



Introduction
Globally, around half of the topsoils contain 15–30% clay and the clay content in ~ 20% of the topsoils is > 30% (Wei et al., Reference Wei, Dai, Duan, Liu and Yuan2014). Depending on factors such as parent material and weathering, the dominant clays and clay minerals vary in different soils. For example, in weathered soils, Al and Fe (oxyhydr)oxides are the dominant clay minerals (Zimmerman & Ahn, Reference Zimmerman, Ahn, Shukla and Varma2011). In young soils developed from volcanic parent materials, allophanes and ferrihydrite are found commonly (Allison, Reference Allison2006). Clays from the illite and mica groups are commonly found in arid and high-latitude regions, while humid tropical soils contain predominantly 1:1 kaolinite groups (Ito & Wagai, Reference Ito and Wagai2017). Fibrous silicates such as palygorskite and sepiolite are found in calcareous and gypsiferous soils in arid regions (Shirvani et al., Reference Shirvani, Khalili, Kalbasi, Shariatmadari and Nourbakhsh2020). Smectite-group minerals are ubiquitous in many soils, especially in the semi-arid and arid regions, in Vertisols, and in continental and oceanic sediments (Ito & Wagai, Reference Ito and Wagai2017; Odom, Reference Odom1984).
The extracellular enzymes are common in soils. They are excreted by soil microbes such as fungi and actinomycetes, and by plants. The extracellular enzymes carry out many critical functions including mineralizing soil nutrients. They are also involved in the formation and decomposition of soil organic matter, the bioremediation of polluted soils, and the nutrient cycling processes such as mineralization and immobilization (Bollag et al., Reference Bollag, Mertz, Otjen, Anderson and Coats1994; Burns et al., Reference Burns, DeForest, Marxsen, Sinsabaugh, Stromberger, Wallenstein, Weintraub and Zoppini2013; Naidja et al., Reference Naidja, Huang and Bollag2000; Zimmerman & Ahn, Reference Zimmerman, Ahn, Shukla and Varma2011). Furthermore, the activity of some enzymes can be used as a soil quality indicator to assess soil health, soil productivity, and ecological functionality (Alkorta et al., Reference Alkorta, Aizpurua, Riga, Albizu, Amézaga and Garbisu2003; Gil-Sotres et al., Reference Gil-Sotres, Trasar-Cepeda, Leirós and Seoane2005).
The secreted, extracellular enzymes in soils are known to be susceptible to adsorption and immobilization by soil minerals. The interaction between soil enzymes and minerals has been discussed for decades, covering a wide range of studies from the investigation of pure layered silicate–enzyme reactions to models mimicking the realistic soil mineral colloid–enzyme interaction (Naidja et al., Reference Naidja, Huang and Bollag2000). Datta et al. (Reference Datta, Anand, Moulick, Baraniya, Pathan, Rejsek, Vranova, Sharma, Sharma, Kelkar and Formanek2017) also summarized some key results on the adsorption process (e.g. driving forces, adsorption isotherm) of enzymes on minerals and on adsorption kinetics. The general effects of some soil parameters on enzyme adsorption and some key factors that affect enzyme activity were also reviewed in the same work.
Phosphatases, as a group of important enzymes in soils, along with their properties and activities, have been studied extensively but only brief discussion about the interaction between phosphatases and clay minerals has been included in reviews with a general topic of enzyme–clay mineral interaction (Nannipieri et al., Reference Nannipieri, Giagnoni, Landi, Renella, Bünemann, Oberson and Frossard2011). More comprehensive reviews focusing on phosphatase behavior and properties during adsorption are still lacking. The objective of the present review, therefore, was to focus specifically on the adsorption of phosphatases on clay minerals and how it alters several important properties of phosphatases.
In recognition of the potential application of immobilized (e.g. adsorbed) enzymes, many reviews discuss potential industrial applications. For instance, Jesionowski et al. (Reference Jesionowski, Zdarta and Krajewska2014) focused on selecting an appropriate carrier as the adsorbent for an enzyme and compared the advantages of different kinds of commonly used carriers (e.g. silicas, metals, clay minerals, and natural and synthetic polymers). Many recent reviews discussed different industrial applications of commonly used clay mineral-immobilized enzymes (e.g. An et al., Reference An, Zhou, Zhuang, Tong and Yu2015; Basso & Serban, Reference Basso and Serban2019; Maghraby et al., Reference Maghraby, El-Shabasy, Ibrahim and Azzazy2023). Application of the interaction between phosphatase and minerals is also discussed in the present review, therefore. As current application of such interactions is still limited, this review also expands the discussion into enzymes that can mineralize other macronutrients as indications of future potential application of phosphatases.
Mineralizable Macronutrient Retention in Soils by Clays and Clay Minerals
Soil minerals play an important role in numerous soil reactions and one of the most important roles is the retention of mineralizable macronutrients (C, N, P, and S). With small particle size and, therefore, large specific surface area, the minerals carry abundant positive and negative charges on their internal and external surfaces, making them effective in interacting with different forms of mineralizable macronutrients, including inorganic anions (e.g. phosphate, nitrate, and sulfate), cations (e.g. NH4 +), and organic compounds (e.g. enzymes, organic acids).
The distribution of charges on the variable-charge mineral surface is not homogeneous, and the two sources of charges are permanent charge, which is an inherent characteristic of the mineral, and pH-dependent charge. For some clay minerals, such as allophane, kaolinite, and Al/Fe (oxyhydr)oxides, the charges are predominantly pH-dependent while a large percentage of permanent charge is often observed in chlorite and 2:1 clays such as smectite and vermiculite (McBride, Reference McBride, Dixon and Weed1989; Sumner, Reference Sumner2000).
Dissolved ions attracted and retained by clays can exchange with free ions in the surrounding soil solution and the cation- and anion-exchange capacities of a specific clay mineral are pH-dependent. pH-dependent charges arise through the adsorption of ions and protonation and deprotonation of the exposed hydroxyl groups on the edges of the mineral surfaces where the mineral unit layer is disrupted (Cross & Yariv, Reference Cross, Yariv, Cross and Yariv1979).
The complexation between ions and the charged mineral surfaces can be divided into two mechanisms: inner-sphere and outer-sphere complexation. In the case of outer-sphere complexation, water molecules are involved and form a bridge between the adsorbed ion and the clay-mineral surface. Outer-sphere complexation is, therefore, rather weak, rapid, and reversible, as it mainly involves electrostatic interactions and occurs only between surfaces and ions that bear the opposite charges (Sparks, Reference Sparks and Sparks2003).
In contrast, the adsorbed ion is bonded directly to the mineral surface in inner-sphere complexation, which provides a relatively strong bonding environment and is often irreversible compared to outer-sphere complexation (Sparks, Reference Sparks and Sparks2003). Depending on the characteristics of the adsorbate and adsorbent, inner-sphere adsorbed ions are less susceptible to being exchanged easily by ions other than the outer-sphere adsorbed ones (Sposito, Reference Sposito1984).
Through ion-exchange reactions, the minerals can store and supply the adsorbed ions, largely affecting the fate and bioavailability of nutrients in soils. For example, a large amount of nitrate has been found to be retained in Oxisols and Ultisols (Kome et al., Reference Kome, Enang, Tabi and Yerima2019; Lehmann et al., Reference Lehmann, Lehmann, Lilienfein, Rebel, do Carmo Lima and Wilcke2004). Ammonium is commonly fixed in the interlayers of 2:1 phyllosilicates (e.g. vermiculite, montmorillonite) (Mamo et al., Reference Mamo, Taylor and Shuford1993). Clays such as Fe and Al (oxyhydr)oxides (e.g. ferrihydrite, gibbsite), and clay minerals such as allophane and imogolite are effective at retaining phosphate and organic phosphorus compounds through adsorption and precipitation (Redel et al., Reference Redel, Cartes, Velásquez, Poblete-Grant, Poblete-Grant, Bol and Mora2016; Sims & Sharpley, Reference Sims and Sharpley2005).
The retention of mineralizable macronutrients may also be a result of the interaction between the minerals and enzymes involved in the mineralization. The enzyme–clay mineral interaction can regulate enzyme activities and alter their kinetic properties (Datta et al., Reference Datta, Anand, Moulick, Baraniya, Pathan, Rejsek, Vranova, Sharma, Sharma, Kelkar and Formanek2017). As the free enzymes are probably prone to rapid denaturation, the bound forms may serve as a reservoir of potential enzyme activity (Burns, Reference Burns1982; Rejsek et al., Reference Rejsek, Vranova, Pavelka and Formanek2012; Shindo et al., Reference Shindo, Watanabe, Onaga, Urakawa, Nakahara and Huang2002). In some cases, the association between enzymes and minerals stabilized the structure of the enzymes, and the lifetime of the enzyme was prolonged while high activity was maintained (Bollag et al., Reference Bollag, Mertz, Otjen, Anderson and Coats1994; Burns et al., Reference Burns, DeForest, Marxsen, Sinsabaugh, Stromberger, Wallenstein, Weintraub and Zoppini2013). The stabilization effect can also protect the enzymes against proteolysis and abiotic processes such as thermal denaturation and dehydration (Zimmerman & Ahn, Reference Zimmerman, Ahn, Shukla and Varma2011). Decreased enzyme activity upon enzyme adsorption, however, has also been documented widely when the adsorption made the active site of the enzyme less accessible or when enzyme deformation was induced (Dick & Tabatabai, Reference Dick and Tabatabai1987; Hughes & Simpson, Reference Hughes and Simpson1978; Makboul & Ottow, Reference Makboul and Ottow1979; Tietjen & Wetzel, Reference Tietjen and Wetzel2003; Zimmerman & Ahn, Reference Zimmerman, Ahn, Shukla and Varma2011). Other properties related to the enzyme activity (e.g. optimal temperature, pH, or substrate affinity) may be largely influenced by adsorption to minerals, which would indirectly affect the mineralizable macronutrient cycles, such as enzymatic degradation of organic C, N, P, and S (Rao et al., Reference Rao, Violante and Gianfreda2000).
Importance of Organic P Mineralization
Of all the macronutrients, P is acknowledged to be rather immobile and unavailable due to precipitation with various metals and the partitioning reaction (e.g. adsorption) with minerals and organic matter (Abel et al., Reference Abel, Ticconi and Delatorre2002; Quiquampiox & Mousain, Reference Quiquampiox, Mousain, Turner, Frossard and Baldwin2005; Vance et al., Reference Vance, Uhde-Stone and Allan2003). Adsorbed phosphates are difficult to exchange due to transformation into more recalcitrant precipitates. Furthermore, the clay-sized colloidal phosphate has also been reported to have enhanced mobility and is prone to loss via surface runoff and subsurface routes such as preferential flow, leading to nutrient loss (Chen & Arai, Reference Chen, Arai and Sparks2020). Phosphorus deficiency, therefore, has been observed in many soils, such as tropical forests and weathered soils, and biomass accumulation and productivity can be severely constrained in P-deficient soils (Oliverio et al., Reference Oliverio, Bissett, McGuire, Saltonstall, Turner and Fierer2020). Furthermore, due to the strong fixation of phosphate in soils, the inefficiency of phosphate fertilizer has been a problem, so an excessive amount is usually applied (Vance et al., Reference Vance, Uhde-Stone and Allan2003; Zhang et al., Reference Zhang, Tang, Feng, Wang, Li, Shen and Zhang2019). A consequence of excessive P fertilization is the loss of P to waters, resulting in eutrophication. By estimation, the inexpensive yet finite and non-renewable source of the widely used rock phosphate fertilizer is likely to be depleted within decades, especially given the increasing requirement for P fertilization and the political instability of some areas where rock phosphate is found (Abelson, Reference Abelson1999; Garske & Ekardt, Reference Garske and Ekardt2021; Richardson et al., Reference Richardson, Lynch, Ryan, Delhaize, Smith, Smith, Harvey, Ryan, Veneklaas, Lambers, Oberson, Culvenor and Simpson2011). As a result, the price of phosphate fertilizer has increased to >$700/ton (with an increase of $100–400/ton since 2009) recently in 2021 following the sharp price increase in 2008 (Beghin & Nogueira, Reference Beghin and Nogueira2021; Cordell & White, Reference Cordell and White2014).
Soils can provide phosphate through weathering of mineral phosphate and mineralization of organic P compounds (Quiquampiox & Mousain, Reference Quiquampiox, Mousain, Turner, Frossard and Baldwin2005). Organic P accounts for 20–80% of total P in soils and it can represent a particularly important potential pool to provide phosphate, especially in soils with no fertilizer input and/or poor phosphate solubility (Tiessen et al., Reference Tiessen, Stewart and Cole1984). Other strategies such as promoted mineralization of the organic P, therefore, could be another option to increase the P availability in soils. Phosphohydrolases are a group of enzymes that can catalyze the hydrolysis of organic P and some inorganic P such as linear polyphosphate and pyrophosphate (Quiquampiox & Mousain, Reference Quiquampiox, Mousain, Turner, Frossard and Baldwin2005). Another group of enzymes that can release P from organic P compounds in soils is lyases involved in the P cycle (Rodríguez et al., Reference Rodríguez, Fraga, Gonzalez and Bashan2006).
Classification of Soil Phosphohydrolases and Phosphate-Releasing Lyases
Phosphohydrolases can catalyze the hydrolysis of different bonds and they can be classified depending on the bond type according to the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) (Fig. 1). The Enzyme Commission (EC) number is used as a numerical classification to categorize enzymes based on the reactions catalyzed. For example, many phosphohydrolases target the ester bonds. Phosphoric monoester hydrolases (EC 3.1.3) are enzymes that can cleave the ester bonds on a phosphomonoester. Of all phosphoric monoester hydrolases, phosphatases have been studied widely, and depending on their optimal pH (below or above pH 7.0), phosphatases can be categorized into acid phosphatases (EC 3.1.3.2) and alkaline phosphatases (EC 3.1.3.1) (Vincent et al., Reference Vincent, Crowder and Averill1992). Depending on substrate specificity, phosphatases can also be grouped into, for example, protein serine/threonine phosphatase (PSTP or PSP), protein tyrosine phosphatase (PTP), histidine phosphatase, lipid phosphatase, sugar phosphatases, and glycerophosphatases (Alef & Nannipieri, Reference Alef, Nannipieri, Alef and Nannipieri1995; Fahs et al., Reference Fahs, Lujan and Kohn2016). Other examples of phosphoric monoester hydrolases include phytases and nucleotidases.
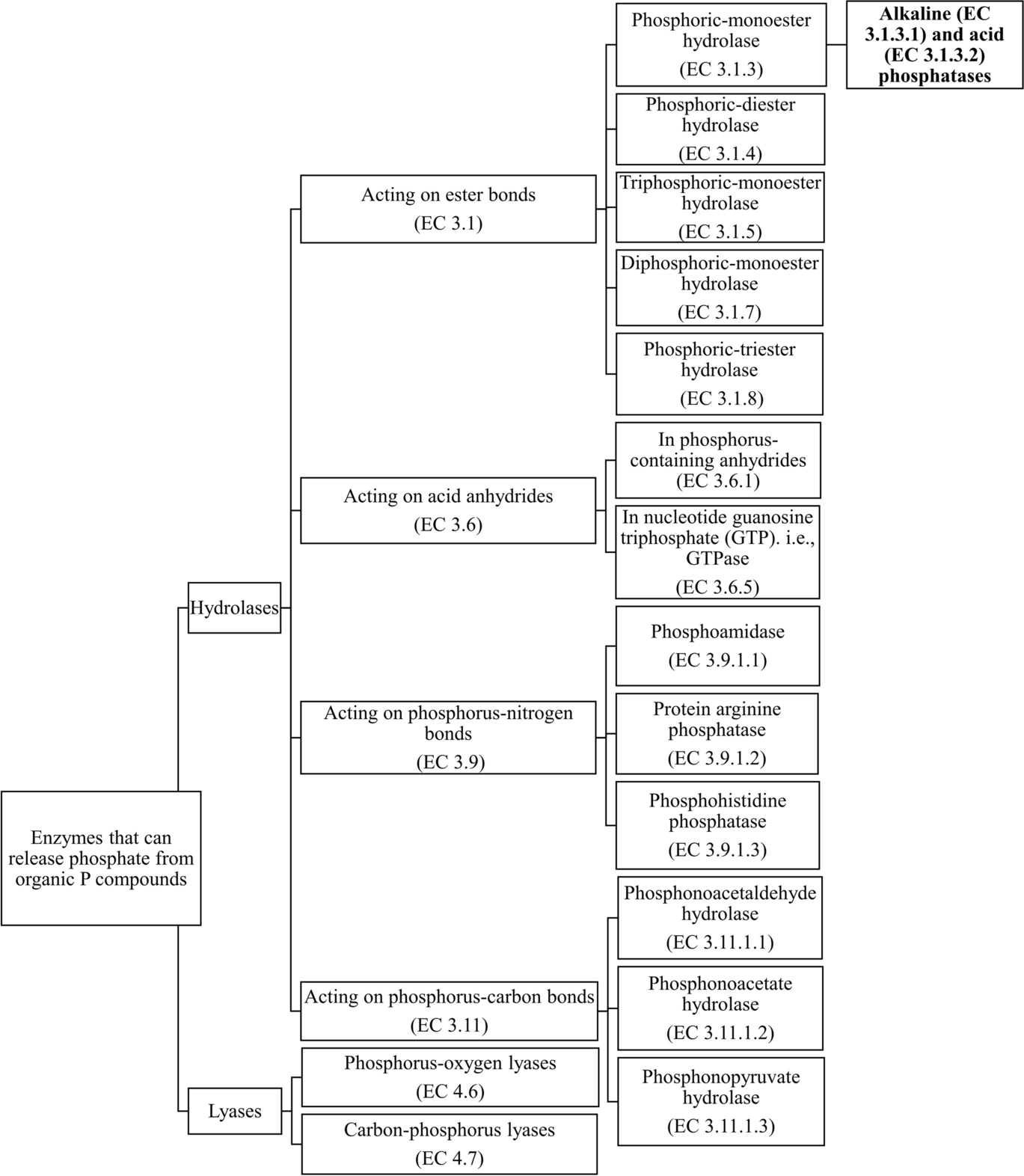
Fig. 1 Classification of enzymes that can release phosphate from organic P compounds, based on the enzyme database from the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Alkaline (EC 3.1.3.1) and acid (EC 3.1.3.2) phosphatases are the main focus of the subsequent sections
Other ester bond-targeting phosphohydrolases consist of phosphoric diester hydrolases (EC 3.1.4) such as phospholipases, triphosphoric monoester hydrolases (EC 3.1.5), diphosphoric monoester hydrolases (EC 3.1.7), and phosphoric triester hydrolases (EC 3.1.8).
Phosphohydrolases also include enzymes acting on guanosine triphosphate (GTP) (EC 3.6.5), enzymes targeting phosphoryl-containing anhydrides (EC 3.6.1) such as pyrophosphotases (or diphosphate phosphohydrolase) (EC 3.6.1.1) which catalyze the hydrolysis of pyrophosphate, phosphoamidases which act on the P–N bonds (EC 3.9.1), and phosphonatases (or phosphonate hydrolases) (EC 3.11) (e.g. phosphonoacetaldehyde hydrolase, phosphonopyruvate hydrolase, and phosphonoacetate hydrolase) which target the C–P bond and hydrolyze C-phosphono groups (Alef & Nannipieri, Reference Alef, Nannipieri, Alef and Nannipieri1995; Chin et al., Reference Chin, McGrath and Quinn2016; Nannipieri et al., Reference Nannipieri, Giagnoni, Landi, Renella, Bünemann, Oberson and Frossard2011; White & Metcalf, Reference White and Metcalf2007).
Lyases are enzymes that can catalyze the cleavage of bonds by means other than hydrolysis or oxidation and they can also be classified according to the bonds they attack. For example, a group of P-O lyases (EC 4.6), the ribonucleases, are usually secreted under phosphate starvation conditions, and they can catalyze the cleavage of P–O bonds in RNAs (Abel et al., Reference Abel, Ticconi and Delatorre2002; Bariola et al., Reference Bariola, Howard, Crispin, Verburg, Jaglan and Green1994). C-P lyases (EC 4.7) have also been found in phosphate-limited environments and they target the C–P bonds in organophosphonates (Oliverio et al., Reference Oliverio, Bissett, McGuire, Saltonstall, Turner and Fierer2020; Tapia-Torres et al., Reference Tapia-Torres, Rodríguez-torres, Elser, Islas, Souza, García-Oliva and Olmedo-Álvarez2016).
Reaction Mechanisms of Phosphohydrolases and Phosphate-Releasing Lyases
The majority of P-containing organic compounds contain the ester bond, and the activity of the phosphomonoesterases is usually greater than that of phosphodi- or phosphotri-esterases in soils (Alef & Nannipieri, Reference Alef, Nannipieri, Alef and Nannipieri1995). Among all phosphomonoesterases in soils, alkaline and acid phosphatases are those studied most widely (Nannipieri et al., Reference Nannipieri, Giagnoni, Landi, Renella, Bünemann, Oberson and Frossard2011).
Alkaline phosphatases have a common conserved core structure containing a serine residue that binds with phosphate on the phosphomonoester during the catalysis, resulting in the formation of a phosphoserine intermediate. The fully active site for each alkaline phosphatase monomer also contains two Zn(II) which are bridged by the phosphate group, and one Mg(II) coordinated with a water molecule that is hydrogen-bonded to the phosphate (Coleman, Reference Coleman1992; Kim & Wyckoff, Reference Kim and Wyckoff1991; Stec et al., Reference Stec, Holtz and Kantrowitz2000). Unlike alkaline phosphatases, a histidine residue acts as the nucleophilic acceptor in the catalytic process of acid phosphatases, and phosphohistidine intermediates are formed. Acid phosphatases also have conserved arginine residues and many of them carry a characteristic RHGXRXP (R: arginine; H: histidine; G:glycine; P: proline; X: any amino acid) motif (some acid phosphatases such as Aspergillus niger acid phosphatase only have the RHG motif) (Ostanin et al., Reference Ostanin, Harms, Stevis, Kuciel, Zhou and Van Ettene1992; Ullah et al., Reference Ullah, Cummins and Dischinger1991). For both alkaline and acid phosphatase, the final hydrolysis products are phosphate and corresponding hydrocarbons such as alcohol or phenol (Fig. 2). Phosphodiesters are converted to corresponding phosphomonoesters through phosphodiesterases, which then go through subsequent hydrolysis via phosphatases and release phosphate (Blake et al., Reference Blake, O’Neil and Surkov2005). Phosphotriesters are hydrolyzed to phosphodiesters which then experience subsequent hydrolysis while releasing phosphate. myo-Inositol hexakisphosphate, or phytic acid, is the predominant source organic P in many soils, accounting for more than half of the organic P pool (Anderson, Reference Anderson, Khasawneh chairman, Sample and Kamprath1980; Liu et al., Reference Liu, Cai, Chen, Mo, Ding, Liang, Liu and Tian2018). The enzymes that hydrolyze phytic acid are referred to as phytases and, depending on catalytic mechanisms, they are generally classified into four categories. The histidine acid phytases (HAPhys) share a similar catalysis mechanism, such as the conserved RHGXRXP motif and the formation of phosphohistidine intermediate (Oh et al., Reference Oh, Choi, Park, Kim and Oh2004; Ullah et al., Reference Ullah, Cummins and Dischinger1991). β-Propeller phytases (BPPhys) have an optimum alkaline pH, and metal ions, especially Ca(II), are found in the active site and contribute to the catalytic activity of BPPhy (Kim et al., Reference Kim, Kim, Bae, Yu and Oh1998). Shin et al. (Reference Shin, Ha, Oh, Oh and Oh2001) classified the active sites on the enzyme and categorized them into “cleavage site” and “affinity site.” Two adjacent phosphate groups on a phytic acid molecule bind simultaneously with Ca(II) in the cleavage site and affinity site, respectively. The phosphate bound to the cleavage site would later be removed, while the other one increases the affinity of enzymes for the remaining myo-inositol polyphosphate intermediate. Cysteine phytases (Cphys) are members of the cysteine phosphatases (CP). The Cphys share some substantial properties with the protein tyrosine phosphatases, another group of enzymes from the CP family. For example, they have a conserved nucleophilic cysteine residue and the catalytic hydrolysis involves the formation of a cysteinyl phosphate intermediate. Furthermore, a characteristic active-site motif of these enzymes is HCXXGXXR(T/S) (C: cysteine; G: glycine; S: serine; T: threonine). The formation of a “cysteinyl-phosphate trigonal–bipyramidal” pentavalent intermediate has been postulated and sequential hydrolysis is facilitated with the reorientation of the intermediate (Chu et al., Reference Chu, Guo, Lin, Chou, Shr, Lai, Tang, Cheng, Selinger and Wang2004; Turner et al., Reference Turner, Richardson and Mullaney2007; Yanke et al., Reference Yanke, Selinger and Cheng1999). Purple acid phytases (PAPhys) are named for to their distinct purple color and they have binuclear metal bridging at the active sites, and most PAPhys are coordinated with five conserved consensus motifs (Dionisio et al., Reference Dionisio, Madsen, Holm, Welinder, Jorgensen, Stoger, Arcalis and Brinch-pedersen2011; Rodríguez, Reference Rodríguez2018). The metal bridge is usually in the form of Fe(III)-divalent metal where Fe(III) is on the chromophoric site and the divalent metal can be Fe(II), Zn(II), or Mn(II) (Faba-Rodriguez et al., Reference Faba-Rodriguez, Gu, Salmon, Dionisio, Brinch-Pedersen, Brearley and Hemmings2022; Hegeman & Grabau, Reference Hegeman and Grabau2001; Nasrabadi et al., Reference Nasrabadi, Greiner, Yamchi and Nourzadeh Roshan2018). The scissile phosphate group binds with the metal ions on the active site through hydrogen bonds. The substrate is then rearranged so that the phosphate group is coordinated directly with metal ions, forming a catalytic complex that facilitates the nucleophilic attack, initiating the hydrolysis (Rodríguez, Reference Rodríguez2018). Depending on the species of phytases, hydrolysis products are phosphate and different myo-inositol polyphosphates.
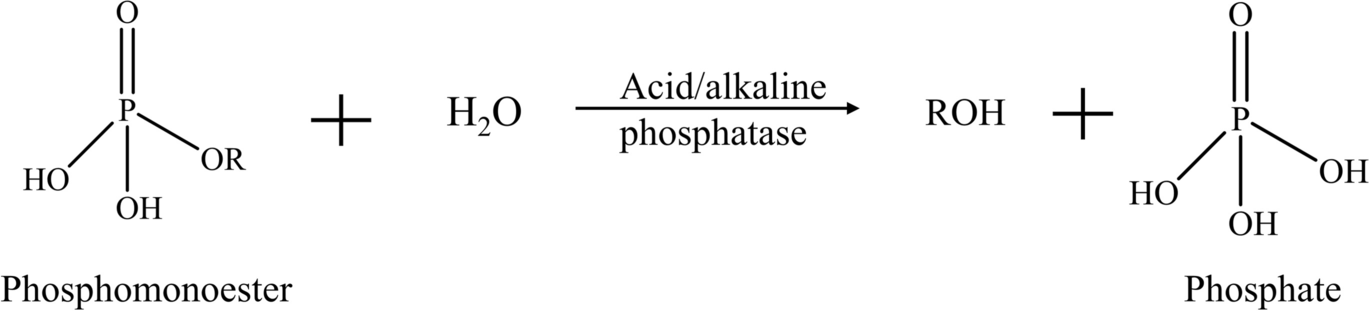
Fig. 2 Phosphomonoester hydrolysis reaction catalyzed by alkaline and acid phosphatases
As well as the compounds containing P in the highest oxidation state (+ 5) such as phosphates and phosphate esters, reduced P forms are also found in soils, such as the organophosphonate, glyphosate, which involves a directly connected C–P bond (Kehler et al., Reference Kehler, Haygarth, Tamburini and Blackwell2021; Ternan et al., Reference Ternan, Mc Grath, Mc Mullan and Quinn1998).
Organophosphonates can be catabolized by phosphonatases or C-P lyases. The characteristic property of the substrate of the phosphonatases is the presence of an electron-withdrawing β-carbonyl group which facilitates the heterolytic cleavage of the C–P bond (Kamat & Raushel, Reference Kamat and Raushel2013). The C-P lyases have a redox-active [4Fe-4S]-cluster and the catalysis process involves a radical-based homolytic cleavage of the C–P bond. The products of the C-P lyase degradation are inorganic phosphate and corresponding hydrocarbon, e.g. alkanes, alkenes, and benzenes from alkyl-, vinyl-, or phenyl-phosphonate degradation (Daughton et al., Reference Daughton, Cook and Alexander1979; Stosiek et al., Reference Stosiek, Talma and Klimek-ochab2020; Wackett et al., Reference Wackett, Shames, Venditti and Walsh1987). More detailed mechanisms of phosphonatases and C-P lyases have been reviewed in the literature (Kamat & Raushel, Reference Kamat and Raushel2013; Stosiek et al., Reference Stosiek, Talma and Klimek-ochab2020).
Enzyme Activity and other Properties Affected by the Formation of the Enzyme–Clay Mineral Complex
Enzyme Adsorption Mechanisms in Clays and Clay Minerals
Protein enzymes bear a variety of functional groups which can interact with the clay mineral surface and the adsorption of enzymes onto the mineral surface can be the result of various forces and interactions including salt linkage, ligand exchange, hydrogen bonding, hydrophobic interaction, conformational entropy, Coulombic force (or electrostatic interaction), and van der Waals forces (Boyd & Mortland, Reference Boyd, Mortland, Bollag and Stotzky1990; Datta et al., Reference Datta, Anand, Moulick, Baraniya, Pathan, Rejsek, Vranova, Sharma, Sharma, Kelkar and Formanek2017; Roth & Lenhoff, Reference Roth and Lenhoff1995; Theng, Reference Theng2012).
Among all of the forces mentioned, electrostatic attraction, as a long-range force, has been proposed by some authors to be the one factor responsible for the first contact between the enzyme and clay (Datta et al., Reference Datta, Anand, Moulick, Baraniya, Pathan, Rejsek, Vranova, Sharma, Sharma, Kelkar and Formanek2017). Positively charged amino acids (e.g. arginine, histidine, and lysine above pH 4) can be attracted electrostatically to a negatively charged mineral surface. Furthermore, ligand exchange between the carboxylic groups of the enzyme and hydroxyl groups of the mineral has been proposed as the mechanism for enzyme adsorption on metal oxide surfaces (Sepelyak et al., Reference Sepelyak, Feldkamp, Moody, White and Hem1984).
Hydrophobic interaction also contributes significantly to the enzyme–mineral reaction. Phyllosilicates such as montmorillonite have hydrophobic siloxane layers and hydrophilic exchangeable ions on the mineral surfaces (Staunton & Quiquampoix, Reference Staunton and Quiquampoix1994). Some amino acids have a hydrophobic side chain (e.g. tryptophan, tyrosine), which would probably engage in hydrophobic interaction with the siloxane layer after the enzyme exchanges with the surface exchangeable ions, and the dehydration from such hydrophobic interaction facilitates the adsorption of most enzymes (Norde, Reference Norde2008). For a hydrophobic mineral surface, even when both the enzyme and surface have charges of the same sign, hydrophobic force may, therefore, overcome the electrostatic repulsion and bring the surface and the enzyme together (Norde, Reference Norde2008; Quiquampoix et al., Reference Quiquampoix, Abadie, Baron, Leprince, Matumoto-Pintro, Ratcliffe, Staunton, Horbett and Brash1995). The adsorption process may be further augmented by enzyme conformational change (Fig. 3), van der Waals forces, and hydrogen bonding.
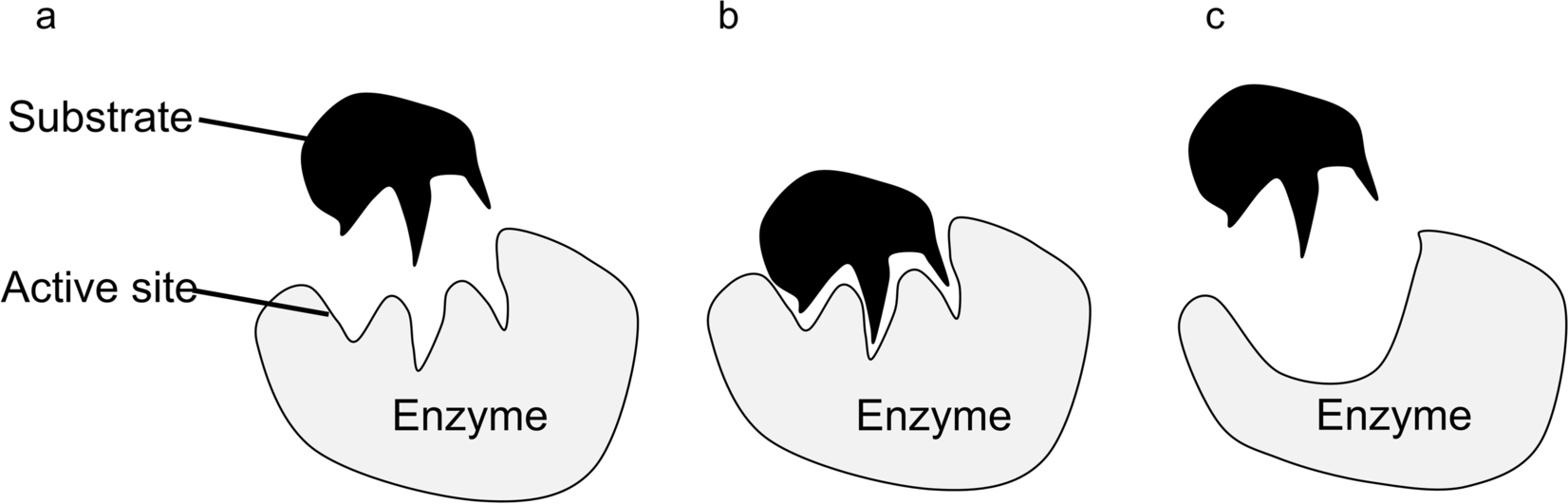
Fig. 3 a The enzyme is in its active conformation and the substrate enters the active site. b Substrate binds to the active site, forming an enzyme–substrate complex. c Conformational change of the enzyme alters the shape of the active site, preventing substrate binding
The adsorption of the enzyme on the mineral surface can induce a conformational change in the enzyme, leading to changes in the enzyme’s conformational entropy and Gibbs free energy of the system. For example, a decrease in the ordered secondary structure of the enzyme due to adsorption would lead to increased conformational entropy, thus reducing the Gibbs free energy of the system. Subsequently, the modification of enzyme structure and adsorption becomes more irreversible (Quiquampiox & Mousain, Reference Quiquampiox, Mousain, Turner, Frossard and Baldwin2005).
Van der Waals interaction consists of three components, permanent dipole-permanent dipole (Keesom force), permanent dipole-induced dipole (Debye force), and induced dipole-induced dipole (London force). Van der Waals forces increase as the distance between the enzyme and mineral surface decreases and as the molecular size increases (Roth & Lenhoff, Reference Roth and Lenhoff1995; Stotzky, Reference Stotzky, Huang and Schnitzer1986). Although the attractive force is rather weak, it can work on all molecules despite charges and can be strengthened through the formation of a salt bridge between enzyme-bound salt ions and the mineral surface or between clay-complexed cations and the enzyme, providing a polarizable layer, or through adjacent van der Waals forces acting together, creating a stronger interaction (Quiquampiox & Mousain, Reference Quiquampiox, Mousain, Turner, Frossard and Baldwin2005; Roth & Lenhoff, Reference Roth and Lenhoff1995; Stotzky, Reference Stotzky, Huang and Schnitzer1986).
Hydrogen bonds can be formed between OH groups or O on the mineral surface, or more importantly, clay-associated water, and amino acid functional groups (e.g. carbonyl, amine) (Stotzky, Reference Stotzky, Huang and Schnitzer1986). Like the van der Waals forces, hydrogen bonding becomes a strong binding force when numerous hydrogen bonds are functioning together (Stotzky, Reference Stotzky, Huang and Schnitzer1986). Hydrogen bonds are generally formed between the enzyme and a polar surface while a nonpolar surface doesn’t interact with the enzyme via hydrogen bonds (Norde, Reference Norde2008).
Factors Affecting the Adsorption of Phosphatase on the Mineral Surface
The adsorption of enzymes on a mineral surface is affected by many factors in various aspects. The general effects of temperature, pH, soil moisture, and dissolved ion concentration on enzyme adsorption have been reviewed by Datta et al. (Reference Datta, Anand, Moulick, Baraniya, Pathan, Rejsek, Vranova, Sharma, Sharma, Kelkar and Formanek2017). The interaction between clay minerals and phosphatases is the most widely studied and the question of how the adsorption onto the clay mineral affects other P-related enzymes (e.g. phytase) has been investigated much less commonly, or not at all. This review, therefore, focused on the influences of several widely studied factors on alkaline and acid phosphatase adsorption and, subsequently, the effects of adsorption on phosphatase activity and other properties.
The electrostatic interaction between an enzyme and the mineral surface depends on the pH, ionic strength of the bathing solution, and background cation charge (Roth & Lenhoff, Reference Roth and Lenhoff1995). At a pH above the point of zero charge of the mineral and below the isoelectric point of the enzyme, i.e. when the enzymes are positively charged, greater adsorption generally occurs with a negatively charged mineral surface (Carrasco et al., Reference Carrasco, Rad and González-Carcedo1995; Datta et al., Reference Datta, Anand, Moulick, Baraniya, Pathan, Rejsek, Vranova, Sharma, Sharma, Kelkar and Formanek2017). pH not only affects the charges on the mineral surface and the enzyme but enzyme conformation is also altered upon pH changes, influencing the adsorption by minerals. For example, a decrease in pH, i.e. an increase in the positive charge on the enzyme, could induce the enzyme to unfold on an electronegative surface (Quiquampoix et al., Reference Quiquampoix, Abadie, Baron, Leprince, Matumoto-Pintro, Ratcliffe, Staunton, Horbett and Brash1995). Furthermore, strongly acidic or alkaline pH could inhibit adsorption by denaturing the enzymes and a near-neutral pH is likely more appropriate for enzyme adsorption (Sarkar & Leonowicz, Reference Sarkar and Leonowicz1989). The effects of ionic strength are less commonly studied, and Leprince and Quiquampoix (Reference Leprince and Quiquampoix1996) found decreased adsorption of acid phosphatases on montmorillonite with increasing ionic strength and they attributed it to the competition between phosphatase and Na+ for negative sites.
The adsorption also depends on the structure of enzymes and the polarity of the mineral surface. Norde (Reference Norde2008) adopted two notions, “hard” and “soft”, to describe proteins and, therefore, enzymes, depending on their behavior upon adsorption. “Hard” enzymes (e.g. rigid or tightly coiled) undergo limited conformational change when adsorbed on the polar mineral surface and the adsorption is only favored in the case of electrostatic attraction. On the other hand, “soft” enzymes undergo structural change upon adsorption and can gain conformational entropy, as discussed in the section Enzyme adsorption mechanisms in clays and clay minerals, via decreased ordered structure, thus facilitating adsorption even on an electrostatically unfavorable surface.
The types of clay minerals may influence their ability to adsorb enzymes and many factors such as swelling, interlayer space, and charge density contribute to their adsorption capacity. Montmorillonite has been found to have a larger adsorption capacity for acid phosphatase than kaolinite, possibly due to large surface area, high charge density, and interlattice fixation of the enzyme in montmorillonite (Gianfreda & Bollag, Reference Gianfreda and Bollag1994; Makboul & Ottow, Reference Makboul and Ottow1979). Greater adsorption of alkaline phosphatase by goethite than by montmorillonite was discovered by Tan et al. (Reference Tan, He, Wang, Li, Kong, Tian, Shen, Megharaj and He2018), while the opposite trend was reported by Zhu et al. (Reference Zhu, Wu, Feng, Liu and Giesy2016). Such discrepancy was attributed to different pH used in the adsorption experiment and, therefore, different charge properties of the phosphatases and minerals (Tan et al., Reference Tan, He, Wang, Li, Kong, Tian, Shen, Megharaj and He2018).
According to Shindo et al. (Reference Shindo, Watanabe, Onaga, Urakawa, Nakahara and Huang2002), the adsorption of an acid phosphatase followed the order: montmorillonite > > kaolinite > Mn oxide > Fe oxide > Al oxide > > allophane. In their study, a positive correlation between the amount of adsorption and mineral-specific surface area could not be established. The charges of the enzyme and mineral surfaces, and thus electrostatic interaction and charge density, mineral microstructure, and ligand exchange capacity of the oxides likely played a larger role in affecting the extent of adsorption. More adsorption of acid phosphatase was observed (Huang et al., Reference Huang, Liang and Cai2005) on goethite than on fine clays; this, in turn, was greater than the adsorption on coarse clays from an Ultisol; and, finally, this, in turn, was also greater than the adsorption on kaolinite. Those authors concluded that large surface area and ion exchange capacity were responsible for greater adsorption.
Even with the same mineral, different saturating cations may influence the extent of adsorption, which was probably due to the difference in how easily the cations can be displaced by the enzyme (Boyd & Mortland, Reference Boyd, Mortland, Bollag and Stotzky1990). For example, Carrasco et al. (Reference Carrasco, Rad and González-Carcedo1995) found that the amount of alkaline phosphatase adsorption followed the order: Ca-sepiolite > Na-sepiolite > H-sepiolite and that H-sepiolite-adsorbed enzyme was desorbed easily under high salinity and pH.
On the contrary, little effect from varying clay minerals has been reported. For instance, Sedaghat et al. (Reference Sedaghat, Ghiaci, Aghaei and Soleimanian-zad2009) found a similar amount of adsorption of an alkaline phosphatase on sepiolite and bentonite despite a much larger specific surface area of sepiolite. They concluded that the enzyme was preferentially immobilized on the external surface of the mineral, instead of penetrating the interlayer. Discrepancies among different literature might be due to different reaction conditions such as pH, the specific microstructure of the mineral, and properties of different enzymes and, therefore, varied electrostatic attraction or repulsion between the mineral surface and the enzyme.
The interaction between clays and organic matter could also affect the adsorption of enzymes because the organic matter may serve as a protective layer on the clay surface, decreasing enzyme adsorption (Quiquampoix et al., Reference Quiquampoix, Abadie, Baron, Leprince, Matumoto-Pintro, Ratcliffe, Staunton, Horbett and Brash1995). Kelleher et al. (Reference Kelleher, Willeford, Simpson, Simpson, Stout, Rafferty and Kingery2004) found less acid phosphatase immobilized on organic molecule-intercalated montmorillonite (i.e. the organic molecules were immobilized between layers of the montmorillonite) than on pure montmorillonite. The opposite trend was also observed. For example, Tang et al. (Reference Tang, Shen, Suib, Coughlin and Vinopal1993) reported more adsorption of alkaline phosphatase by protamine-intercalated bentonite at pH 10.4. Both the phosphatase and mineral surface were negatively charged while the protamine was positively charged, acting as a bridge to connect the enzyme and mineral. Excess protamine decreased phosphatase adsorption due to steric blockage of active sites, however. Huang et al. (Reference Huang, Liang and Cai2005) found that within the same size fraction of clays, natural clays that contained organic matter expressed more enzyme adsorption than inorganic clays (natural clays with organic matter removed by H2O2). According to those authors, the enhanced adsorption was attributed to the additional adsorption of phosphatase by humic substances as well as their ability to trap the phosphatase within the “macromolecular net” of humic acids.
Enzyme Activity and Kinetics Parameters Affected by its Adsorption
Upon adsorption by clay minerals, phosphatase activity was mostly inhibited, while enhanced catalytic activity has seldom been observed (Table 1). Interestingly, enhanced phosphatase activity was more commonly observed when adsorbed on poorly crystalline minerals such as allophane and ferrihydrite. The mesoporosity of allophanes was considered to be responsible for making the minerals more suitable supports in biocatalytic processes (Calabi-Floody et al., Reference Calabi-Floody, Velásquez, Gianfreda, Saggar, Bolan, Rumpel and Mora2012; Wang, Reference Wang2006). From the literature, possible contributors of enhanced enzyme activity upon adsorption could be: greater concentrations of the enzyme and the substrate on the clay mineral surface, conformational change, or stabilization of the enzyme structure induced by adsorption (Allison, Reference Allison2006; An et al., Reference An, Zhou, Zhuang, Tong and Yu2015; Tietjen & Wetzel, Reference Tietjen and Wetzel2003). Inhibited enzyme activity is mostly attributed to conformational change or even deformation of the enzyme, the incorporation of enzymes into the internal structure of the clay, steric hindrance, the interaction between amino acids on the active site and the mineral surface, or diffusional limitation, leading to restricted access of the substrate to the enzyme (Datta et al., Reference Datta, Anand, Moulick, Baraniya, Pathan, Rejsek, Vranova, Sharma, Sharma, Kelkar and Formanek2017; Dick & Tabatabai, Reference Dick and Tabatabai1987; Olagoke et al., Reference Olagoke, Kalbitz and Vogel2019; Tietjen & Wetzel, Reference Tietjen and Wetzel2003; Zimmerman & Ahn, Reference Zimmerman, Ahn, Shukla and Varma2011).
Table 1 Experimental conditions and changes in phosphatase properties in phosphatase-clay mineral sorption studies
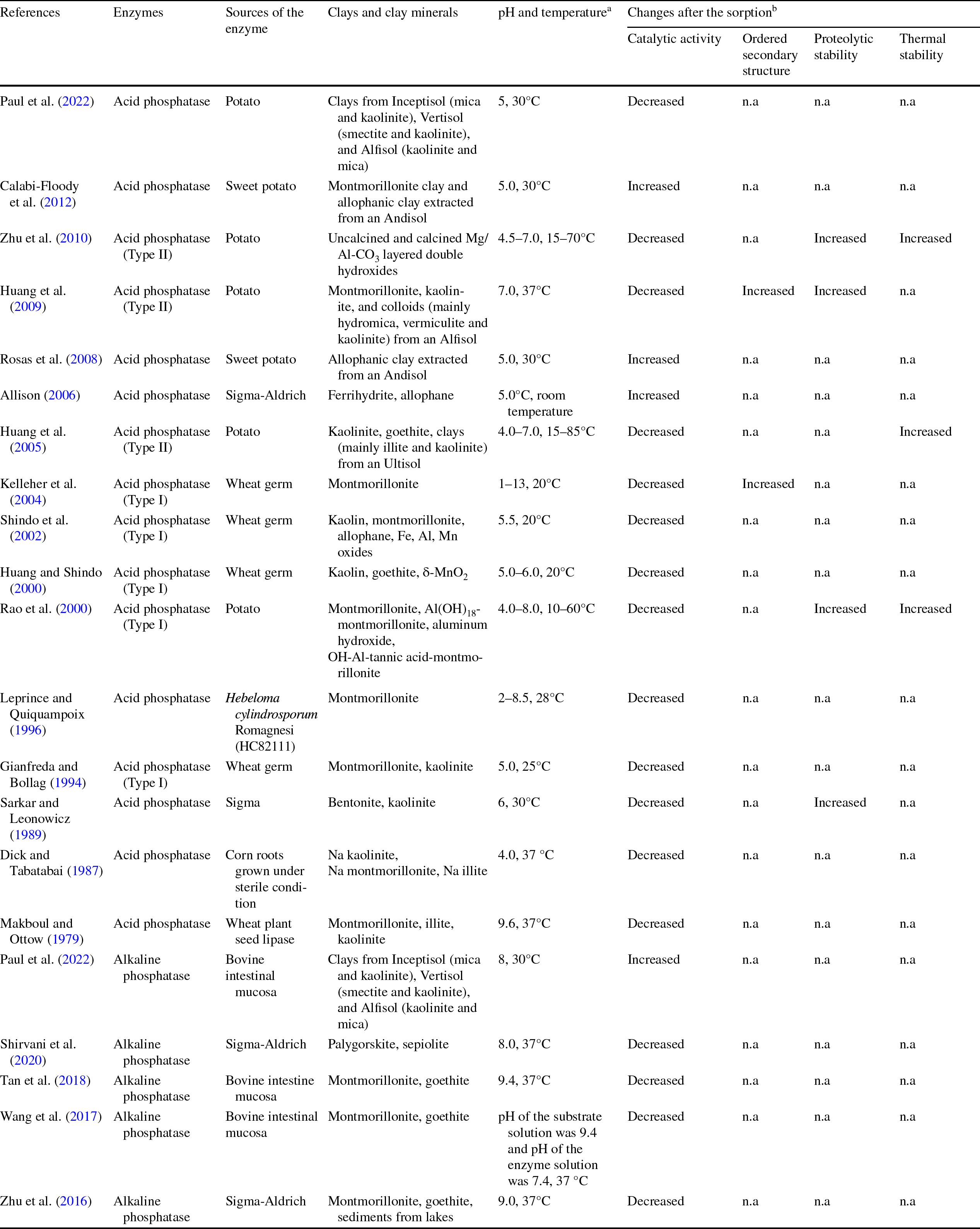
a The pH and temperature are the conditions used for enzyme activity measurement. RT: room temperature
b Compared to respective properties of the free enzyme
The most commonly used model to study enzyme kinetics is the Michaelis–Menten equation:
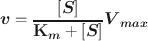
where v is the velocity of the reaction, [S] the substrate concentration, Km the Michaelis–Menten constant, and V max the maximum velocity. The mechanism of how a clay mineral, i.e. the inhibitor, inhibits the enzyme activity can be represented by changes in Km and V max (Johnson & Goody, Reference Johnson and Goody2011). Four types of reversible inhibition mechanisms are generally acknowledged: competitive inhibition, non-competitive inhibition, uncompetitive inhibition, and mixed inhibition. Competitive inhibition is observed with unchanged V max and increased Km, and the inhibitor competes with the substrate generally in a mutually exclusive form, and often for the same active site on the enzyme. The noncompetitive inhibitor binds with the enzyme or the enzyme–substrate complex with the same affinity, thus leading to unchanged Km and decreased V max. The uncompetitive inhibitor binds only to the enzyme–substrate complex to prevent product formation and, therefore, decreases both the V max and Km. Mixed inhibition is often observed with increased or decreased Km and decreased V max, and the inhibitor can bind to the enzyme or the enzyme–substrate complex with different affinity (Copeland, Reference Copeland2000; Cornish-Bowden, Reference Cornish-Bowden and Cornish-Bowden1979; Fange et al., Reference Fange, Lovmar, Pavlov and Ehrenberg2011).
Mixed inhibition is the most widely observed mechanism for clay minerals with regard to their influence on phosphatase activity (Quiquampiox & Mousain, Reference Quiquampiox, Mousain, Turner, Frossard and Baldwin2005; Shirvani et al., Reference Shirvani, Khalili, Kalbasi, Shariatmadari and Nourbakhsh2020). For instance, kaolinite, kaolin, mica, montmorillonite, tannic acid-montmorillonite complex, goethite, allophane, Fe/Al/Mn oxides, uncalcined and calcined Mg/Al-CO3 layered double hydroxide, and δ-MnO2 have been reported to be inhibitors for many acid phosphatases (Gianfreda & Bollag, Reference Gianfreda and Bollag1994; Huang & Shindo, Reference Huang and Shindo2000; Makboul & Ottow, Reference Makboul and Ottow1979; Paul et al., Reference Paul, Datta, Bera, Math, Bhattacharyya and Dahuja2022; Rao et al., Reference Rao, Violante and Gianfreda2000; Shindo et al., Reference Shindo, Watanabe, Onaga, Urakawa, Nakahara and Huang2002; Zhu et al., Reference Zhu, Huang, Pigna and Violante2010), and palygorskite, sepiolite, goethite, and montmorillonite for some alkaline phosphatases (Ghiaci et al., Reference Ghiaci, Aghaei, Soleimanian and Sedaghat2009; Sedaghat et al., Reference Sedaghat, Ghiaci, Aghaei and Soleimanian-zad2009; Shirvani et al., Reference Shirvani, Khalili, Kalbasi, Shariatmadari and Nourbakhsh2020; Wang et al., Reference Wang, Li, Tan, He, Xie, Megharaj and Wei2017; Zhu et al., Reference Zhu, Wu, Feng, Liu and Giesy2016). Discrepancies and other inhibition mechanisms have also been reported. For example, illite and montmorillonite can be uncompetitive inhibitors for some acid phosphatases (Kelleher et al., Reference Kelleher, Willeford, Simpson, Simpson, Stout, Rafferty and Kingery2004; Makboul & Ottow, Reference Makboul and Ottow1979), but noncompetitive inhibitors for other acid phosphatases (Dick & Tabatabai, Reference Dick and Tabatabai1987). Montmorillonite has also been found to be an uncompetitive inhibitor for alkaline phosphatase (Wang et al., Reference Wang, Li, Tan, He, Xie, Megharaj and Wei2017).
Other Properties Affected by the Adsorption
After being adsorbed, the enzymes experience a different microenvironment (e.g. altered diffusion, charge, or steric hindrance) from that experienced by free enzymes (Zimmerman & Ahn, Reference Zimmerman, Ahn, Shukla and Varma2011). Therefore, in addition to catalytic activity, other properties including optimum pH, enzyme conformation, and thermal and proteolytic stability can be influenced.pH-dependent phosphatase adsorption has been observed by several researchers. For instance, the optimum pH of acid phosphatase was shifted to more alkaline values when they were adsorbed on negatively charged mineral surfaces, which was also attributed to the pH-dependent modification of the enzyme conformation (Leprince & Quiquampoix, Reference Leprince and Quiquampoix1996). Instead of exhibiting an optimum pH, the activity of a montmorillonite-adsorbed alkaline phosphatase continued to decrease with pH increasing from 4.0 to 8.0 (Rao et al., Reference Rao, Violante and Gianfreda2000). No change in the optimum pH was reported when an alkaline phosphatase was immobilized on sepiolite, bentonite, and protamine-intercalated bentonite in some studies. This was possibly due to high ionic strength and the compression of the diffuse double layer of the mineral, leading to less local pH variation on the clay mineral surface from the solution pH (Carrasco et al., Reference Carrasco, Rad and González-Carcedo1995; Ghiaci et al., Reference Ghiaci, Aghaei, Soleimanian and Sedaghat2009; Sedaghat et al., Reference Sedaghat, Ghiaci, Aghaei and Soleimanian-zad2009; Tang et al., Reference Tang, Shen, Suib, Coughlin and Vinopal1993). Adsorption of acid phosphatase on goethite, kaolinite, and clay minerals from an Ultisol failed to affect significantly the enzyme’s optimum pH, and the adsorbed phosphatases expressed less sensitivity toward pH changes (Huang et al., Reference Huang, Liang and Cai2005). Unlike the more widely investigated reasons for shifted optimum pH, the factors contributing to unchanged phosphatase optimum pH are poorly understood and have not been studied extensively; studies in the past decade did not probe the optimum pH of adsorbed phosphatase.
Conformational changes of adsorbed enzymes have been recognized (Table 1, Fig. 3). The attractive electrostatic force between minerals and the enzyme could induce the unfolding of the enzyme, exposing the hydrophobic chain, and leading to a hydrophobic interaction with the siloxane layers of phyllosilicates (Staunton & Quiquampoix, Reference Staunton and Quiquampoix1994). Hydrophobic interaction between the mineral and enzyme can also induce a conformational change, as discussed in the section Enzyme adsorption mechanisms in clays and clay minerals. The extent of conformational change depends on the balance between several factors including the intramolecular forces of the enzyme, the effects from the mineral surface, and the solvent molecule (Boyd & Mortland, Reference Boyd, Mortland, Bollag and Stotzky1990; Datta et al., Reference Datta, Anand, Moulick, Baraniya, Pathan, Rejsek, Vranova, Sharma, Sharma, Kelkar and Formanek2017). Some enzyme structures, e.g. fibrous proteins, are more likely to unfold due to more interaction with the mineral surface upon adsorption (Boyd & Mortland, Reference Boyd, Mortland, Bollag and Stotzky1990).
An increased ordered secondary structure of phosphatase upon mineral adsorption has been reported. The conformational change of an acid phosphatase when adsorbed on montmorillonite, kaolinite, and inorganic and organic colloids from an Alfisol were studied by Huang et al. (Reference Huang, Zhu, Qiao, Cai, Rong, Liang and Chen2009). This change increased the ordered secondary structure of the phosphatase and the 2:1 clay led to a more significant change than the 1:1 clay. Such a difference was attributed to the difference in the hydrophobicity between kaolinite and montmorillonite. All of the external planar surfaces of montmorillonite are siloxane surfaces which are typically hydrophobic. As for kaolinite, half of the external planar surface is siloxane and, therefore, the kaolinite surface is considered to be less hydrophobic. The more hydrophobic surface of montmorillonite promoted the formation of intramolecular hydrogen bonds.
Adsorption of enzymes on clay minerals can serve as a means to protect the enzymes against, for example, proteolysis, heat, light, and other inhibitors (Huang et al., Reference Huang, Zhu, Qiao, Cai, Rong, Liang and Chen2009; Rao et al., Reference Rao, Violante and Gianfreda2000; Sarkar & Leonowicz, Reference Sarkar and Leonowicz1989). Different minerals vary in their ability to affect the sensitivity of phosphatases toward proteolysis (Table 1). For example, adsorption by montmorillonite was found to provide more proteolytic protection than kaolinite (Huang et al., Reference Huang, Zhu, Qiao, Cai, Rong, Liang and Chen2009). Increased proteolytic resistance was attributed to the conformational change of the enzyme when immobilized, and to the alteration of the cleavage site, disturbing the recognition from the proteinase. A more significant extent of the conformational change induced by montmorillonite than kaolinite led to greater resistance of montmorillonite-adsorbed phosphatase. In another study, increased hydrolytic stability of an acid phosphatase was induced in the order: adsorption on calcined Mg/Al-CO3 layered double hydroxide (LDH) > adsorption on uncalcined Mg/Al-CO3 LDH > enzyme-free (Zhu et al., Reference Zhu, Huang, Pigna and Violante2010). The latter authors suggested that the enzymes penetrated into the pores on the porous surface of LDH, rendering them less accessible for protease and, therefore, the greater porosity of calcined minerals accounted for more elevated proteolytic resistance of the enzyme. The difference in the proteolytic stability may be a result of the specific mineral site where the enzyme is adsorbed. If the enzyme is fixed in the interlayer space, it would be inaccessible to the protease; hence there is an increased proteolytic resistance. On the other hand, if the enzyme is immobilized on the surface or edges of the mineral which can act as a support for concentrated enzymes and proteases, the proteolysis would be enhanced, resulting in decreased proteolytic stability of immobilized enzyme (Boyd & Mortland, Reference Boyd and Mortland1985).
As for the effects on thermal stability, less temperature sensitivity from montmorillonite-adsorbed acid phosphatase was reported by Rao et al. (Reference Rao, Violante and Gianfreda2000). The acid phosphatase adsorbed on kaolinite and goethite had greater thermal stability than the free enzyme (Huang et al., Reference Huang, Liang and Cai2005). The greater thermal stability of kaolinite-adsorbed than goethite-adsorbed acid phosphatase was probably a result of tighter binding on kaolinite as less desorption was detected from kaolinite. They also reported greater thermal stability of an organic clay-adsorbed acid phosphatase, which was attributed to the protective effect of organic matter. Zhu et al. (Reference Zhu, Huang, Pigna and Violante2010) observed enhanced thermal stability of uncalcined and calcined Mg/Al-CO3 LDH-adsorbed acid phosphatase than the free enzyme, which was attributed to the loss of enzyme conformational flexibility. Increased thermal stability was also observed for the alkaline phosphatase adsorbed on a bilayer-surfactant-covered sepiolite (Sedaghat et al., Reference Sedaghat, Ghiaci, Aghaei and Soleimanian-zad2009). Decreased thermal stability of Na-sepiolite-adsorbed alkaline phosphatase was attributed to a rapid loss of moisture from the clay during the incubation, which affected the immobilized enzyme (Carrasco et al., Reference Carrasco, Rad and González-Carcedo1995). No significant changes in the thermal stability of an alkaline phosphatase immobilized on bentonite were observed (Ghiaci et al., Reference Ghiaci, Aghaei, Soleimanian and Sedaghat2009). Light sensitivity has been studied less, and Tietjen and Wetzel (Reference Tietjen and Wetzel2003) found less light sensitivity and photodegradation of montmorillonite- and lake basin clay-immobilized alkaline phosphatase.
Adsorption on clay minerals may protect the enzyme against other inhibitors, e.g. metal cations (aq) or oxyanions. For instance, a decreased inhibitory effect of Cd2+ on the activity of palygorskite-, sepiolite-, goethite-, and montmorillonite-adsorbed alkaline phosphatase than free phosphatase has been observed (Shirvani et al., Reference Shirvani, Khalili, Kalbasi, Shariatmadari and Nourbakhsh2020; Tan et al., Reference Tan, He, Wang, Li, Kong, Tian, Shen, Megharaj and He2018). Wang et al. (Reference Wang, Li, Tan, He, Xie, Megharaj and Wei2017) reported less sensitivity of goethite- and montmorillonite-adsorbed alkaline phosphatase toward the inhibitory effects of arsenate. Rosas et al. (Reference Rosas, Luz Mora, Jara, López, Rao and Gianfreda2008) reported the protective effect of an allophanic clay on an acid phosphatase against Mn2+ and Mo7O4 – inhibition. Huang and Shindo (Reference Huang and Shindo2000) observed different effects from kaolin, goethite, and δ-MnO2 in protecting an acid phosphatase against Cu2+ inhibition. At the same CuCl2 concentration, the goethite-adsorbed enzyme showed greater activity than the δ-MnO2-adsorbed enzyme and this, in turn, was greater than the activity of the kaolin-adsorbed enzyme; the activity of the latter was similar to that of the free enzyme. When copper citrate was used, however, its inhibitory effect was more significant on the clay-adsorbed enzymes than on the free ones. The observed difference induced by varied copper forms was postulated to be a result of the interaction between the carboxylic group of the citrate and the hydroxyl group on the mineral surface, modifying the enzyme conformation.
The protection was attributed to the adsorption of the inhibitors by clay minerals, resulting in lowered inhibitor concentration in the solution and decreased interaction between inhibitors and enzyme active sites. Another factor contributing to the diminished inhibition effect from the inhibitors may be the conformational change of the enzyme after adsorption, burying the active site, which would limit access by the inhibitors (Shirvani et al., Reference Shirvani, Khalili, Kalbasi, Shariatmadari and Nourbakhsh2020; Wang et al., Reference Wang, Li, Tan, He, Xie, Megharaj and Wei2017). The extent of protection depends on many factors, including the concentration and identity of the inhibitor, exposure time, order of addition of the inhibitor, and clay mineral type. For example, adsorption on montmorillonite and goethite diminished the sensitivity of an alkaline phosphatase toward Cd2+ inhibition with an exposure time of >1 h. The opposite effect was observed when the exposure to Cd2+ was <1 h (Tan et al., Reference Tan, He, Wang, Li, Kong, Tian, Shen, Megharaj and He2018). When Mn2+ was added together with allophane to an acid phosphatase, its activity decreased with increasing Mn2+ concentration. When added to allophane-adsorbed acid phosphatase, Mn2+ did not affect significantly the enzyme activity. On the contrary, the opposite trend was observed with Mo7O4 − (Rosas et al., Reference Rosas, Luz Mora, Jara, López, Rao and Gianfreda2008).
The adsorption of phosphatases on clay minerals is, therefore, usually a tradeoff between decreased activity and protection against environmental stress (Fig. 4). From the literature, it is not surprising to find that even with the same mineral, the inhibition mechanism and the way in which the enzyme properties are affected can be different. Effects of different forces in the adsorption and the variety of enzyme sources and, thus, enzyme species and behavior (e.g. conformation, orientation on the mineral surface) with respect to different reaction conditions make enzyme adsorption a complex process and increase the difficulty of predicting the results. Specific enzyme structure and characteristics and reaction conditions including pH and temperature could all lead to different properties and behavior of the mineral surface and enzyme. If one were to investigate or compare the effects of a certain mineral on an enzyme, careful attention should, therefore, be paid to clay and enzyme species and individual reaction conditions.
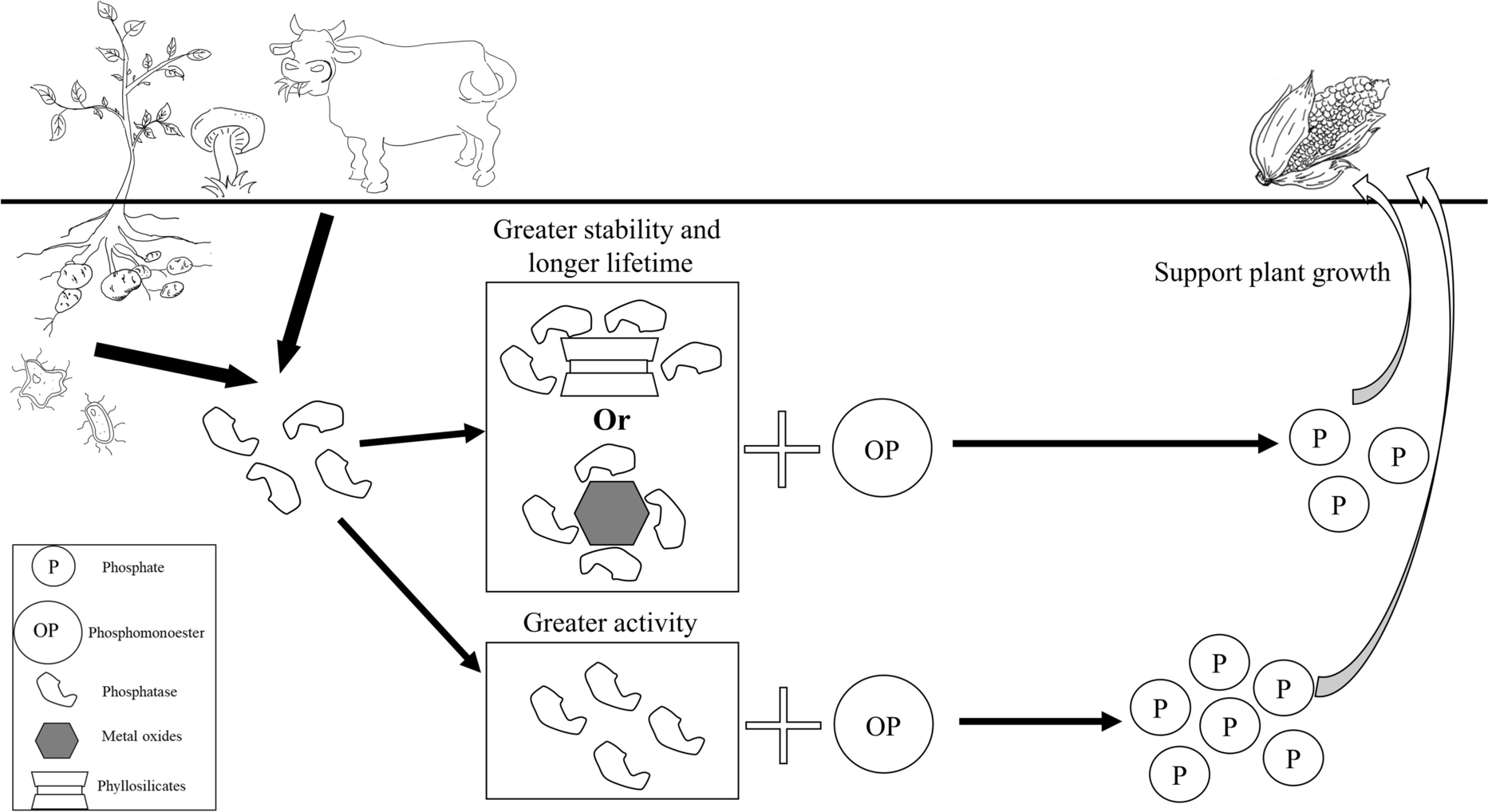
Fig. 4 Phosphatase activities in the terrestrial environment. The role of clays and clay minerals as phosphatase sinks is highlighted. The interaction could suppress P mineralization and enhance enzyme stability
The Phosphatase–Mineral Interaction affects the Fate of P in Soils
The fate of P in soils is dependent on the biological and geochemical activities in the soil and, from the aspect of biology, the release of bioavailable P is achieved through mineralization (e.g. by phosphatase) and solubilization (e.g. by phosphorus-solubilizing microorganisms) (Alori et al., Reference Alori, Glick and Babalola2017; Hussain et al., Reference Hussain, Phillips, Hu, Frey, Geuder, Edwards, Lapen, Ptacek and Blowes2021). Up to 85% of the total P in soils may exist as organic P and a meta-analysis study that summarized results from 149 soils showed that ~30–60% of the organic P in the studied soils can be mineralized by phosphatases and/or phytases (Bünemann, Reference Bünemann2008; Dalal, Reference Dalal1977; Tarafdar et al., Reference Tarafdar, Yadav and Meena2001).
An enhanced phosphatase activity promotes the mineralization of organic P and the release of phosphate (Fig. 2), which is a more bioavailable fraction. For instance, Tarafdar et al. (Reference Tarafdar, Yadav and Meena2001) observed a positive linear relationship between the activity of acid phosphatase and the amount of phosphate released from different organic P compounds. Zou et al. (Reference Zou, Binkley and Caldwell1995) found a positive correlation between phosphatase activity and gross P mineralization rate. Accordingly, depletion of organic P was usually observed with increasing phosphatase activity in the bulk soil (McConnell et al., Reference McConnell, Kaye and Kemanian2020; Schaap et al., Reference Schaap, Fuchslueger, Hoosbeek, Hofhansl, Martins, Valverde-Barrantes, Hartley, Lugli and Quesada2021). An enhanced depletion of different organic P forms (e.g. sodium hydroxide-extractable, bicarbonate-extractable) was also found to be related to increased activity of phosphatases in the rhizosphere of rape, onion, wheat, clover, barley, Norway spruce, and radiata pine, indicating the important role of phosphatases in the organic P mineralization and in providing bioavailable P for plants (Asmar et al., Reference Asmar, Singh, Gahoonia and Nielsen1995; Chen et al., Reference Chen, Condron, Davis and Sherlock2002; Häussling & Marschner, Reference Häussling and Marschner1989; Liu et al., Reference Liu, Loganathan, Hedley and Skinner2004; Tarafdar & Jungk, Reference Tarafdar and Jungk1987).
As well as for phosphate, other P forms are also affected by phosphatase activities. Margalef et al. (Reference Margalef, Sardans, Fernández-Martínez, Molowny-Horas, Janssens, Ciais, Goll, Richter, Obersteiner, Asensio and Peñuelas2017) collated data from 183 studies from 378 sites around the world and found that some P forms may be correlated to the activity of phosphatases in the soil. They found a positive correlation between alkaline phosphatase activity and Olsen-P (the “Olsen-P test” provides estimation on the available P content in the soil), and between organic P and acid phosphatase activity. Yu et al. (Reference Yu, He, Stoffella, Calvert, Yang, Banks and Baligar2006) observed significant correlations between acid and/or alkaline phosphatase activity and different P fractions, including hydrochloric acid-extractable P, Olsen P, and total P, and significant correlations between neutral, as well as natural phosphatase activity (i.e. measured at soil pH) and P fractions such as Olsen P, water-extractable inorganic P, and sodium bicarbonate-extractable organic P. Compton and Cole (Reference Compton and Cole2001) observed a positive relationship between phosphatase activity in the bulk soil and some organic P forms (e.g. the organic P after sodium bicarbonate extraction and labile organic P) at pH 5, which might be attributed to the stimulation of phosphatase excretion induced by high organic P content.
The promoted release of phosphate and other labile P by phosphatases, while more bioavailable, are also prone to leaching. Yu et al. (Reference Yu, He, Stoffella, Calvert, Yang, Banks and Baligar2006) found natural phosphatase activity to be related to the concentration of total P, total dissolved P, and phosphate in surface runoff. A negative correlation was observed between alkaline phosphatase activity and total P and total dissolved P. No correlations were found between acid phosphatase activity and any P fractions, which was probably due to the neutral to slightly alkaline nature of the runoff water. Yu et al. (Reference Yu, He, Stoffella, Calvert, Yang, Banks and Baligar2006) proposed, therefore, that soil natural phosphatase activity may be an index for the P-loss potential by surface runoff. A positive correlation was observed between neutral phosphatase activity and total P in the surface water of a rice paddy field, indicating a possible enhanced P runoff loss (Wang et al., Reference Wang, Liang, Chen, Luo, Liang, Li, Huang, Li, Wan, Li and Shao2012).
The interaction between soil minerals and phosphatases clearly affects phosphatase activity, which in turn influences both soil P availability and loss. Depending on which results are desired, either promoting or suppressing the adsorption of phosphatases on the mineral surfaces could be the goal of soil management. For instance, in strongly weathered soils, the organic P and secondary minerals (e.g. sasaite, wavellite, crandallite, cacoxenite) are usually the predominant P forms and these soils are often observed with limited P availability (Kovács et al., Reference Kovács, Farics, Szabó and Sajó2020; Margalef et al., Reference Margalef, Sardans, Fernández-Martínez, Molowny-Horas, Janssens, Ciais, Goll, Richter, Obersteiner, Asensio and Peñuelas2017; Rocha et al., Reference Rocha, Menegale, Rodrigues, de M Gonçalves, Pavinato, Foltran, Harrison and James2019; Wang et al., Reference Wang, Xie, Zhang and Wang2023). Furthermore, with the long-term development of ecosystems (e.g. forest), available P is gradually lost through surface runoff, for example (Turner et al., Reference Turner, Lambers, Condron, Cramer, Leake, Richardson and Smith2013). Phosphorus depletion and limitation in agricultural soils not only affect yield and decrease biomass but also influence plant community composition and diversity (Turner & Condron, Reference Turner and Condron2013). For such soils, mineralization by extracellular enzymes and maintaining their stability (e.g. through clay adsorption) and activity are particularly crucial in providing bioavailable P (Fig. 4). Some studies observed enhanced phosphatase activity when adsorbed on the mineral surface, which can lead to useful application. Many heavy metals (e.g. Cd, Pb, Cu, Hg, As, W) inhibit phosphatase activity and in some heavy metal-polluted soils, the inhibitory effect, therefore, becomes a severe problem (Hong et al., Reference Hong, Kim, Lee, Yang and Kim2020; Huang & Shindo, Reference Huang and Shindo2000; Mao et al., Reference Mao, Tang, Feng, Gao, Zhou, Xu and Wang2015; Yang et al., Reference Yang, Liu, Zheng and Feng2006). Phosphatase–clay interaction may be used to enhance organic P mineralization or to protect phosphatase against inhibitors or environmental stress, but the real-world application is still limited. For example, an analysis of the inhibition constants and ecological dose of arsenate (Tian et al., Reference Tian, Zhao, Megharaj and He2018) showed that the adsorbents (montmorillonite or soil) protected alkaline phosphatase from arsenate deactivation. Li and Xu (Reference Li and Xu2018) found an increase in phosphatase activity and available P level in a Cd-polluted rice field when sepiolite was added. Although this could also be a result of Cd immobilization by clay minerals and, thus, decreased Cd, the addition of adsorbed or immobilized enzymes to soils still has the potential as one of the applications of enzyme–clay mineral association.
Industrial Application of Enzyme-Mineral Complexes
Many natural and anthropogenic activities contribute to the contamination of soils and waters, and the application of free enzymes or adsorbed enzymes has been developed as a means of bioremediation. One of the main requirements for effective bioremediation by enzymes is the stability of the enzyme under the environmental conditions of the contaminated site (Gianfreda & Rao, Reference Gianfreda and Rao2004). The indigenous extracellular enzymes or free enzymes added to soils are prone to rapid denaturation or degradation due to, for example, microbial and enzymatic degradation or chemical-mediated denaturation, thus generally having a short lifetime (Gianfreda et al., Reference Gianfreda, Rao, Sannino, Saccomandi and Violante2002; Sarkar & Leonowicz, Reference Sarkar and Leonowicz1989). The association of minerals and enzymes, however, can largely contribute to the stabilization and persistence of the extracellular enzymes in soils (Boyd & Mortland, Reference Boyd, Mortland, Bollag and Stotzky1990). As mentioned above, greater thermal stability, proteolysis stability, storage stability, and protection against other inhibitors may result from the adsorption to minerals. Upon adsorption and with increased stability, although usually at a cost of reduced activity, the enzymes can survive longer with long-term retention of activity and contribute to promoting the degradation of xenobiotic substances in aqueous and terrestrial environments to produce less toxic compounds (Gianfreda & Rao, Reference Gianfreda and Rao2004; Gianfreda et al., Reference Gianfreda, Rao, Sannino, Saccomandi and Violante2002; Hoehamer et al., Reference Hoehamer, Mazur and Wolfe2005; Meng et al., Reference Meng, Jiang, Li, Huang, Zhai, Zhang, Guan, Cai and Xiangru2019). Compared to the use of free enzymes, other advantages of applying adsorbed enzymes include improved enzyme reusability, enhanced enantioselectivity, and they can usually be recovered at the end of the process (Burns & Dick, Reference Burns and Dick2002; Chen et al., Reference Chen, Guo, Xin, Gu, Zhang and Guo2023; Gianfreda & Rao, Reference Gianfreda and Rao2004).
Organophosphorus pesticides (e.g. glyphosate), accounting for ~40% of the global pesticide market, have been used commonly in agriculture and forestry (Chen et al., Reference Chen, Guo, Xin, Gu, Zhang and Guo2023). They are highly toxic and disposal methods might be inefficient and produce toxic pollution (Chen et al., Reference Chen, Guo, Xin, Gu, Zhang and Guo2023). As an alternative, using enzymes as a bioremediation method could be a more sustainable, renewable, and green approach that is more environmentally friendly and less invasive (Dzionek et al., Reference Dzionek, Wojcieszynska and Guzik2016; Somu et al., Reference Somu, Narayanasamy, Gomez, Rajendran, Lee and Balakrishnan2022). For instance, organophosphorus hydrolases (e.g. phosphotriesterase, parathion hydrolase), organophosphate acid anhydrase, and methyl parathion hydrolase can mineralize organophosphorus compounds such as (methyl) paraoxon, (methyl) parathion, and soman. Nitrilases can be used for degrading herbicides, polymers, plastics, and cyanide (Mousavi et al., Reference Mousavi, Hashemi, Iman, Ravan, Gholami, Lai, Chiang, Omidifar, Yousefi and Behbudi2021). Enzymes such as tyrosinase, phenoloxidases, and peroxidases have been used for the removal of phenols, aromatic amines, and related compounds (Gianfreda et al., Reference Gianfreda, Rao, Sannino, Saccomandi and Violante2002; Mousavi et al., Reference Mousavi, Hashemi, Iman, Ravan, Gholami, Lai, Chiang, Omidifar, Yousefi and Behbudi2021; Somu et al., Reference Somu, Narayanasamy, Gomez, Rajendran, Lee and Balakrishnan2022).
The application of free and immobilized enzymes has, therefore, become more common. Daumann et al. (Reference Daumann, Larrabee, Ollis, Schenk and Gahan2014) found that glycerophosphodiesterases immobilized on functionalized magnetite nanoparticles had prolonged storage time and can be used for bioremediation of organophosphate pollution. Laccases have wide substrate specificity and can be used for degrading a variety of contaminants such as dyes, pesticides, benzenediol, and polycyclic aromatic hydrocarbons (Mousavi et al., Reference Mousavi, Hashemi, Iman, Ravan, Gholami, Lai, Chiang, Omidifar, Yousefi and Behbudi2021). Montmorillonite- and kaolinite-adsorbed laccase and peroxidase have been used to transform and detoxify 2,4-dichlorophenol, a degradation product from a herbicide (Gianfreda & Bollag, Reference Gianfreda and Bollag1994; Ruggiero et al., Reference Ruggiero, Sarkar and Bollag1989). The removal efficiency has been confirmed both with and without the presence of soils. The authors also reported that the immobilized enzyme can be separated from the reaction mixture and used repeatedly. Masaphy et al. (Reference Masaphy, Fahima, Levanon, Henis and Mingelgrin1996) used a crude parathion-degrading enzyme extract from Xanthomonas sp. to degrade parathion, an organophosphate pesticide. To simulate the soil environment, the enzyme was added to a Na-montmorillonite suspension for parathion degradation. Their study suggested the potential of using the enzyme as an approach in soil decontamination.
Many enzymes have also been used for the bioremediation of haloorganics and heavy metal (e.g. Se, As, Cr, Hg) pollution, which can be an indication of the future application of phosphatases (Burns & Dick, Reference Burns and Dick2002). With the enzyme stabilization brought about via enzyme–clay mineral interaction, the beneficial influences such as eco-friendly remediation and, in some cases, enhanced mineralization can be prolonged with reusable enzymes. To further induce the excretion of enzymes and, therefore, to improve the remediation effect and mineralization efficiency, methods such as adjusting soil properties (e.g. moisture, pH, and fertilizer N amendment) can be applied (Arenberg & Arai, Reference Arenberg, Arai and Sparks2019).
Enzymes adsorbed on the mineral surfaces are also applied in other fields such as the biomedical, biosensor, biocatalyst, food, pharmaceutical, textile, and detergent industries, and in wastewater treatment (An et al., Reference An, Zhou, Zhuang, Tong and Yu2015; Basso & Serban, Reference Basso and Serban2019; Jesionowski et al., Reference Jesionowski, Zdarta and Krajewska2014; Maghraby et al., Reference Maghraby, El-Shabasy, Ibrahim and Azzazy2023). Adsorption can increase thermal stability and protect enzymes against denaturation during the purification process in the food industry (Hassan et al., Reference Hassan, Yang, Xiao, Liu, Wang, Cui and Yang2019). In the pharmaceutical industry, a loss of activity during shelf life can be a problem, and enzymes adsorbed to, e.g. silica surfaces, with greater stability may help alleviate the problem (Norde, Reference Norde2008). Amorphous clay minerals such as allophane can adsorb water from the surrounding environment, providing an aqueous environment for adsorbed enzymes, which can be applied to the biotechnological field (Calabi-Floody et al., Reference Calabi-Floody, Velásquez, Gianfreda, Saggar, Bolan, Rumpel and Mora2012).
Future Perspectives
Previous studies of the phosphatase–mineral interaction investigated the macroscopic behavior of mineralization, such as phosphatase activity and its thermal and/or proteolysis stability. The results showed that the interaction between phosphatases and minerals was a trade-off between mostly inhibited enzyme activity and protection against the environmental stress. Various and complex phosphatase behavior in the literature was attributed to the variety of specific properties of the enzymes and minerals involved. The wide application of adsorbed enzymes, the complexity of the phosphatase–mineral reactions, and the significant role of phosphatase in affecting P fate in soils have, however, made it important to further investigate the microscopic characteristics of the interaction and reaction mechanisms (e.g. the three-dimensional conformational change of the enzyme, effects on detailed biochemical pathway) between phosphatases and clay minerals. Spectroscopic techniques such as nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR), and in situ attenuated total reflectance Fourier-transform infrared (ATR-FTIR) are robust in probing such characteristics, and their recent applications have provided more insights into the microscopic aspects of the protein–clay mineral interactions (Reardon et al., Reference Reardon, Chacon, Walter, Bowden, Washton and Kleber2016; Schmidt & Martínez, Reference Schmidt and Martínez2016; Ustunol et al., Reference Ustunol, Coward, Quirk and Grassian2021). The future microscopic study of phosphatase–clay mineral interaction could, therefore, benefit from these techniques. Furthermore, description of the specific reaction conditions and well-characterized macro- and microscopic properties of the specific phosphatase and minerals could be very helpful when investigating their interaction and comparing results from different studies.
Acknowledgements
The authors gratefully acknowledge the USDA National Institute of Food and Agriculture (Hatch 875-967) for supporting this project financially.
Data Availability
Data available on request from the authors.
Declarations
Conflict of Interest
The authors declare no conflict of interest.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.





