenBank to find all the published sequences of the 18S and 28S belonging to the class Trematoda (both Digenea and Aspidogastrea). The criteria for the selection of taxa that were included in the analyses were as follows: (1) the sequence has a length greater than 400 bp; (2) the sequence belongs to taxa identified up to the level of species, and therefore those taxa determined only up to the genus or family are omitted; and (3) the sequence is from adult specimens, which might guarantee proper taxonomic identification. However, some exceptions were made to achieve the highest representation of families during the sequence selection. If a sequence of a certain genus was the only representative of a family but was not identified up to species level (e.g. Acanthochasmus sp., Amphiorchis sp., etc.), or the identification was based on larval forms (cercariae or metacercariae), then the sequence was included. Finally, the sequences of species determined as ‘cf’, ‘aff.’ or ‘sp.’ (of different lineages or possible new species according to the original publication) were also included (e.g. Euparyphium cf murinum, Phyllodistomum cf symmetrorchis).
Fifty-one novel sequences of 28S were generated for specimens collected from different vertebrate species in localities across Mexico (table 1). Sampled specimens were processed either for morphological examination to achieve the species identification, or for DNA extraction. Genomic DNA was extracted from each individual using the REDExtract-N-Amp Tissue PCR kit (Sigma, St. Louis, MS, USA) following the manufacturer's instructions. The 28S gene was amplified using polymerase chain reaction (PCR). The use of primers for PCR amplification, the thermo-cycling profile and sequencing followed the protocols described in greater detail by Hernández-Mena et al. (Reference Hernández-Mena, García-Varela and Pérez-Ponce de León2017). No new sequences of the 18S rRNA gene were obtained in our study.
Table 1. Species of digeneans for which 28S rDNA sequences were generated in the present study, ordered alphabetically by family.

A, amphibian; R, reptilian; B, bird; M, mammal; F, fish.
Alignments were built for each nuclear marker and analysed separately. Newly generated sequences of digeneans were aligned with all the sequences available in GenBank, along with those of some aspidogastreans that were used as outgroups for rooting the trees. A first alignment was built in each case to discard taxa with sequences that were not homologous, or to delete duplicate sequences. Alignments were performed with the software SATé, under the default setting SATé-II-fast (Aligner = MAFT, Merger = MUSCLE, Tree Estimator = RAXML, Model = GTRCAT; Liu et al., Reference Liu, Raghavan, Nelesen, Linder and Warnow2009, Reference Liu, Warnow, Holder, Nelesen, Yu, Stamatakis and Linder2012), implementing 200 iterations. After the iterations, the best alignments were used for phylogenetic analyses. Nucleotide substitution was estimated with jModelTest v2 (Darriba et al., Reference Darriba, Taboada, Doallo and Posada2012). The two alignments are available in supplementary material S1 and S2 as fasta files.
Phylogenetic trees were generated using the maximum likelihood (ML) and BI, and the model of nucleotide evolution GTR + GAMMA + I was employed for both phylogenetic inference methods. The ML analysis was performed using raxmlGUI v. 1.3 (Silvestro & Michalak, Reference Silvestro and Michalak2012); the tree with the best fit was obtained after 100 repetitions, and 10,000 bootstrap replicates were run to estimate the nodal support. MrBayes v. 3.2.1 (Ronquist et al., Reference Ronquist, Teslenko and van der Mark2012) was employed to perform the BI analysis, and four independent MCMC runs of 40 million generations each were performed. For each run, four chains with a heating parameter value of 0.9 were employed, and the tree topologies were sampled every 1000 generations (printfreq = 1000 samplefreq = 1000 diagnfreq = 10,000). Burn-in periods were set to the first 1500 generations. A 50% majority-rule consensus tree was obtained, and nodal support was estimated as posterior probability values. The phylogenetic trees obtained from both analyses for each molecular marker were visualized in FigTree v.1.4.3. (Rambaut, Reference Rambaut2016).
Phylogenetic analyses of 28S
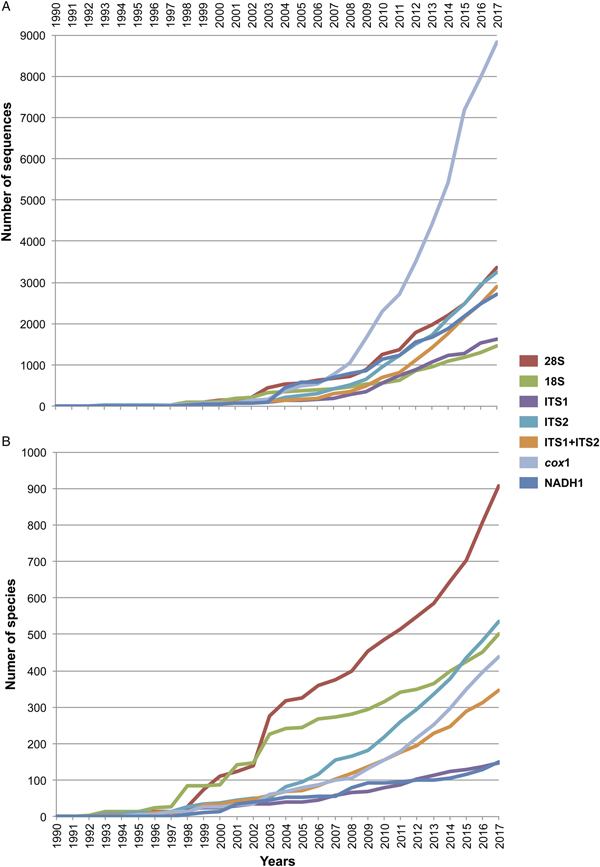
The final alignment of the 28S dataset consisted of 1077 terminals and was 1983 bp long; the nucleotide frequencies were A = 0.215, C = 0.216, G = 0.316 and T = 0.253. The phylogenetic tree obtained from the ML analysis had a likelihood of −198,962.8. Phylogenetic analyses inferred through ML and BI yielded an overall similar topology in terms of the inter-relationships among species and among genera allocated in families; however, the phylogenetic relationships among superfamilies were somewhat different in the ML and BI analyses. In fact, the 50% majority rule consensus tree of BI showed that the relationships among some superfamilies were not fully resolved, particularly within the Plagiorchiida. Figures 2 and 3 depict the condensed phylogenetic trees showing families as terminals (species and genera were collapsed into a single branch), and the arrangement of these families in superfamilies and suborders is presented according to the proposal of Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003). The two known and widely accepted orders within Digenea, i.e. Diplostomida and Plagiorchiida La Rue, 1957, were recovered as monophyletic groups with high bootstrap support for the ML analysis (100 and 83 for Diplostomida and Plagiorchiida, respectively), and moderate posterior probability supported the values from the BI analysis (0.75 and 0.75 for Diplostomida and Plagiorchiida, respectively) (figs 2 and 3). Supplementary figs S3 and S4 depict the ML and BI phylogenetic trees for individual sequences, respectively.
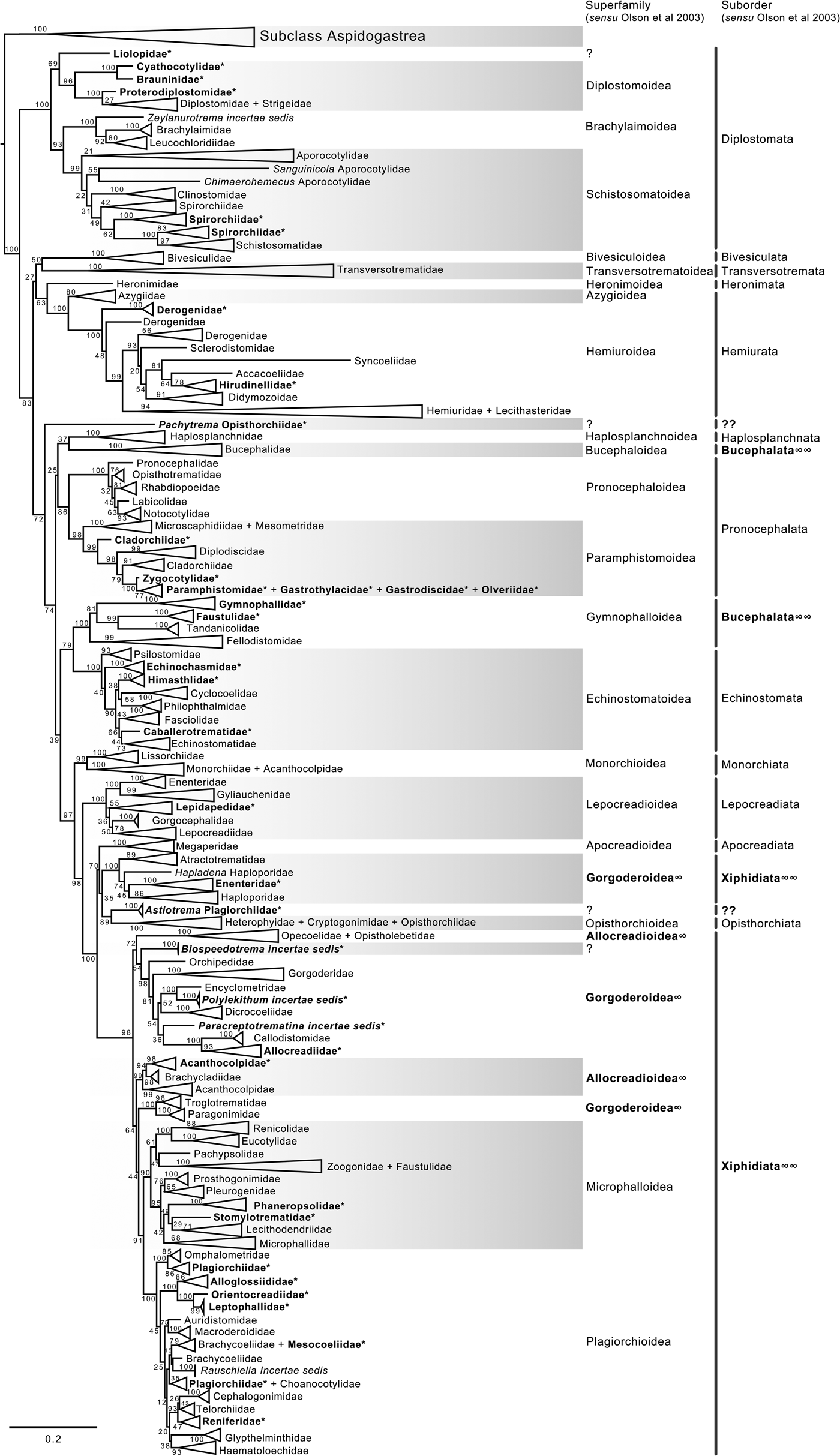
Fig. 2. Condensed tree based on the maximum-likelihood phylogenetic analysis constructed based on the partial (D1–D3) large subunit ribosomal gene (lsrDNA = 28S rDNA) of 1079 species of digeneans included in 106 families; species of the subclass Aspidogastrea were used as outgroups. The tree has a likelihood of −198,962.897393. Bootstrap support values for ML are provided at the nodes. The phylogenetic classification follows that of Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) for superfamilies and suborders of the subclass Digenea. * refers to the genera and families not included in Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) analysis. These taxa are in bold. ∞ indicates non monophyletic subfamilies and suborders.

Fig. 3. Condensed tree based on the Bayesian inference constructed based on the partial (D1–D3) large subunit ribosomal gene (lsrDNA = 28S rDNA) of 1079 species of digeneans included in 106 families; species of the subclass Aspidogastrea were used as outgroups. Posterior probability support values for BI are provided at the nodes. The phylogenetic classification follows that of Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) for superfamilies and suborders of the subclass Digenea. * refers to the genera and families not included in Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) analysis. These taxa are in bold. ∞ indicates non monophyletic subfamilies and suborders.
The phylogenetic classification of Digenea proposed by Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) was highly supported by our phylogenetic analyses at the level of superfamily and suborder. The inter-relationships within the Diplostomida, the less diverse order of Digenea, were consistent in both phylogenetic trees, with three major groups making up three superfamilies: (1) Diplostomoidea Poirier, 1886 (including the paraphyletic diplostomids + strigeids, in addition to proterodiplostomids, cyathocotylids and brauninids), and both analyses suggested that liolopids could be considered members of the Diplostomoidea; (2) Brachylaimoidea Joyeux and Foley, 1930 (including brachylaimoids, leucochloridiids, and Zeylanurotrema Crusz & Sanmugasunderam, 1973); and (3) Schistosomatoidea Stiles and Hassal, 1898, including the blood flukes (aporocotylids, spirorchiids, and schistosomatids) and clinostomids. Two families were recovered as paraphyletic, i.e. Aporocotylidae Odhner, 1912 and Spirorchiidae Stunkard, 1921. Instead, the inter-relationships of the Plagiorchiida were less consistent as several groupings were supported by low nodal support values. Two of the superfamilies (Gorgoderoidea Looss, 1901 and Allocreadioidea Looss, 1902) and two of the suborders (Bucephalata La Rue, 1926 and Xiphidiata Olson, Cribb, Tkach, Bray and Littlewood, 2003) within the order Plagiorchiida as recognized by Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) were not recovered as monophyletic in our analyses (see figs 2 and 3). Our 28S phylogenetic tree of Digenea included genera and species representatives of 106 families. Six families were recovered either as paraphyletic or polyphyletic assemblages, including Derogenidae Nicoll, 1910, Cladorchiidae Fischoeder, 1901, Acanthocolpidae Lühe, 1906, Faustulidae Poche, 1926, Enenteridae Yamaguti, 1958, Plagirochiidae Lühe, 1901 and Brachycoeliidae Looss, 1899 (fig. 2). Additionally, several groupings at the family level showed that the representative genera and species were intermingled, i.e. Diplostomidae Poirier, 1886 + Strigeidae Railliet, 1919; Hemiuridae Looss, 1899 + Lecithasteridae Odhner, 1905; Microscaphidiidae Looss, 1900 + Mesometridae Poche, 1926; Paramphistomidae Fischoederr, 1901 + Gastrothylacidae Travassos, 1934 + Gastrodiscidae Monticelli, 1892 + Olveriidae Yamaguti, 1958; Monorchiidae Odhner, 1911 + Acanthocolpidae Lühe, 1906; Megaperidae Manter, 1934; Heterophyidae Leiper, 1909 + Cryptogonimidae Ward, 1917 + Opisthorchiidae Looss, 1899; Opecoelidae Ozaki, 1925 + Opistholebetidae Fukui, 1929; Zoogonidae Odhner, 1902 + Faustulidae; Brachycoeliidae Looss, 1899 + Mesocoeliidae Dullfus, 1933; and Plagiorchiidae + Choanocotylidae Jue Sue & Platt, 1998 (see fig. 2). Furthermore, at least five additional families had to be erected to assign genera classified as inserta sedis after the phylogenetic analysis of the 28S rRNA gene: Zeylanurotrema (considered either a member of the Brachylaimidae Joyeux & Foley or UrotrematidaePoche, 1926); Biospeedotrema Bray, Waeschenbach, Dyal, Littlewood & Morand, 2014 (considered a member of the Opecoelidae); Polylekithum Arnold, 1934 and Paracreptotrematina Amin & Myer, 1982 (considered members of the Allocreadiidae Looss, 1902), and Rauschiella Babero, 1951 (considered a member of the Plagiorchiidae).
Within the Plagiorchiida, the ML analysis revealed two major clades, one including members of five superfamilies, i.e. Bivesiculoidea Yamaguti, 1934, Transversotrematoidea Witenberg, 1944, Heronimoidea Ward, 1918, Azygioidea Lühe, 1906 and Hemiuroidea Looss, 1899. The other 14 superfamilies were included in the second major clade (fig. 2). In our analyses, Pachytrema calculus Looss, 1907, a parasite of the marine bird Tringa nebularia Gunnerus from the Danube delta, Odessa region (GenBank accession AF151942, Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003), was recovered as the sister taxa of this large group of superfamilies, albeit with a moderate bootstrap support value (72%). Members of the genus Pachytrema Looss, 1907 allegedly belong to the family Opisthorchiidae Looss, 1899 (WoRMS, 2018a), although the position of this genus within the Plagiorchiida requires further scrutiny because opistorchiids were not monophyletic (see fig. 2). Because of the basal polytomy, the BI tree did not provide information about the position of Pachytrema (fig. 3). This family will require the erection of a superfamily and suborder, unless further phylogenetic analyses reveal its relationships with other superfamilies of plagiorchiids. The suborder Bucephalata was recovered as polyphyletic because the Bucephalidae were not grouped with Gymnophalloidea. By contrast, even though the Xiphidiata was recovered as paraphyletic because haploporids and atractotrematids (Gorgoderoidea sensu Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003) were not nested with other members of the Gorgoderoidea, most xiphidiatans showed a highly supported monophyletic group (98% bootstrap support and 0.73 posterior probabilities), including representatives of four large superfamilies of digeneans, i.e. Allocreadioidea (sensu Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003), Gorgoderoidea (sensu Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003), Microphalloidea Ward, 1901 and Plagiorchioidea.
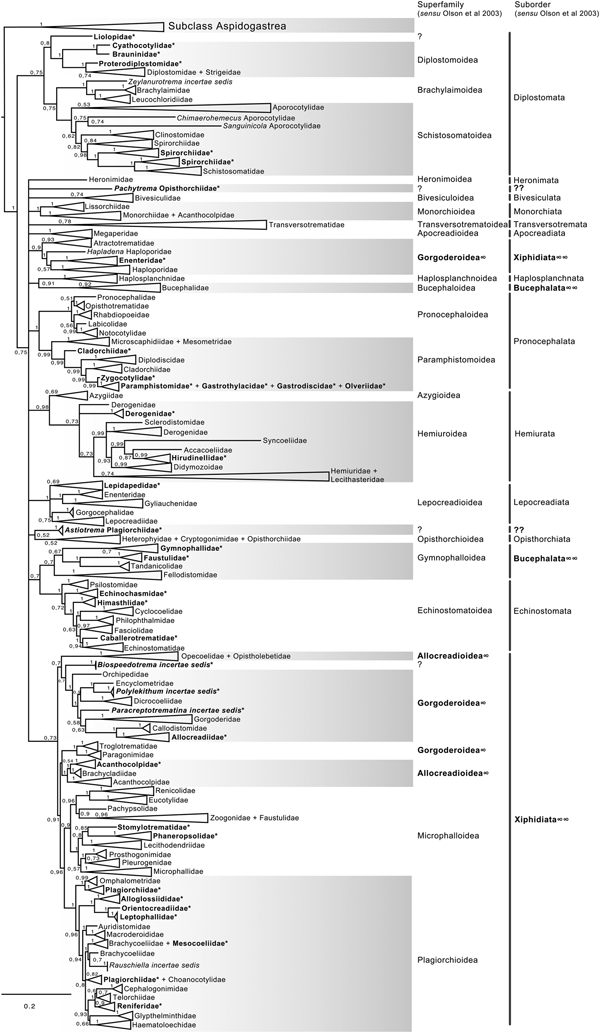
Phylogenetic analyses of 18S
The final alignment of the 18S dataset consisted of 419 terminals and was 2290 bp long; the nucleotide frequencies were A = 0.236, C = 0.218, G = 0.286 and T = 0.260. The phylogenetic tree obtained from the ML analysis had a likelihood of –73,815.4. Phylogenetic analyses inferred using ML and BI yielded a similar topology in terms of recovery of the same groups at the family and superfamily level. Additionally, even though both trees contained fewer terminals than the 28S dataset, the topology also recovered the orders Diplostomida and Plagiorchiida as monophyletic assemblages, with support values of 100 and 92 and 0.74 and 0.74 for ML and BI, respectively (figs 4 and 5). Supplementary figs S5 and S6 show the ML and BI phylogenetic trees for individual sequences, respectively. Figures 4 and 5 depict the condensed phylogenetic tree showing families as terminals, as well as the arrangement of these families in superfamilies and suborders following the proposal of Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003). Although the topology of both phylogenetic trees is similar, the BI analysis also showed a lower resolution for the inter-relationships among families of Plagiorchiids because they formed a polytomy, as the tree represents the 50% majority rule consensus tree. In the 18S analysis, the superfamily Allocreadioidea and two suborders, i.e. Bucephalata, and Xiphidiata, were recovered as polyphyletic. The inter-relationships among families showed some differences with respect to the 28S trees, but the major focus of our paper is on the superfamily and suborder levels. For example, in the 18S tree, the families included in Gymnophalloidea Ward, 1901, i.e. Gymnophallidae Odhner, 1905, Tandanicolidae Johnaton, 1927, and Fellodistomidae Nicoll, 1909, were nested in a group with transversotrematids, azygioids and hemuiroids. Instead, in the 28S trees, gymnophalloids were closely related to echinostomatoids. These and other relationships among families require further scrutiny.
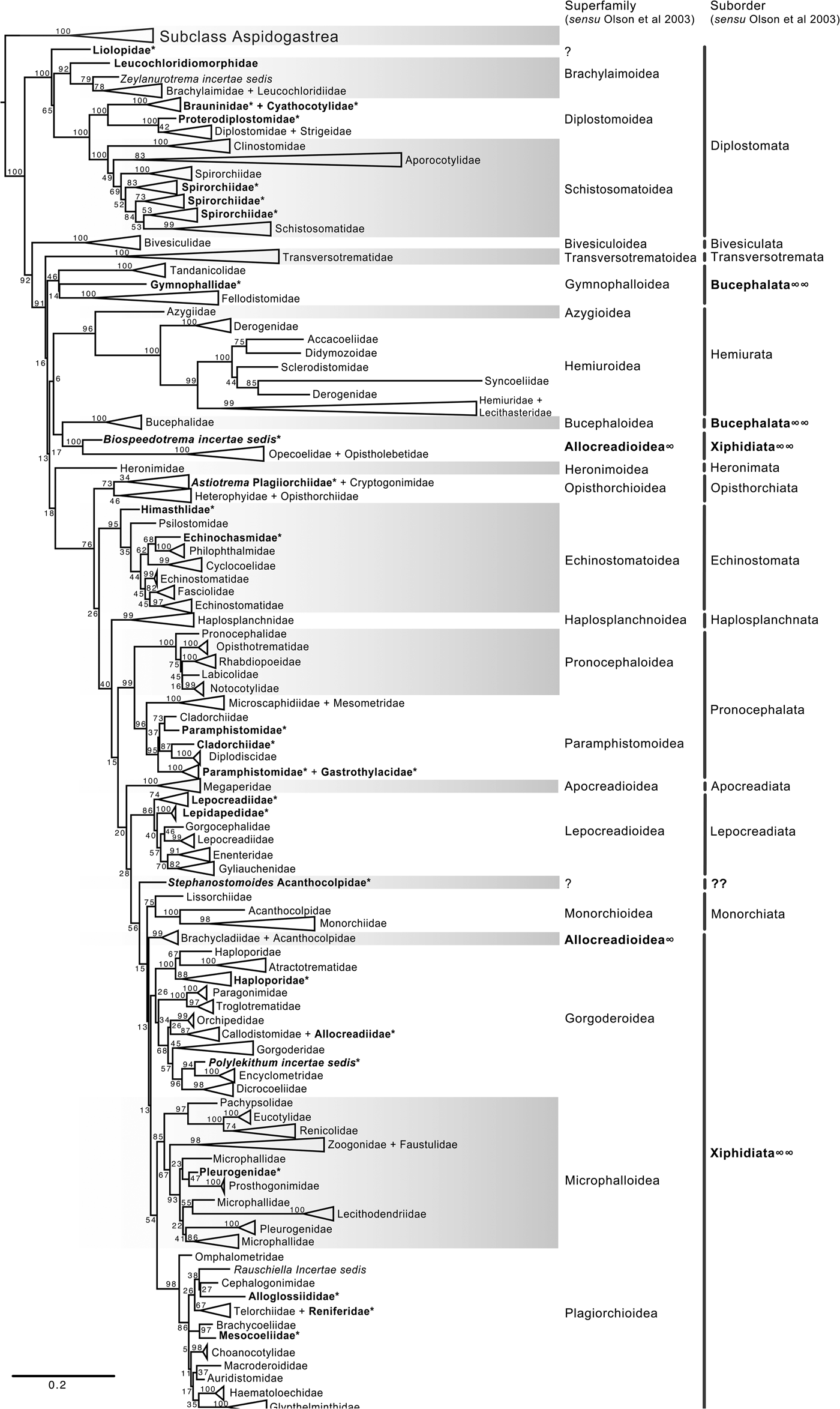
Fig. 4. Condensed tree based on the maximum-likelihood phylogenetic analysis constructed based on the complete small subunit of the ribosomal RNA gene (ssrDNA = 18S rDNA) of 419 species of digeneans included in 98 families; species of the subclass Aspidogastrea were used as outgroups The tree has a likelihood of −73,815.447087. Bootstrap support values for ML are provided at the nodes. The phylogenetic classification follows that of Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) for superfamilies and suborders of the subclass Digenea. * refers to the genera and families not included in Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) analysis. These taxa are in bold. ∞ indicates non monophyletic subfamilies and suborders.
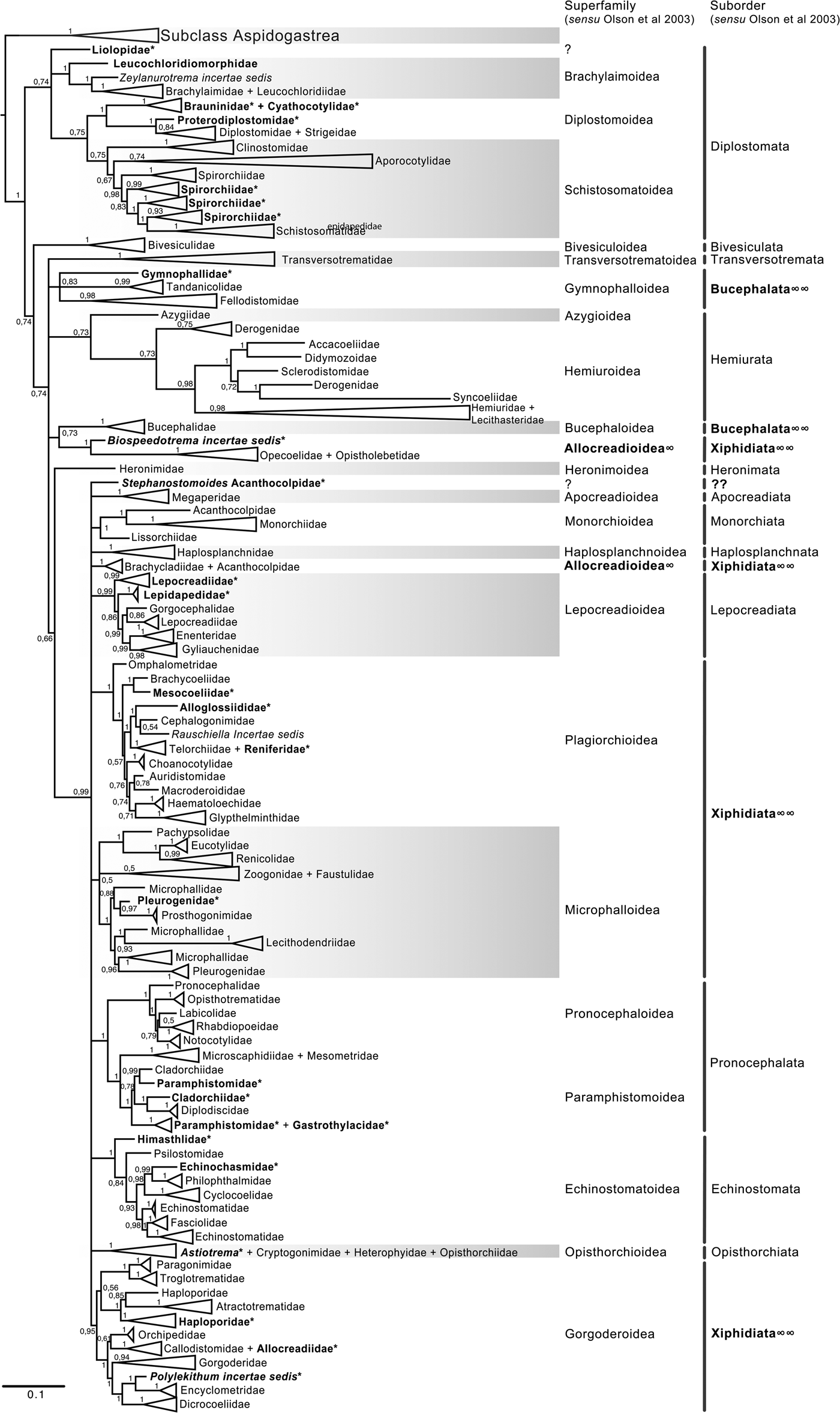
Fig. 5. Condensed tree based on the Bayesian inference constructed based on the complete small subunit of the ribosomal RNA gene (ssrDNA = 18S rDNA) of 419 species of digeneans included in 98 families; species of the subclass Aspidogastrea were used as outgroups. Posterior probability support values for BI are provided at the nodes. The phylogenetic classification follows that of Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) for superfamilies and suborders of the subclass Digenea. * refers to the genera and families not included in Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) analysis. These taxa are in bold. ∞ indicates non monophyletic subfamilies and suborders.
Revisiting the higher-level classification
The inter-relationships of Digenea are better explained through the analysis of the 28S rRNA gene because it is a molecular marker with a higher representation in the number of species and the number of families, providing phylogenetic signal to resolve sister group relationships not only among species and genera but also among higher-level groups, such as families, superfamilies and suborders. Our phylogenetic analysis consisted of 1077 taxa allocated in 106 nominal families for this molecular marker. Using a conservative estimation of a digenean species richness of c. 18,000 species (Cribb et al., Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a; Pérez-Ponce de León, Reference Pérez-Ponce de León2001; Gibson et al. Reference Gibson, Jones and Bray2002; Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003; Kostadinova & Pérez-del Olmo, Reference Kostadinova, Pérez-del Olmo, Toldedo and Fried2014; Littlewood et al., Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015), the genetic library of 28S included approximately only 5.5% of the species included in the subclass Digenea. Still, the sample size of our analysis for the 28S rRNA gene included almost 900 more taxa and 29 more families than those considered in the molecular phylogenetic analyses by Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003). However, it should also be noted that despite a smaller number of species represented in the 18S data set, the phylogenetic analyses using this gene were in general terms congruent with the 28S trees since they yielded overall similar higher-level groups (families, superfamilies and suborders). Additionally, despite the accomplishment of a large sequencing effort in the last 15 years for this group of parasitic Platyhelminthes, our results are largely consistent with the topology of the trees obtained in previous phylogenetic analyses (see Olson et al. Reference Olson, Cribb, Tkach, Bray and Littlewood2003; Littlewood et al., Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015). These findings indicate that the current classification system at higher levels of the taxonomic hierarchy of the group using nuclear markers through conventional Sanger sequencing methods is stable.
With the exception of the suborder Xiphidiata and Bucephalata and the superfamilies Gorgoderoidea and Allocreadioidea (sensu Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003), all major groupings were recovered as monophyletic in our analyses, indicating the robustness and stability of the higher-level classification of Digenea using 28S. A different scenario is shown, however, at the inter- and intra-family levels. Phylogenetic relationships among genera within a family, or even families within superfamilies are not fully resolved, most likely due to an incipient representation of taxa. The branch topology at the family level might be a reflection of the increase in representation of families in the analyses, but this might also be a result of the dynamic progress made in the taxonomy of particular digenean groups through molecular phylogenetic analyses. For instance, Tkach et al. (Reference Tkach, Pawlowski and Mariaux2000) used 28S rDNA sequences to study the inter-relationships of the suborder Plagiorchiata, one of the derived and most diverse groups of Digenea. These authors determined that the suborder, as traditionally conceived at that moment, was not monophyletic, and they proposed that it consisted of eight families included in two superfamilies: Plagiorchioidea and Microphalloidea. Two years later, Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) included in the analysis more representative species of the order Plagiorchiida and proposed a new suborder, Xiphidiata, to include the superfamilies contained in Plagiorchiata (sensu Tkach et al. Reference Tkach, Pawlowski and Mariaux2000) plus the superfamilies Gorgoderoidea and Allocreadioidea. Our analyses were consistent with Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003), except that Xiphidiata was not recovered as monophyletic because members of the families Atractotrematidae, Haploporidae and Cadenatella Dollfus, 1946 were not nested within the group in the 28S dataset (figs 2 and 3); the same results were obtained using the 18S dataset. Bray et al. (Reference Bray, Waeschenbach, Cribb, Weedall, Dyal and Littlewood2009, Reference Bray, Cribb, Waeschenbach and Littlewood2014) used molecular evidence to demonstrate that Cadenatella belongs in the superfamily Haploporoidea and not in Enenteridae. Still, haploporoids were not recovered as members of the Xiphidiata in our analyses. Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) erected the suborder Xiphidiata to include digeneans with cercariae bearing a stylet, a feature that is absent in members of the Haploporidae. In our analyses, haploporids and atractotrematids were nested in a clade of the paraphyletic Gorgoderoidea (following Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003 classification), although the Haploporoidea have been recognized as a valid superfamily that includes species of Haploporidae and Atractotrematidae (see Jones et al., Reference Jones, Bray and Gibson2005; Littlewood et al., Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015). In the World Register of Marine Species, the superfamily Haploporoidea is also considered valid, but it is included in the suborder Xiphidiata. Based on our phylogenetic results, we propose the name Haploporata nom. nov. for the suborder that includes several families included in Haploporoidea, a superfamily that is recognized as valid (see Littlewood et al., Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015; WoRMS, 2018b). In this case, the cercariae of the members of the group lack a stylet, and the synapomorphy that defines the group is the presence of a hermaphroditic sac, as defined in the key to superfamilies in Bray et al. (Reference Bray, Gibson and Jones2008). The addition of Haploporata nom. nov. at the subordinal level also resolves the polyphyly of Xiphidiata in the 28S dataset (fig. 2).
The suborder Bucephalata was recovered as polyphyletic in our analyses of 28S (and also in the 18S analyses) because the suborder include families of the superfamily Gymnophalloidea, i.e. Gymnophallidae, Tandanicolidae, Fellodistomidae and Faustulidae in part (see Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003; WoRMS, 2018c). Cribb et al. (Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a) provided the first molecular evidence to show that Bucephalidae (superfamily Bucephaloidea) is not closely related to Fellodistomidae and Tandanicolidae. Our phylogenetic analyses confirmed the results of Cribb et al. (Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a) because bucephalids are nested in a separate clade from the sister taxa of haplosplachnids (superfamily Haplosplachnoidea). Members of the superfamily Bucephaloidea have been diagnosed in the key to the superfamilies proposed by Bray (Reference Bray, Bray, Gibson and Jones2008) by having a mouth well apart from the anterior extremity; caecum single, elongate to saccular; ventral sucker absent; and genital pore close to the posterior end of body. The superfamily is considered to include only the Bucephalidae Poche 1907 (Overstreet & Curran, Reference Overstreet, SS, Gibson, Jones and Bray2002). Based on the phylogenetic analyses, we propose the name Gymnophallata nom. nov. as the suborder that includes members of the Gymnophalloidea. Furthermore, Bray (Reference Bray, Gibson, Jones and Bray2002) considered Gymnophalloidea to contain species of the families Gymnophallidae, Fellodistomidae and Tandanicolidae. Our phylogenetic analyses showed that Faustulidae in part were also nested within the Gymnophalloidea, although the family was recovered as a polyphyletic group, supporting the findings obtained by Cutmore et al. (Reference Cutmore, Bray and Cribb2018). Previous molecular evidence provided by Hall et al. (Reference Hall, Cribb and Barker1999) and Cribb et al. (Reference Cribb, Bray, Littlewood, Pichelin, Herniou, Littlewood and Bray2001a) has shown that the Faustulidae are distantly related to Fellodistomidae and closely related to the Zoogonidae. Our analyses also confirm these findings. The species of faustulids nesting within the Gymnophalloidea are Pseudobacciger cheyenae Sun, Bray, Yong, Cutmore and Cribb, 2014 and Bacciger astyanactis Lunaschi, 1988 (added to the analysis of the 28S phylogenetic tree). A recent phylogenetic analysis using 28S and ITS DNA sequences has demonstrated that even though P. cheyenae is currently recognized within the Faustulidae, this species is not closely related to the other faustulid genera, such as Antorchis Linton, 1911, Bacciger Nicoll, 1924, Paradiscogaster Yamaguti, 1934 and Trigonocryptus Martin, 1958; Cutmore et al. (Reference Cutmore, Miller, Bray and Cribb2014) demonstrated that these genera are associated with the Zoogonidae in the Microphalloidea; instead, P. cheyenae is nested within Gymnophalloidea as a sister taxa to the Tandanicolidae (Sun et al. Reference Sun, Bray, Yong, Cutmore and Cribb2014). Based on these results, the species Bacciger astyanactis must be re-allocated because it does not belong in Bacciger and should probably be included within Pseudobacciger. This nomenclatural change requires further scrutiny and a detailed study of the morphological traits used to diagnose the genus. Additionally, even though it remains unknown whether the two species belong in the same genus, they still require the erection of a new family. Still, according to the phylogenetic analysis using the 28S rRNA gene by Cutmore et al. (Reference Cutmore, Bray and Cribb2018), Bacciger might be retained in the Microphallidae. These authors sequenced two new species of Bacciger (not included in our analysis), and both species nested as the sister group of Pseudobacciger cheyenae, also rendering Bacciger polyphyletic as a member of the Tandanicolidae.
In their revised classification of Digenea, Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) recovered Gorgoderoidea and Allocreadioidea as monophyletic groups. However, these two superfamilies were recovered as non-monophyletic assemblages in our 28S phylogenetic trees (figs 2 and 3). Gorgodedoidea should be considered valid for the monophyletic group that includes Orchipediidae, Encyclometridae, Dicrocoeliidae, Gorgoderidae, Callodistomidae and Allocreadiidae (in addition to the three genera uncovered as incertae sedis, i.e. Biospeedotrema, Polylekithum and Paracreptotrematina, all of which necessitate the erection of a new family after detailed morphological scrutiny). In our analyses, Haploporidae + Atractotrematidae (and Cadenatella) were monophyletic and should be considered as Haploporoidea, not Gorgoderoidea. Interestingly, the family Allocreadiidae was a member of Gorgoderoidea, not Allocreadioidea. The family was erected in 1902 by Looss; however, following the International Code of Zoological Nomenclature, the name Gorgoderidae gained priority because it was erected in 1899 by Looss to include the type-genus Gorgodera Looss, 1899. Instead, Allocreadioidea is recognized as a superfamily that contains two large groups in the phylogenetic trees reported by Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003), one containing Opecoelidae + Opistholebetidae and the other Brachycladiidae + Acanthocolpidae. In our opinion, based on the new phylogenetic tree, the superfamily Allocreadioidea should not be recognized as valid; the grouping of Opecoelidae and Opistholebethidae (the latter recognized as a synonym in WoRMS) should be included in their own superfamily, for which the name Opecoelioidea should be used as in Littlewood et al. (Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015). Additionally, for the monophyletic group containing Brachycladiidae + Acanthocolpidae, the name Brachycladioidea should be used. Curran et al. (Reference Curran, Tkach and Overstreet2006) were the first using the superfamily name Brachycladoidea after recognizing that Allocreadioidea of Olson et al. was not sustainable. The family group name Brachycladiidae Odhner, 1905 predates Acanthocolpidae Lühe, 1906.
Several other studies have also illustrated the dynamic progress achieved in the taxonomy of particular digenean groups through 28S rDNA phylogenetic analyses. These studies have resulted in more stable classification schemes of genera within families and families within superfamilies of the subclass Digenea. For example, at the superfamily level, the studies of Microphalloidea (Tkach et al., Reference Tkach, Littlewood, Olson, Kinsella and Swiderski2003), Lepocreadioidea Odhner, 1905 (Bray et al. Reference Bray, Waeschenbach, Cribb, Weedall, Dyal and Littlewood2009), Brachylaimoidea (Heneberg et al., Reference Heneberg, Sitko and Bizos2016) Echinostomatoidea Looss, 1899 (Tkach et al., Reference Tkach, Kudlai and Kostadinova2016), and the recent study of Hemiuroidea by Sokolov et al. (Reference Sokolov, Atopkin, Urabe and Gordeev2018), can be highlighted. Additionally, at the family level, several studies have determined the inter-relationships among genera and the re-organization of subfamilies, such as the study by Tkach et al. (Reference Tkach, Gradda-Kazubska and Swiderski2001) with Omphalometridae Looss, 1899 by Choudhury et al. (Reference Choudhury, Rosas-Valdez, Johnson, Hoffman and de León G2007) with Allocreadiidae Looss, 1902 by Cremonte et al. (Reference Cremonte, Gilardoni, Pina, Rodrigues and Ituarte2015) with Gymnophallidae Odhner, 1905 by Nolan et al. (Reference Nolan, Curran, Miller, Cutmore, Cantacessi and Cribb2015) with Bucephalidae Poche, 1907 by Bray et al. (Reference Bray, Cribb, Littlewood and Waeschenbach2016) with Opecoelidae Ozaki, 1925 by Hernández-Mena et al. (Reference Hernández-Mena, Mendoza-Garfias, Ornelas-García and de León G2016) with Macroderoidiidae McMullen, 1937 by Curran et al. (Reference Curran, Pulis, Andres and Overstreet2018) and by Atopkin et al. (Reference Atopkin, Besprozvannykh, Ha, Nguyen, Nguyen and Chalenko2019) with Haploporidae. Still, many other families remain in need of revision through the use of molecular 28S rDNA sequences (and other informative molecular markers) to test the morphology-based classification schemes and to generate a more stable classification at that level of the taxonomic hierarchy.
Kostadinova & Pérez-del Olmo (Reference Kostadinova, Pérez-del Olmo, Toldedo and Fried2014) noted that, in general terms, the molecular phylogenetic analysis of Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) was congruent with the classification system of Digenea in the three volumes of Keys to the Trematoda (Gibson et al., Reference Gibson, Jones and Bray2002; Jones et al., Reference Jones, Bray and Gibson2005; Bray et al. Reference Bray, Gibson and Jones2008), with some exceptions in the superfamilial placement, although the molecular phylogeny did not support the traditional classification of the group in three suborders. Kostadinova & Pérez-del Olmo (Reference Kostadinova, Pérez-del Olmo, Toldedo and Fried2014) also provided arguments about important omissions in the molecular phylogeny of Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) because the study was limited in the diversity of taxa, such as the type-families of superfamilies, for example, Allocreadioidea, Gymnophalloidea and Paramphistomatoidea. These omissions were actually acknowledged by Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003), and these authors discussed that the lack of sequencing data for some families was crucial to the full elucidation of the digenean phylogeny. As previously discussed in our study, the previous two decades have witnessed an increased effort to sample and characterize morphologically and molecularly representative species of digeneans from different families and superfamilies. The augmented representation of species and genera of digeneans allowed us to conduct new phylogenetic analyses and to test the phylogenetic classification of Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003). Overall, the results were consistent, even though the sampling effort was increased by almost four orders of magnitude, and by almost two orders of magnitude comparison with the taxon sampling in the analysis by Littlewood et al. (Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015). Interestingly, our analysis yielded the same 24 superfamilies recovered by Littlewood et al. (Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015), although the resolution was slightly higher, polytomies were resolved and nodal support was relatively higher for most superfamily groupings, at least in the ML analysis. As previously argued, the 28S rDNA gene provides an adequate phylogenetic signal and overall topological stability. Based on our results (ML analysis of the 28S rRNAgene), we slightly modified the phylogeny-based classification of the Digenea proposed originally by Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003):
Class Trematoda Rudolphi, 1808
Subclass Aspidogastrea Faust and Tang, 1936
Subclass Digenea Carus, 1863
Order Diplostomida Olson, Cribb, Tkach, Bray & Littlewood, 2003
Suborder Diplostomata Olson, Cribb, Tkach, Bray & Littlewood, 2003
Superfamily Brachylaimoidea Joyeux and Foley, 1930
Superfamily Diplostomoidea Poirier, 1886
Superfamily Schistosomatoidea Stiles and Hassall, 1898
Order Plagiorchiida La Rue, 1957
Suborder Bivesiculata Olson, Cribb, Tkach, Bray & Littlewood, 2003
Superfamily Bivesiculoidea Yamaguti, 1934
Suborder Transversotremata Olson, Cribb, Tkach, Bray & Littlewood, 2003
Superfamily Transversotrematoidea Witenberg, 1944
Suborder Heronimata Skrjabin and Schulz, 1937
Superfamily Heronimoidea Ward, 1918
Suborder Hemiurata Skrjabin and Guschanskaja, 1954
Superfamily Azygioidea Lühe, 1909
Superfamily Hemiuroidea Looss, 1899
Suborder Haplosplanchnata Olson, Cribb, Tkach, Bray & Littlewood, 2003
Superfamily Haplosplanchnoidea Poche, 1925
Suborder Bucephalata La Rue, 1926
Superfamily Bucephaloidea Poche, 1907
Suborder Pronocephalata Olson, Cribb, Tkach, Bray & Littlewood, 2003
Superfamily Pronocephaloidea Looss, 1899
Superfamily Paramphistomoidea Fischoeder, 1901
Suborder Gymnophallata nom. nov.
Superfamily Gymnophalloidea Odhner, 1905
Suborder Echinostomata La Rue, 1926
Superfamily Echinostomoidea Looss, 1902
Suborder Monorchiata Olson, Cribb, Tkach, Bray & Littlewood, 2003
Superfamily Monorchioidea Odhner, 1911
Suborder Lepocreadiata Olson, Cribb, Tkach, Bray & Littlewood, 2003
Superfamily Lepocreadioidea Odhner, 1905
Suborder Apocreadiata* Olson, Cribb, Tkach, Bray & Littlewood, 2003
Superfamily Apocreadioidea* Skrjabin, 1942
Suborder Haploporata nom. nov.
Superfamily Haploporoidea Nicoll, 1914
Suborder Opisthorchiata La Rue, 1957
Superfamily Opisthorchioidea Braun, 1901
Suborder Xiphidiata Olson, Cribb, Tkach, Bray & Littlewood, 2003
Superfamily Opecoelioidea Ozaki, 1925.
Superfamily Brachycladioidea Odhner, 1905.
Superfamily Gorgoderoidea Looss, 1901
Superfamily Microphalloidea Ward, 1901
Superfamily Plagiorchioidea Lühe, 1901
*The suborder Apocreadiata, and the superfamily Apocreadioidea require further scrutiny. In a recent revision of the Megaperidae Manter, 1934 based on morphological characters, the family was recognized as valid, although the family Apocreadiidae was considered a synonym of the latter (Blend et al., Reference Blend, Karar and Dronen2017).
Ribosomal vs. complete mitochondrial genome phylogenies
The advancement of molecular technologies is linked to the generation of massive amounts of molecular data for taxonomic studies. NGS technology has augmented our capacity to obtain large amounts of data in a short time, contrasting with partial DNA sequences of nuclear or mitochondrial genes obtained using conventional Sanger sequencing methods. In parasites, and particularly in the subclass Digenea, the use of this technology is still incipient. The first digenean mitogenomes were generated for species of Schistosoma Weinland, 1858, a human parasite (see Le et al., Reference Le, Humair, Blair, Agatsuma, Littlewood and McManus2001; Littlewood et al., Reference Littlewood, Lockyer, Webster, Johnston and Le2006; Zarowiecki et al., Reference Zarowiecki, Huyse and Littlewood2007). Littlewood et al. (Reference Littlewood, Lockyer, Webster, Johnston and Le2006) used the mitogenome data to analyse the evolutionary history of mitochondrial genomes among parasitic platyhelminths; Lawton et al. (Reference Lawton, Hirai, Ironside, Johnson and Rollinson2011) later used mitogenomic information to study the evolution, speciation and divergence of schistosomes around the world. However, in the last two decades, the complete mitochondrial genome of only c. 48 species of digeneans has been sequenced (GenBank database, accessed 17 November 2018). More recently, certain studies have analysed the phylogenetic relationships among species of digeneans using mitogenomes (e.g. Webster & Littlewood, Reference Webster and Littlewood2012; Brabec et al., Reference Brabec, Kostadinova, Scholz and Littlewood2015; Briscoe et al., Reference Briscoe, Bray, Brabec and Littlewood2016; Chen et al., Reference Chen, Feng, Chen, Wang, Feng, Yang, Mughal and Fang2016). Some of these studies actually challenged the stability of the current phylogenetic classification based on nuclear rRNA genes, suggesting, for example, that the order Diplostomida was paraphyletic because two diplostomidan superfamilies were nested within the order Plagiorchiida; the significance of the basal dichotomy of Digenea was questioned. For instance, Brabec et al. (Reference Brabec, Kostadinova, Scholz and Littlewood2015) generated the mitogenome of two species of diplostomids, and their phylogenetic analyses using amino acids and nucleotides recovered Diplostomidae as the sister group of the Plagiorchiida, although those relationships were supported by a low nodal support value. In a recent study, Locke et al. (Reference Locke, Van Dam, Caffara, Pinto, López-Hernández and Blana2018) generated the complete mitochondrial genome of seven additional diplostomoids representing three families and performed a phylogenetic analysis with mitogenomes. To choose between the topologies from the mitochondrial and nuclear phylogenetic analyses, these authors also analysed hundreds of ultra-conserved elements (UCEs) obtained by shotgun sequencing. Even though the mitogenome phylogenetic tree yielded Diplostomida as paraphyletic because strigeids, diplostomids and clinostomids were recovered as sister groups of Plagiorchiida, not Diplostomida, the UCE phylogeny supported the monophyly of the order. All previous phylogenetic analyses of Digenea through nuclear rRNA genes have supported the monophyly of the two large orders within the subclass (Olson et al., Reference Olson, Cribb, Tkach, Bray and Littlewood2003; Littlewood et al. Reference Littlewood, Bray, Waeschenbach, Morand, Krasnov and Littlewood2015; this work). However, insufficient taxon sampling for mitogenomes might still be the cause of the discordance between the two sources of molecular data. Locke et al. (Reference Locke, Van Dam, Caffara, Pinto, López-Hernández and Blana2018) discussed the possible causes of the mitogenome topology and noted that most discordance occurred in short internal branches, which could be related to a rapid radiation and incomplete lineage sorting. Therefore, with the still limited number of digenean species for which complete mitochondrial genomes have been sequenced, it is not possible to determine the power of the phylogenetic signal of mitogenomes to resolve phylogenetic relationships at deeper levels of the classification of Digenea, in comparison with the resolution power of single nuclear rDNA sequences. However, as discussed by Sanderson (Reference Sanderson, Olson, Hughes and Cotton2016), scaling up molecular phylogenetic datasets is methodologically challenging because the amount of data needed to resolve phylogenetic inter-relationships must increase with the number of taxa included. Our study illustrate a case study where the results derived from the use of a ‘traditional’ (few loci) molecular phylogenetic analysis are in contrast with a NGS-driven analyses where just a few representative species have been sequenced using the new technologies. Future studies will determine whether that addition of taxa or characters will provide a better resolution for the inter-relationships of Digenea, and will demonstrate the utility of mitogenomes in providing a more stable classification system, with higher nodal support.
Concluding remarks
In this study, we employed large taxon sampling of digeneans to test the consistency of the molecular classification system proposed by Olson et al. (Reference Olson, Cribb, Tkach, Bray and Littlewood2003) through nuclear rDNA sequences. In that comprehensive analysis, species allocated to several families were not represented. Still, the addition of more species included in a wide variety of genera and families indicates that the classification of Digenea is robust and stable. However, inter-relationships among higher levels of the hierarchy, such as superfamilies and families, still require further scrutiny as well as the strategic selection of additional species to incorporate representatives of other families. Inconsistencies regarding the placement and inter-relationships among superfamilies, or the lack of nodal support, require the addition of more taxa; likewise, the reciprocal monophyly and nodal support for most superfamilies shows that the placement of some groups on the tree will not change with the inclusion of more taxa and quite certainly will not change with the use of other molecular markers, such as mitogenomes. In particular, low nodal support for some groupings highlights the need to add more samples (species) to the analyses. Definitively, we pose that higher-level phylogenetic analyses should consider the use of the variable regions D1–D3 of the 28S rRNA gene. Undoubtedly, this nuclear rRNA gene has the necessary phylogenetic signal to establish inter-relationships among species, as well as among higher-level groups, and can be regarded as reliable for establishing digenean inter-relationships. Then, within particular groups, the addition of other nuclear and mitochondrial genes will be necessary to supply another source of information, which in combination with morphology, host association, host-specificity and biogeography, will provide robust explanations to re-assess not only the species composition within genera, genera within families, and so on but also to generate a well-supported phylogenetic tree that can be used to build robust classifications. Several authors have argued that the addition of more data should lead to a stronger phylogenetic hypothesis (see Nylander, Reference Nylander2001 and references therein); still a debate does exist about what is the most important factor in phylogenetic analysis, characters or taxa. A well-justified concern rises then about the ratio between the number of characters and the number of taxa for a robust analysis. The strategy of our study was to increase the number of taxa and to test the performance of the 28S rRNA gene in resolving the inter-relationships of the Digenea. We acknowledge that there are theoretical considerations involved in the reconstruction of large phylogenies, and that the information content of the selected nuclear gene in our study is over the limit regarding the number of taxa and considering the overall size of the new dataset; it also has practical considerations because large data matrices require more computational time; also, it is well-known that increasing the number of taxa increases the number of possible phylogenetic trees (Felsenstein, Reference Felsenstein1978). We believe that future advancement on building a robust classification system for this parasitic group will require a strategic sampling and selection of both, taxa and characters. If the D1–D3 most variable region of the 28S rRNA gene continues to be used as the marker of reference to build a higher-level classification of the Digenea, then the number of terminals will have be reduced looking at the representation of families rather than species and genera within each family, thus reducing the number of species on the tree. An alternative option is to keep increasing the number of sequenced taxa of digeneans, i.e. the number of species representing as many families as possible, but also increasing the number of characters by adding other nuclear genes, such as the 18S, which has shown to be valuable in reconstructing phylogenies of digeneans; however, as shown in fig. 1, the number of species sequenced for this marker is less than half than the number of species sequenced for the 28S, and this will require a large sequencing effort. Brabec et al. (Reference Brabec, Kostadinova, Scholz and Littlewood2015) characterized the complete transcribed region of the nuclear rRNA operon for two species of diplostomids (c. 9000 bp); even though this could represent an alternative to increase the number of characters, sequencing effort to obtain the rRNA operon for representative species of a larger number of families of digeneans seems unlikely. Actually, Brabec et al. (Reference Brabec, Kostadinova, Scholz and Littlewood2015) discovered that the alignment of the rRNA operons of the two species revealed remarkable high levels of nucleotide conservation with coding regions (see Brabec et al., Reference Brabec, Kostadinova, Scholz and Littlewood2015). In brief, we propose that future phylogenetic analyses should be based on the 28S rRNA gene and sampling size of the data matrix should be strategic to avoid sequencing a large number of species for a relatively restricted number of characters from the D1–D3 variable regions. More taxa representing families that have not been yet sequenced should be included, and most probably, as sequences become available, the sequence of only one species for each genus should be analyses looking for a balance between the number of taxa and characters in the 28S data matrix.
The explanatory power of mitogenomes remains controversial until more species are included in the analysis to overcome the incomplete taxon sampling. For instance, we agree with Locke et al. (Reference Locke, Van Dam, Caffara, Pinto, López-Hernández and Blana2018) that sequencing the mitogenome of early divergent lineages of diplostomoids, such as brachylaimids, liolopids, aporocotylids and spirorchiids, as well as plagiorchiids, such as bivesiculids, is absolutely necessary to resolve the conflict regarding the position of diplostomoids within the order Plagiorchiida; finally, sequencing the mitogenome of an aspidogastrean for use as an outgroup for the phylogenetic analysis of Digenea may contribute to the identification of congruent results between nuclear rRNA genes and mitogenome phylogenies, rendering an even higher stability to the classification system of the group. Novel data generated through NGS technology are becoming highly necessary in taxonomic studies and are required to accomplish the ‘next-generation’ Tree of Life (see Sanderson, Reference Sanderson, Olson, Hughes and Cotton2016); transiting into the NGS era is becoming easier and cheaper every day (Olson et al., Reference Olson, Hughes and Cotton2016). It is possible then that the near future will witness a dramatic change in the use of massive amounts of molecular data to resolve taxonomic problems, from species delimitation to phylogenetic relationships. In parasites, due to their implications in human and animal health, the NGS era has undergone rapid development. The International Helminth Genomes Consortium (2019) combined 36 published genomes of medically important nematodes and platyhelminths into a genome comparison with non-parasitic worms, allowing the identification of gene families at key nodes of the phylogeny that are relevant for the parasitic way of life; the authors identified gene families that modulate host immune responses or genes that enable parasite migration through host tissues genomics. In our opinion, we cannot stop sequencing fragments of nuclear and mitochondrial genes to resolve the taxonomic problems of parasites. We must increase sampling efforts and conduct more phylogenetic analyses to try to reach a stable classification system for the entire subclass Digenea (and other groups of parasitic helminths). This needs to be in parallel with the development of NGS methods. Whether or not the results from NGS will produce more accurate estimations of the ‘true’ phylogenetic history of the Digenea than the traditional nuclear markers remain to be seen.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0022149X19000191
Author ORCIDs
Gerardo Pérez-Ponce de León, 0000-0001-6472-5113.
Acknowledgements
We thank Laura Márquez Valdelamar for technical assistance with the automatic sequencer. We also thank Miguel Rubio, Rogelio Aguilar, Jorge López and Valeria Salinas for the donation of digenean specimens for DNA sequencing.
Financial support
This research was supported by a grant from the Programa de Apoyo a Proyectos de Investigación e Inovación Tecnológica (PAPIIT-UNAM) IN202617 to GPPL. DIHM thanks the support of the Programa de Posgrado en Ciencias Biológicas, UNAM and CONACYT for the scholarship to complete his PhD programme.
Conflicts of interest
None.
Ethical standards
Specimens of digeneans collected in vertebrates of Mexico were obtained under the Cartilla Nacional de Colector Científico (FAUT 0057) issued by the Secretaría del Medio Ambiente y Recursos Naturales (SEMARNAT), to GPPL.