


Inulin is a mixture of fructans consisting of monomers linearly linked by means of β(2-1) bonds with different degrees of polymerization. This prebiotic cannot be hydrolysed by digestive enzymes in the upper intestinal tract of humans (Schneemann, Reference Schneemann1999). Therefore, the non-digestible carbohydrates reach the colon where they are fermented by Bifidobacterium spp. and other lactic acid-producing bacteria. This results in enhanced concentrations of these bacteria in the gut which beneficially affect the hosts (Ellgard et al. Reference Ellgard, Andersson, Bosaeus and Bosaeus1997; Klinder et al. Reference Klinder, Forster, Caderni, Femia and Pool-Zobel2004a). In particular, a number of different experimental studies have shown that the fermentation products of inulin contribute to colon cancer-preventing properties, as has been summarized in a recent review (Pool-Zobel, Reference Pool-Zobel2005). For example, in vitro it beneficially modulated markers of tumour progression in a human colon tumour cell model (Klinder et al. Reference Klinder, Gietl and Hughes2004b). In vivo, several studies have shown that the addition of inulin to the diet of azoxymethane-treated rats reduced the yield of aberrant crypt foci (Pool-Zobel, Reference Pool-Zobel2005). The effects of inulin were dose related, and also dependent on the chain length of the inulins (Verghese et al. Reference Verghese, Rao, Chawan and Shackelford2002, Reference Verghese, Walker, Shackelford, Chawan and Van Loo2003).
The consumption of dietary fibre may be indirectly protective in the colon by elevating faecal volume and enhancing defecation, both of which reduce exposure of colonocytes to cancer risk factors. In addition, protection may be a result of the gut flora-mediated fermentation of dietary fibre. The fermentation products of inulin may be protective in early stages of cancer onset since the number of apoptotic cells per crypt was higher in rats fed oligofructose and long-chain inulin (Hughes & Rowland, Reference Hughes and Rowland2001). Poulsen et al. (Reference Poulsen, Molck and Jacobsen2002) also reported a decreased cell proliferation and therefore a reduced cell turnover which may be indicative of cancer suppression also at later stages of the progression process.
Some types of dietary fibre including inulin, however, could be protective via the formation of SCFA during fermentation by the gut flora (Cummings, Reference Cummings1981). In non-transformed cells, the SCFA, butyrate, is utilized as an energy source (Roediger, Reference Roediger1989), and in tumour cells, butyrate reduces survival by inducing their apoptosis and inhibiting proliferation (Kruh, Reference Kruh1982). Another mechanism of protection by fermentation products, especially by butyrate, has been hypothesized to be the induction of glutathione S-transferases (GST) which are phase II enzymes of biotransformation that detoxify many carcinogens (Turesky et al. Reference Turesky, Lang, Butler, Teitel and Kadlubar1991; Manus et al. Reference Manus, Adang, Ambergen, Brägelmann, Armbrecht and Stockbrügger1997). The increased cellular levels of such enzyme systems has been shown to protect against food-derived genotoxic compounds such as 4-hydroxynonenal in tumour-derived cell lines (Ebert et al. Reference Ebert, Beyer-Sehlmeyer, Liegibel, Kautenburger, Becker and Pool-Zobel2001). Similar mechanisms occurring in non-transformed cells may very well reduce cancer initiation, and thus be considered an effective means of primary cancer chemoprevention (Johnson et al. Reference Johnson, Williamson and Musk1994; Pool-Zobel et al. Reference Pool-Zobel, Veeriah and Bohmer2005b) since GST are capable of detoxifying endogenous and food-derived carcinogens like 4-hydroxynonenal or benzo(a)pyrene.
It is now of interest to explore whether complex fermentation samples (containing butyrate, other SCFA and additional compounds) may have similar activities. Therefore, we investigated fermentation samples from inulin using human non-transformed primary colon cells representing the more appropriate target cells of chemoprevention. We compared the effects to those of butyrate and to a mixture of SCFA composed according to the fermentation sample. The fermentation products studied here were generated in vitro using anaerobic procedures that simulate the physiological conditions of the human gut. This presents an experimental approach to analyse the biological activities of different dietary fibres and their resulting complex fermentation products (Wang & Gibson, Reference Wang and Gibson1993). The results were expected to enhance our understanding of the chemopreventive properties of prebiotics' fermentation supernatants from diet in terms of reducing colorectal cancer risks (Lupton, Reference Lupton2004).
Materials and methods
Fermentation of inulin-derived prebiotics
The carbohydrate source used in the fermentation experiments was the fructan Synergy1, a commercially available 1 : 1 mixture of inulin (Femia et al. Reference Femia, Luceri, Dolara, Giannini, Biggeri, Salvadori, Clune, Collins, Paglierani and Caderni2002) enriched with oligofructose (ORAFTI, Tienen, Belgium). The fermentation of this inulin-type fructan mixture was conducted in vitro under anaerobic conditions (80 % N2, 10 % CO2, 10 % H2 at 37°C) in a batch-culture system with faecal inoculum of different donors (Manderson et al. Reference Manderson, Pinart, Tuohy, Grace, Hotchkiss, Widmer, Yadhav, Gibson and Rastall2005; Van Loo et al. Reference Van Loo, Clune, Bennett and Collins2005). Each tube was mixed well and incubated at 37°C for 24 h during which it was shaken manually at intervals. After placing the suspensions on ice to stop the fermentation, the samples were centrifuged at 6000 g at 4°C for 30 min. The fermentation supernatants were stored at − 20°C and filter-sterilized (pore size 0·22 μm) before use in the experiments.
Determination of SCFA
The content of the SCFA of the fermentation supernatant and the corresponding faeces control was determined using GC (Kiessling et al. Reference Kiessling, Schneider and Jahreis2002).
In brief, the sample was weighed and an internal standard solution (2-methyl valeric acid, 0·01 %) was added. The solution was acidified with 0·5 ml H2SO4 (pH < 2) and extracted by shaking with 2·0 ml diethyl ether and subsequent centrifugation (10 min at 2000 rpm). The ether phase was injected directly on to the oven-heated (150°C) column (BT21-FFAPP, 25 m × 0·53 mm × 0·5 mm; Achrom NV/SA, Mechelen, Belgium), the carrier gas used was N2 and detection temperature was set at 230°C (GC VEGA 6000; Carlo Elba, Milan, Italy). Peaks were integrated automatically using Atlas software (ThermoLab Systems, Breda, The Netherlands).
Preparation of synthetic SCFA mixtures
The analytical data of the SCFA determination were taken as a basis to compose the synthetic Synergy1 fermentation supernatant (SFS) mixture. This was prepared to mimic the amounts and proportions of SCFA found in the inulin-type fructan fermentation supernatant. The concomitant investigation of the synthetic mixture of the major SCFA acetate, propionate and butyrate was expected to reveal which proportion of biological activity was due to the SCFA in the fermentation supernatant.
For this we dissolved sodium butyrate, sodium propionate and sodium acetate in the determined molar concentrations in cell culture medium and prepared 50-fold concentrated stock solutions. Aliquots were stored at − 20°C and diluted to the end concentrations applied in the experiments before use.
Primary colon tissue preparation and isolation of cells
Primary colon cells were isolated from colon tissue obtained during surgery of colorectal tumours, diverticulitis and colon polyps from patients who had given their informed consent. The tissue specimens were parts of the non-tumorous tissue which was co-removed for medical indications. The university ethics committee approved the study. Mean age of the three donors of colon cells for incubation and RNA isolation was 66 (sd 20) years. One of the donors was male, two were female. The mean age of the six donors whose cells were used for determining metabolic activity was 57 (sd 19) years. Four of the donors were male, two were female. The cells used for incubation and cytosol preparation (GST activity) were taken from two male and one female subject (mean age 69 (sd 12) years). The tissue was prepared as described previously (Schäferhenrich et al. Reference Schäferhenrich, Sendt, Scheele, Kuechler, Liehr, Claussen, Rapp, Greulich and Pool-Zobel2003). These epithelial stripes were used for incubation and subsequent analysis of gene expression using conditions that had been determined as optimal before. After 12 h treatment, the cells were isolated from the epithelial stripes as described before (Schäferhenrich et al. Reference Schäferhenrich, Sendt, Scheele, Kuechler, Liehr, Claussen, Rapp, Greulich and Pool-Zobel2003). Viability and cell yields were determined with trypan blue. We also digested the tissue stripes directly to yield a single cell suspension which was seeded and incubated for the metabolic activity assay.
Measurement of the metabolic activity
Single cells were seeded into ninety-six-well microtitre plates (50 000 cells/well) and were incubated in minimal essential medium enriched with 20 % FCS, 2 mm-glutamine, 1 % penicillin/streptomycin, 100 μg/ml gentamycin, 2·5 μg/ml fungizone, 10 ng/ml epidermal growth factor, 5 μg/ml insulin, 5 μg/ml transferrin and 5 ng/ml sodium selenite (Rogler et al. Reference Rogler, Daig, Aschenbrenner, Vogl, Schlottmann, Falk, Gross, Scholmerich and Andus1998).
Primary colon cells were incubated for 4, 12 and 24 h with SFS (0–15 %) and butyrate (0–50 mm, positive control) to determine the biologically effective concentration ranges. A faeces blank supernatant (fermentation without substrate) was included in each experiment as the negative control. A mixture of the SCFA (acetate, propionate and butyrate) was diluted in culture medium at concentrations which mimicked the content of SCFA in the SFS. This mixture was also included as a positive control. To measure the metabolic capacity of the cells, they were incubated in ninety-six-well microtitre plates with the dye resazurin which is reduced into resorufin only by viable cells (CellTiter-Blue® assay; Promega, Mannheim, Germany). The product is highly fluorescent and was detected with Ex/Em 520/595 nm after 2 h incubation with the reagent. Mean values and standard deviations were calculated of at least three independent experiments.
Gene expression studies
Using concentrations determined to be subtoxic, human colon epithelium stripes were incubated with SFS (10 %), a synthetic mixture of SCFA, a faeces supernatant control, medium or butyrate. The chosen butyrate concentration (10 mm) mimics possible physiological concentrations in the gut lumen (Hass et al. Reference Hass, Busche, Luciano, Reale and Engelhardt1997). We incubated small tissue pieces in Petri dishes (35 mm) to ensure the greatest possible surface for contact with the medium and to maintain the highest possible viability. After allowing the epithelial stripes to settle for 15 min, they were subjected to treatment with the test compounds.
Total RNA was isolated from the cells (up to 6 × 106 cells) with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions and stored at − 20°C. The ratio A 260/A 280 and concentration of total RNA were determined spectrophotometrically (Eppendorf BioPhotometer, Hamburg, Germany) for protein or phenol contamination followed by formaldehyde denaturing RNA gel electrophoresis (1·5 %) to check the integrity of the ribosomal RNA and possible DNA contamination.
Expression analysis of 112 human genes (sixteen reference spots, and ninety-six genes related to drug metabolism) was performed with cDNA gene macroarrays (GEArray Q Series Human Drug Metabolism Gene Array HS11; SuperArray® Bioscience Corporation, Frederick, MD, USA). Genes were classified into functional categories, representing phase I enzymes (cytochrome p450 family, epoxide hydrolases), phase II enzymes (acetyltransferases, GST, sulphotransferases and miscellaneous others which included UDP-glucuronosyl transferases), and metallothioneins and p-glycoproteins. A detailed gene list is available at the company's website (www.superarray.com). Three arrays each were used for RNA isolated from the medium controls and the treated samples of primary colon cells. The array was performed according to the manufacturer's protocol and as described previously (Pool-Zobel et al. Reference Pool-Zobel, Selvaraju, Sauer, Kautenburger, Kiefer, Richter, Soom and Wolfl2005a). Raw data were normalized between 0 and 100 % expression whereas the signals of the negative controls were calculated as 0 % and the means of the signals of the positive controls were set to equal 100 %. Thus, the data shown here represent mean expression levels relative to negative and positive reference genes as described previously (Pool-Zobel et al. Reference Pool-Zobel, Selvaraju, Sauer, Kautenburger, Kiefer, Richter, Soom and Wolfl2005a).
Relative quantification of glutathione S-transferase expression with real-time PCR
The expression of several GST isoforms (GSTA4, GSTM2, GSTM5, GSTP1, GSTT2) found to be modulated according to the array analysis was also studied using quantitative real-time PCR (SYBR Green I system). Total RNA (1 μg) was reverse transcribed (SuperScript II, First-Strand cDNA Synthesis System; Invitrogen, Karlsruhe, Germany) in 20 μl buffer with oligo-(dT)15 primers according to the manufacturers' instructions. cDNA (5 μl; 33·3 ng total RNA equivalent) was used in a 25 μl PCR amplification reaction containing 2 × iQ SYBR Green supermix® (100 mm-KCl, 40 mm-Tris-HCl, pH 8·4, 0·4 mm each dNTP, 50 U/ml iTaq DNA polymerase, 6 mm-MgCl2, SYBR Green I, 20 nm-fluorescein, stabilizers) and 10 pmol gene-specific primers for the target GST genes and the reference (GAPDH) gene. The following primer sequences were used to amplify a region of GSTA4, GSTM2, GSTM5, GSTP1, GSTT2 and GAPDH mRNA:
iGSTA4_F 5′-ccg gat gga gtc cgt gag atg g-3′
iGSTA4_R 5′-cca tgg gca ctt gtt gga aca gc-3′
iGSTM2_F 5′-agc cgt atg cag ctg gcc aaa c-3′
iGSTM2_R 5′-cca caa agg tga tct tgt ccc ca-3′
iGSTM5_F 5′-ttg cag gag aca aga tca cct ttg-3′
iGSTM5_R 5′-gat ctt ctt caa acc ctc aaa gcg-3′
iGSTP1_F 5′-ctg cgc atg ctg ctg gca gat c-3′
iGSTP1_R 5′-ttg gac tgg tac agg gtg agg tc-3′
iGSTT2_F 5′-tga cac tgg ctg atc tca tgg cc-3′
iGSTT2_R 5′-gcc tcc tgg cat agc tca gca c-3′
iGAPDH_F 5′-cca ccc atg gca aat tcc atg gc-3′
iGAPDH_R 5′-agt gga ctc cac gac gta ctc ag-3′
PCR cycles included one cycle of 95°C for 2 min followed by forty cycles each of 94°C for 30 s, annealing temperature 60°C for 30 s and 72°C for 40 s, and a final extension step of 72°C for 10 min (iCycler iQ® Real-Time PCR Detection System; Biorad GmbH, Munich, Germany). Product-specific amplification was confirmed by melting curve analysis and agarose gel electrophoresis. All experiments were performed in duplicate. The fluorescence threshold value (C T) was calculated using the iCycler iQ® optical v3·0a system software. The relative quantification of GST-mRNA expression was calculated with the comparative ΔΔC T (ΔΔC T = ΔC Tcontrol − ΔC Texperiment) method. For normalization, ΔC T values were calculated by subtracting the average of the C T value in the control for the reference gene from the average of the C T value for the target gene and subtracting the average of the C T value in the treated sample of the reference gene from the target gene. Then the difference between the ΔC T values of control and treatment (ΔΔC T) was calculated. The fold change was calculated according to the efficiency method (ΔΔC T method) where it is assumed that the PCR efficiency is 100 % (E = 2; fold change = E difference; Pfaffl, Reference Pfaffl2001; Pfaffl et al. Reference Pfaffl, Horgan and Dempfle2002).
Preparation of cytosol, measurement of total glutathione S-transferase activity and determination of cytosolic protein
Epithelial stripes were incubated for 12 h with 10 % SFS, 10 mm-butyrate and controls, and then single cells were isolated. The cells were washed and resuspended in cold homogenizing buffer consisting of 250 mm-sucrose, 20 mm-Tris-HCl, 1 mm-dithiothreitol and 1 mm-Pefabloc (Roth, Karlsruhe, Germany; pH 7·4) and homogenized using ultrasound (Bandelin Electronics, Berlin, Germany). After centrifugation (16 000 g, 60 min, 4°C), the supernatant was aliquoted and frozen at − 80°C until use. Total GST activity was determined spectrophotometrically at 340 nm and 30°C using 1-chloro-2,4-dinitrobenzene as substrate. Total protein content was measured using the method of Bradford with bovine serum albumin as standard protein.
Statistical evaluation
The new batch of cells for each experiment was isolated from a different donor. The cells were divided and were then treated with the test compounds. All experiments were conducted independently at least three times (three batches of cells). Means and standard deviations were calculated from at least three independently reproduced experiments. Data of at least three (n ≥ 3) experiments were evaluated to establish two-sided significance levels of independently reproduced determinations. Differences due to different treatments were calculated with GraphPad Prism software version 4 (GraphPad Software Inc., San Diego, CA, USA) using one-way ANOVA with Dunnett's multiple comparison post-test or two-way ANOVA with Bonferroni's post-test. Where appropriate, Microsoft Office Excel 2003 (Microsoft Corp., Redmond, WA, USA) was used to detect differences using unpaired t test.
Results
Analysis of the fermentation supernatants
Table 1 shows the concentrations of SCFA in the supernatants used in the present study. In the SFS, the total SCFA amounted to 94·4 mmol/l whereas only 35·9 mmol/l were detected in the faeces control. Butyrate increased from 4 mmol/l in the faeces control to a concentration of 10 mmol/l in the inulin-derived fermentation sample.
Table 1 Concentration and molar ratios of the SCFA (mmol/l) in the fermentation sample determined using GC†
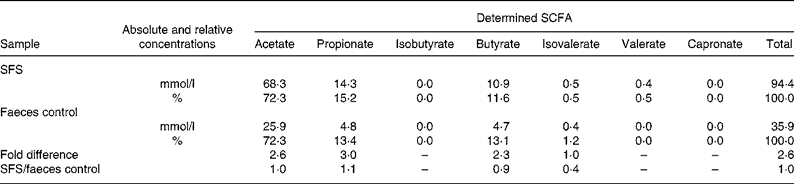
SFS, Synergy1 fermentation supernatant.
† Acetate, propionate and butyrate more than doubled after fermentation of Synergy1 whereas the ratio between the SCFA remained constant.
Metabolic activity
In order to estimate the effect of the fermentation supernatant and of SCFA on viability of the primary colon cells, we measured kinetics of metabolic activity. For this, primary single cells could only be incubated for relatively short periods ( < 12 h) since pilot studies had shown that their viabilities (determined with trypan blue) decrease after 12 h from 89 (sd 3) % to 60 (sd 9) %. Table 2 shows relative values after setting the medium control to equal 100 % since the basic levels of metabolic activity varied highly between the different donors despite similar starting viabilities (90·7 (sd 5·6) %). Absolute values are shown in the footnote to Table 2 which shows that after 4 h of incubation with the test compounds, neither SFS, synthetic SCFA mix nor the faeces control impaired the cells' metabolic activities. Only the highest tested dose of butyrate (50 mm) significantly reduced metabolic activity (85·5 (sd 7·2) %) in comparison to the medium control, which was set to equal 100 %.
Table 2 Metabolic activity in human primary colon cells measured after 4, 12 and 24 h†(Mean values and standard deviations)

a,b Mean values with alike superscript letters were significantly different (P < 0·05; two-way ANOVA, Bonferroni's post-test).
Mean values were significantly different from those of the control (medium) group: *P < 0·05; **P < 0·01 (one-way ANOVA, Dunnett's multiple comparison test).
† The absolute metabolic activity of the controls decreased time-dependently from 8314 (sd 2372) fluorescence units (FU) to 3829 (sd 2257) FU after 12 h and to 1107 (sd 404) FU after 24 h in the medium controls (n 3).
‡ Concentrations of 10 % yielded treatment doses of 1·1 mm-butyrate and 9·4 SCFA in culture medium.
§ Concentrations of 10 % yielded treatment doses of 0·5 mm-butyrate and 3·6 mM SCFA (see also Table 1).
After 12 h of treatment, the metabolic activity was significantly increased by 15 % (v/v) SFS (123·5 (sd 5·4) %) and by 15 % (v/v) of the faeces control (133·8 (sd 20·4) %), indicating trophic effects due to the treatments. In contrast, corresponding concentrations of the SCFA mixture (15 %, v/v) and of butyrate (~1 mm) did not have these effects. Very high amounts of butyrate (50 mm) continued to significantly reduce the metabolic activity to 79·9 (sd 6·6) %, after 12 h, and further down to 67·0 (sd 9·1) % after 24 h. After the 24 h treatment period, the opposing effects of SFS which increased metabolic activity, and pure SCFA which had no effect, were still apparent and again pointed to trophic effects by the complex SFS on survival of primary cells. After 24 h of exposure, the faeces control was neither toxic nor trophic. Since the absolute values of metabolic activity in the medium controls at 4 h were decreased by 53·9 % after 12 h treatment and by 86·7 % after 24 h treatment, the complex SFS seems to compensate this loss of viability and retained metabolic activity on account of yet unidentified fermentation products.
RNA isolation and glutathione S-transferase gene expression
Gene expression was analysed using the human drug metabolism macroarray HS11 (Superarray) on which ninety-six genes for enzymes of the biotransformation are spotted on a nylon membrane. For standardization, 1 μg total RNA was used for each array. Twelve hours was the largest possible duration to incubate primary tissue in vitro and to recover sufficiently viable cells (76 (sd 15) %; trypan blue exclusion test) and enough intact RNA (7·6 (sd 2·8) μg/treated sample and 3·6–5·5 μg RNA/1 × 106 cells, respectively) to perform gene expression analysis. The same RNA was also used for confirmatory real-time quantitative PCR (qPCR) experiments.
The ‘fold change’ values were calculated from the normalized data. These were based on the corresponding values in the faeces control (complete fermentation supernatant) or the medium control (SCFA mixture). The changes were considered to be biologically significant if the ratio was ≤ 0·5 or ≥ 2·0 (Pool-Zobel et al. Reference Pool-Zobel, Selvaraju, Sauer, Kautenburger, Kiefer, Richter, Soom and Wolfl2005a). Additionally, statistical significance was analysed using a two-sided t test. Regulation by the SFS is summarized in Table 3 and a detailed presentation of all data on expression of GST in primary cells is shown in Table 4.
Table 3 Summary of those genes related to drug-metabolizing enzymes which were up- or down-regulated in primary colon cells treated with the fermentation sample in comparison to the faeces control† (Mean values and standard deviations)
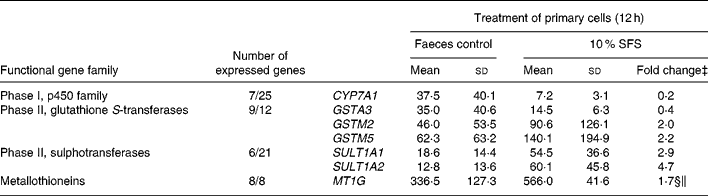
SFS, Synergy1 fermentation supernatant.
† Only those genes which reached an expression level with a signal over the cut-off level ( ≥ 15) in one of the treatments were evaluated (except the glutathione S-transferases, where all genes were considered).
‡ Fold changes ≥ 2·0 and ≤ 0·5 were considered as well as were those genes which are significantly different from the faeces control and as marked with symbols (§two-way ANOVA, Bonferroni's post-test; ‖unpaired t-test).
Table 4 Expression levels and modulation (fold change) of glutathione S-transferases by the Synergy1 fermentation supernatant (SFS) in comparison to the faeces control and in comparison to the medium control in primary colon cells† (Mean values and standard deviations)

SynMix, synthetic SCFA mixture.
† The results for butyrate are already published in Pool-Zobel et al. (Reference Pool-Zobel, Veeriah and Bohmer2005b). All genes of this family were evaluated. Mean changes (n 3) with a fold change ≥ 2·0 and ≤ 0·5 were considered to be different from the medium control. The genes which met these criteria are written in bold.
There were large variations in the levels of gene expression between the different donors for several of the analysed genes. The changes due to treatments, however, pointed to many of the genes in the same direction. Otherwise, they were not considered for additional assessment.
Seven genes were altered by the fermentation supernatant in comparison to the faeces control. CYP7A1 was reduced whereas two sulphotransferases were enhanced. Expression of MT1G was also increased. Table 4 shows the differential expression of GST genes based on the faeces control. It is apparent that GSTM2 and GSTM5 were up-regulated whereas there was a clear reduction of GSTA2 and GSTA3 expression. The expression of GSTM2 and GSTM5 were subjected to confirmatory analysis by real-time qPCR. GSTA4 was additionally included as a negative control. Real-time qPCR analysis confirms the directional changes of the macroarray since GSTM2 was induced 1·4-fold (2-fold in the array) and GSTM5 2·0-fold (2·2-fold in the array). The ‘negative’ control GSTA4 which was not induced according to the array analysis (0·9-fold) was also not altered according to real-time qPCR (1·0-fold).
The comparison of the faeces control (Table 4) to the medium control also shows that a number of genes were differentially regulated. There was, for instance, a down-regulation of MGST3 (0·4-fold), GSTM2 and GSTM5 (each 0·2-fold), but a marked up-regulation of GSTA1, GSTA2 and GSTA4 (2–2·5-fold) by the faeces control. The responsible factors are probably certain bacterial metabolites (other than SCFA) which, however, have not yet been identified. According to real-time qPCR analysis there was also an induction of GSTA4 (1·3-fold) compared to the medium control, but the decreases of GSTM2 (0·8-fold) and GSTM5 (0·9-fold) were not as strong as observed in the array analysis (Fig. 1). From Table 4 it is apparent that the SFS when compared to the medium control seems to counteract the GSTM2, GSTM5 and MGST3 reduction, but is not as efficient in enhancing GSTAs.

Fig. 1 Quantitative analysis of glutathione S-transferase (GST) mRNA by real-time PCR in primary cells treated with the faeces control (a), butyrate (b) and the SCFA mixture (c) for 12 h. We present here results also for the butyrate treatment verifying array results published in Pool-Zobel et al. (Reference Pool-Zobel, Veeriah and Bohmer2005b). Values are means with their standard deviations depicted by vertical bars (n 3). The relative gene expression analysis showed a 1·9-fold up-regulation for GSTA4, a 2·9-fold increase for GSTT2 (n 6) and a 1·4-fold change for GSTP1 (n 3) after butyrate treatment compared to medium-treated (Med) control cells (unpaired t-test, Welch's correction). GADPH, glyceraldehyde-3-phosphate dehydrogenase.
Confirmatory analysis of the up-regulation of GSTA2 and GSTA3 by the faeces control in comparison to the medium control could not be performed. Due to high homology between the A1, A2 and A3 isoforms, the available GSTA2 and GSTA3 cDNA primer sequences were not specific enough to yield specific PCR products.
Table 4 also compares the effects of the synthetic SCFA mixture to previously published data on butyrate (Pool-Zobel et al. Reference Pool-Zobel, Selvaraju, Sauer, Kautenburger, Kiefer, Richter, Soom and Wolfl2005a). When compared to the medium control the butyrate treatment of primary colon cells results in an up-regulation of GSTA2, GSTA3, GSTA4 and GSTT2, whereas the SCFA mixture seems to down-regulate GSTM2 and MGST1. This indicates that the SCFA combination (delivering ~10 mm-butyrate, acetate and propionate to the cell suspension) is far less effective than butyrate (10 mm). The respective confirmatory analysis for butyrate treatment was per-formed for GSTA4 and GSTT2 and for the moderately altered GSTP1 since these had been studied in the macroarray analysis (2·7-, 2·0- and 1·4-fold, respectively), but had not previously been confirmed. Fig. 1 shows that these changes in expression levels by butyrate were largely confirmed by real-time qPCR. Expression results for GSTM2 and GSTM5 after treatment with the SCFA mixture were confirmed (0·4- and 0·8-fold in the microarray, respectively). Fig. 1 shows that the results for GSTA4 also confirm the data of the macroarray analysis, whereas the reduction of GSTM2 was not detected with real-time PCR.
Quantification of total glutathione S-transferase activity
GST activity was measured as a functional characteristic of gene expression using 1-chloro-2,4-dinitrobenzene as substrate. A number of GST isoenzymes utilize this compound as substrate and are capable of catalysing the conjugation reaction. In particular, there was a distinct correlation between GSTP1 protein expression and GST activity in HT29 colon cells (Ebert et al. Reference Ebert, Klinder, Schäferhenrich, Peters, Sendt, Scheele and Pool-Zobel2003). There was already a loss of basal GST activity in primary colon cells treated only with medium for 12 h. The initial activity of 211 (sd 79) nmol/min per mg protein was reduced to 146 (sd 77·6) nmol/min per mg protein after this period. The treatment of colon cells with the different test compounds slightly reduced the enzyme activity even more, as shown in Table 5. Both complex samples (SFS and faeces blank) were inhibitory resulting in only 73 (sd 12) and 81 (sd 6) % of the activity in the medium control which was set to 100 %. In comparison, the SCFA mixture and butyrate were less inhibitory. When compared to the faeces control (which was set to equal 100 %), GST activity was hardly altered by the SFS. However, the observed differences were not significant.
Table 5 Glutathione S-transferase (GST) activity after 12 h treatment with the fermentation product and corresponding controls (Mean values and standard deviations)
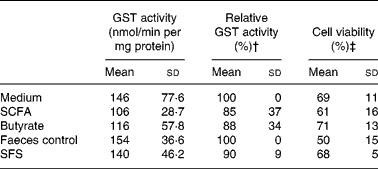
SFS, Synergy1 fermentation supernatant.
† Relative total GST activity was calculated separately for each experiment.
‡ Cell viability decreased in comparison to the basal viability of 90 (sd 4) %, but there were no significant differences between the treatments and the medium control (n 3, one-way ANOVA, Dunnett's multiple comparison test).
Discussion
Inulin enriched with oligofructose (Synergy1) is a source of dietary fibre which yields high amounts of butyrate due to fermentation by gut bacteria. Here, the in vitro fermentation of inulin enriched with oligofructose almost tripled the concentration of SCFA in the supernatant. These in vitro concentrations of SCFA in the SFS correspond to absolute amounts and molar ratios found after high consumption of dietary fibre in vivo (Beyer-Sehlmeyer et al. Reference Beyer-Sehlmeyer, Glei, Hartmann, Hughes, Persin, Bohm, Rowland, Schubert, Jahreis and Pool-Zobel2003). Several in vitro and in vivo rat studies have shown that inulin or the complex fermentation products may exert protective effects in colon cells. Since so far only little is known in non-transformed human colon cells we have developed techniques to measure a modulated state of gene expression and functional consequences leading to chemoprotection in normal colon cells.
As shown in previous studies, butyrate and complex fermentation supernatants are potent inhibitors of cell growth in colon tumour cells. Here the results show that primary human colon cells tolerated butyrate and fermentation supernatants in high amounts during short-term incubations. The loss of metabolic activity throughout the experiments was time dependent and was barely influenced by increasing concentrations. The increase of metabolic activity by the SFS after 12 and 24 h pointed to nutrient effects and indicated that the remaining cells were more active after treatment with the SFS. Only butyrate reduced metabolic activity at 50 mm in the tissue specimens used in the present study. This effect may be donor-specific since the same treatments in specimens of other donors (n 3) indicated trophic effects (increased metabolic activity) at 50 mm after 4 h (J Sauer, KK Richter & BL Pool-Zobel, unpublished results).
The results from the metabolic assay and from the gene array analysis revealed that undefined compounds in the faeces supernatant also have effects on the cells. These factors could include bile acids (e.g. lithocholic acid and desoxycholic acid) and other unidentified bacterial products (Roberfroid, Reference Roberfroid2005). The compounds seem to suppress cell growth in tumour and adenoma cell lines since SFS were more active for inhibiting survival of cell lines than butyrate alone (J Kiefer & D Scharlau, unpublished results). In contrast, the same SFS enhanced metabolic activity in the primary cells, pointing to nutritive effects by products formed during the fermentation of inulin in non-transformed primary cells. Moreover, this normal cell type seemed to be less susceptible to the treatment with the fermentation supernatant than colon cancer cell lines.
Butyrate, as one of the main active fermentation products, is known to modulate gene expression. Previously, we were able to show that important enzymes of biotransformation are inducible in colon cell models. For instance, butyrate can enhance the expression of GST in human colon tumour cells (Ebert et al. Reference Ebert, Beyer-Sehlmeyer, Liegibel, Kautenburger, Becker and Pool-Zobel2001, Reference Ebert, Klinder, Schäferhenrich, Peters, Sendt, Scheele and Pool-Zobel2003; Pool-Zobel et al. Reference Pool-Zobel, Selvaraju, Sauer, Kautenburger, Kiefer, Richter, Soom and Wolfl2005a). The present study investigated the potential of fermentation products derived from inulin to modulate genes related to drug metabolism in primary human colon cells. This is considered to be an important mechanism leading to chemoresistance of the affected cells. Altogether, the treatment with the SFS in comparison to the faeces control altered seven genes. SFS induced GSTM2 and GSTM5 which are capable of detoxifying electrophilic compounds that include carcinogens or environmental toxins. Additionally, these GSTM isoforms possess peroxidase activity (Hayes & Strange, Reference Hayes and Strange2000; Hayes et al. Reference Hayes, Flanagan and Jowsey2005). Moreover, products of oxidative stress can be quenched by conjugation with glutathione. Expression of CYP7A1 was lowered which might result in a reduced metabolic activation of xenobiotics by monooxygenases. In the liver, CYP7A1 plays a key role in cholesterol degradation. It binds cholesterol and converts it to 7α-hydroxycholesterol (Mast & Pikuleva, Reference Mast and Pikuleva2005). Thus, CYP7A1 primarily regulates the pathway through which cholesterol is converted into bile acids. Two sulphotransferases, SULT1A1 and SULT1A2, were enhanced. Sulphate conjugation is an important pathway in the bio-transformation of many exogenous and endogenous compounds. These enzymes catalyse sulphate conjugation of many phenolic or catechol drugs and other xenobiotics as well as endogenous compounds (e.g. oestrogens; Carlini et al. Reference Carlini, Raftogianis, Wood, Jin, Zheng, Rebbeck and Weinshilboum2001) and can therefore enhance the cellular detoxification capacities. But it is also described that sulphation can result in activation or metabolic activation of sulphate acceptor substrates (Falany, Reference Falany1997). These functional consequences (good or bad) will depend on types of exposure that occur. The induction of MT1G, however, might mainly be a favourable effect since this family of metal-binding proteins can scavenge metal ions, free radicals, toxins and activated xenobiotics (Coyle et al. Reference Coyle, Philcox, Carey and Rofe2002). Altogether, the observed shift in gene expression patterns could be possibly related to chemoprevention since by enhancing the detoxification capacity it is possible to reduce exposure to carcinogenic compounds.
The faeces blank was also able to alter gene expression when compared to the medium control, again pointing to yet unidentified factors in the faecal matrix that are responsible. The changes in gene expression by the SFS are not only attributable to the content of SCFA since the synthetic mixture did not cause the same effects. The SCFA combination was less effective than butyrate (Pool-Zobel et al. Reference Pool-Zobel, Selvaraju, Sauer, Kautenburger, Kiefer, Richter, Soom and Wolfl2005a) which could be due to concentration-related effects. The mixture contained only approximately 1 mm-butyrate, but approximately 10 mm-total SCFA. The difference in effects is therefore possibly based on combination effects. Alternatively, the detected effects are only attributable to butyrate. Butyrate itself can directly act on gene expression because of butyrate responsive factors (e.g. butyrate responsive factor 1) on promoters (Maclean et al. Reference Maclean, McKay and Bustin1998; Patel et al. Reference Patel, Nankova and LaGamma2005) or as a potent histone deacetylase inhibitor via histone acetylation (Boffa et al. Reference Boffa, Lupton, Mariani, Ceppi, Newmark, Scalmati and Lipkin1992). Since butyrate is oxidized by the colonocytes its metabolites may play a role in exerting genetic effects, but the role of its metabolites is still unexplained.
In the study presented here, there was also a pronounced variation of expression levels, e.g. of the GSTM isoforms between different donors, which we were not able to explain by diagnosis, gender or determined GST polymorphisms.
Moreover, we found a reduction of GST activity after 12 h treatment, and the lowest values were observed in cells treated with the faeces blank and SFS. This reduction may reflect the decrease of GSTM2 and GSTM5 on transcriptional level. The induction of GSTA isoforms which have only a moderate affinity for 1-chloro-2,4-dinitrobenzene (Eaton & Bammler, Reference Eaton and Bammler1999), and which are not abundant in the colon cells, would probably not be detected by the GST activity measurement. In contrast, butyrate tended to induce GST gene expression, but this could also not be confirmed by the measurement of GST activity. The reasons for this may be that one of the most induced GST forms, namely GSTT2, does not use 1-chloro-2,4-dinitrobenzene as a substrate (Eaton & Bammler, Reference Eaton and Bammler1999). Another reason may be that the 12 h incubation was too short to detect changes in protein expression levels.
Other in vitro studies in HT29 cells also did not show that a complex fermentation sample (produced from different dietary fibre sources) induced GST activity which was explained by too low butyrate concentrations and by the presence of other inhibitory substances in the faeces (Beyer-Sehlmeyer et al. Reference Beyer-Sehlmeyer, Glei, Hartmann, Hughes, Persin, Bohm, Rowland, Schubert, Jahreis and Pool-Zobel2003). GST activity, however, was enhanced in HT29 human colon cells after 72 h treatment with the major fermentation product butyrate (Ebert et al. Reference Ebert, Beyer-Sehlmeyer, Liegibel, Kautenburger, Becker and Pool-Zobel2001). This supports the conclusion that the butyrate concentration in the fermentation supernatants is responsible for effects in cellular systems although other compounds like propionate could additionally contribute to selected activities (Beyer-Sehlmeyer et al. Reference Beyer-Sehlmeyer, Glei, Hartmann, Hughes, Persin, Bohm, Rowland, Schubert, Jahreis and Pool-Zobel2003).
All in all, the present experiments substantiate the hypothesis that butyrate and complex fermentation supernatants may result in favourable effects in non-transformed colon cells. Ongoing studies will lead to a better understanding on how inulin-type fructans and the resulting fermentation supernatants can affect primary colon cells by modulating gene expression and how these changes are connected with a protection from colon cancer development.
Conclusions
The present studies have shown that fermentation products derived from inulin enriched with oligofructose favourably modulated profiles of genes related to xenobiotic metabolism in primary human colon cells. The cells retained more of their metabolic activity than cells not treated with the sample which implies the presence of compounds with nutrient functions. The present studies support the decisive role of butyrate in terms of modulating gene expression (Pool-Zobel et al. Reference Pool-Zobel, Selvaraju, Sauer, Kautenburger, Kiefer, Richter, Soom and Wolfl2005a) in primary healthy human colon cells, but the butyrate concentrations were possibly only suboptimal in the fermentation supernatant as investigated here. The faeces samples also had effects on metabolic activity and gene expression with high interindividual variability. It will be an important goal in the future to find the underlying factors responsible for the variation. The present study provides insight into how fermentation products of dietary fibre, particularly butyrate, can affect non-transformed primary human colon cells. It allows the conclusion that the fermentation products of inulin might have chemopreventive activities in non-transformed human colon cells.
Acknowledgements
We acknowledge Prof. Jahreis, Department of Nutritional Physiology, Friedrich-Schiller-University Jena for analytical assistance. We thank all donors of colon tissue samples for giving their informed consent and supporting our studies. This work was carried out with the financial support of the German Research Association (Deutsche Forschungsgemein-schaft, PO 284/8-1 and 8-2) and of PRECANTOO (Orafti, Tienen, Belgium).
Note
Supplementary information accompanies this paper on the Journal's website (http://www.nutritionsociety.org).
Supplementary Table 1 Summary of those genes related to drug metabolizing enzymes which were up- or down-regulated in primary colon cells treated with the test samples in comparison to the medium control. The cells were treated for 12 h with the fermentation supernatant derived from Synergy and human faeces. Synthetic mixtures of SCFA (composed to mimic the complex SFS), medium or faeces controls were investigated in parallel. Only those genes which reached an expression level with a signal over the cut off level (≥15) in one of the treatments were evaluated (except the GSTs, where all genes were considered). Fold changes ≥2·0 and ≤0·5 were considered in the evaluation as were those genes which are statistically significantly different from the medium, as are marked with symbols (*=two way ANOVA, Bonferroni's post-test; §= unpaired t test; #= fold change calculation).







