Introduction
CVD is one of the leading causes of mortality worldwide. High blood pressure (BP) or hypertension, defined as a BP of >140/90 mmHg(1), is a major risk factor for CVD, with subjects with uncontrolled hypertension being at three times greater risk of developing CVD compared with normotensives(Reference Wang, Lee and Fabsitz2). A reduction in diastolic BP (DBP) of 5–6 mmHg over a 5-year period is associated with a 38 and 23 % reduction in risk from stroke and CHD, respectively(Reference Collins, Peto and MacMahon3). There is no single cause of hypertension, but age, obesity, diet, physical activity and genetic factors are thought to play a key role.
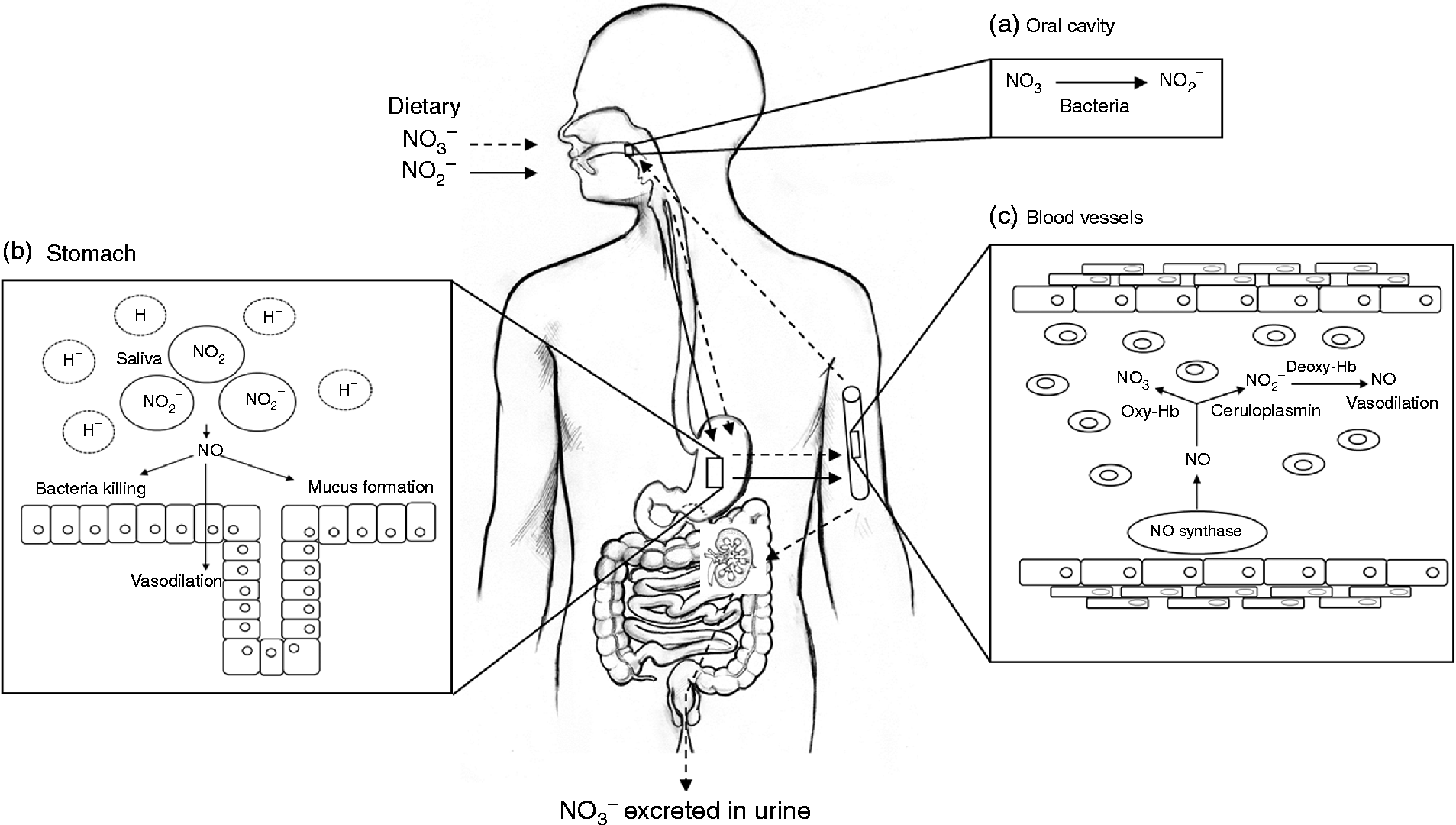
NO is a simple diatomic gas and free radical that is synthesised endogenously by a family of enzymes, namely the NO synthases (NOS)(Reference Schulz, Kelm and Heusch4, Reference Singh and Evans5). The physiological generation of NO has an important role in maintaining vascular homeostasis, and reduced NO production or bioavailability is associated with a number of cardiovascular and metabolic diseases(Reference Cannon6). Evidence has accumulated in recent years to suggest that NO may not be solely generated by NOS, but via a mechanism that relies on the bioconversion of nitrate to nitrite, by oral bacteria and further NO in the vasculature(Reference Lundberg, Weitzberg and Cole7). The nitrate used in this pathway originates from the oxidation of NOS-derived NO and the diet, particularly vegetables (Fig. 1).

Fig. 1 Schematic diagram of nitrate metabolism from vegetables via the nitrate–nitrite–nitric oxide (NO) pathway. A proportion of ingested nitrate (NO3−, - - -▸) is converted directly to nitrite (NO2−, → ) by facultative anaerobic bacteria, that reside on the dorsum of the tongue, during mastication in the mouth (a); the remainder is swallowed and is rapidly absorbed from the upper gastrointestinal tract. Approximately 25 % is taken up from the circulation and concentrated in the salivary glands and re-secreted into the mouth, where it is reduced to nitrite (adapted from Lundberg et al. (Reference Lundberg, Weitzberg and Gladwin10)). Some of the salivary nitrite enters the acidic environment of the stomach once swallowed (b), where NO is produced non-enzymically from nitrite after formation of nitrous acid (HNO2) and then NO and other nitrogen oxides. The NO generated kills pathogenic bacteria and stimulates mucosal blood flow and mucus generation. The remaining nitrite is absorbed into the circulation; in blood vessels (c) nitrite forms vasodilatory NO after a reaction with deoxygenated Hb (deoxy-Hb). Approximately 60 % of ingested nitrate is excreted in urine within 48 h. Oxy-Hb, oxygenated Hb.
The aim of the present review was to consider the current body of evidence for potential beneficial effects of dietary nitrate on BP and endothelial function, with emphasis on evidence from acute and chronic human intervention studies. Furthermore, mechanisms for dietary nitrate-mediated effects on BP and endothelial function have been explored, using evidence from animal and experimental studies.
Sources of dietary nitrate
Exposure estimates from national dietary surveys show average daily nitrate intakes in the USA and Europe to be 0·5–3·0 and 0·6–1·6 mmol/d, respectively(Reference Gangolli, van den Brandt and Feron8, Reference Mensinga, Speijers and Meulenbelt9). The vegetarian diet has been shown to contain approximately 4·3 mmol nitrate per d, almost four times greater than the average diet(Reference Lundberg, Weitzberg and Gladwin10). Vegetables are the main source of nitrate, contributing around 85 % of daily nitrate intake(Reference Gangolli, van den Brandt and Feron8, Reference White11, Reference Knight, Forman and Al-Dabbagh12). The remaining nitrate comes from drinking water, although this varies considerably since in many countries the levels are stringently regulated(Reference Knight, Forman and Al-Dabbagh12), and other foods such as cured meats, which contain nitrate or nitrite as a preservative to prevent the development of botulinum toxin(Reference Binkerd and Kolari13). In vegetables, nitrate is taken up from the soil and is transported via the xylem to the leaf where it accumulates. This explains the variation in the nitrate content of vegetables: leaf>stem>root(Reference Santamaria, Elia and Serio14, Reference Meah, Harrison and Davies15). Nitrate is important for plant nutrition and function and is the main limiting factor for growth(Reference Chen, Wang and Li16, Reference Krouk, Crawford and Coruzzi17).
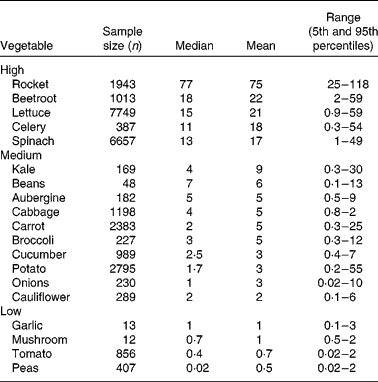
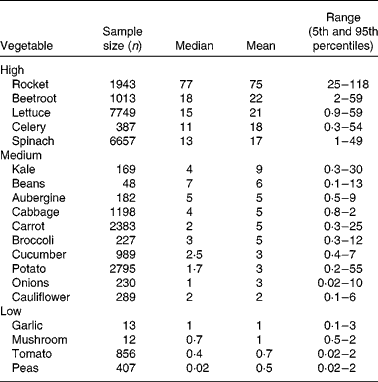
Vegetables can be grouped according to their nitrate content (see Table 1). High-nitrate-accumulating vegetables (>16 mmol/kg) belong to families of Brassicaceae (rocket), Chenopodiaceae (beetroot, spinach), Asteraceae (lettuce) and Apiaceae (celery)(Reference Santamaria, Elia and Serio14, Reference Santamaria18–20). Vegetables such as carrots, cabbage, onions and potatoes are characterised as medium-nitrate-accumulating vegetables (2–16 mmol/kg) and tomatoes, mushrooms and peas as low-nitrate-accumulating vegetables ( < 2 mmol/kg). There are several factors that affect nitrate uptake and accumulation in vegetables, for example genetic, environmental and agricultural factors(Reference Santamaria18). The main environmental factors are humidity, water content, temperature and sunlight. Vegetables grown during summer have reduced nitrate contents compared with vegetables grown during winter. This is because during winter the rate of photosynthesis is reduced, which results in increased accumulation of nitrate in cellular fluids(Reference Ysart, Miller and Barrett21). Therefore, the nitrate content of vegetables grown under low light conditions is often higher than those grown in the presence of increased light. The main agricultural factors are N fertilisation(Reference Santamaria, Elia and Parente22), degree of N fixation of atmospheric N2(Reference Bhattacharjee, Singh and Mukhopadhyay23) and nitrate reductase activity in the root(Reference Pate24), which varies markedly within vegetable species, cultivars and genotypes(Reference Blom-Zandstra25).
Acceptable daily intake
The acceptable daily intake (ADI) set by the European Food Safety Authority for nitrate is 3·7 mg/kg (0·06 mmol/kg). This equates to 260 mg/d for a 70 kg adult (4·2 mmol). The WHO first set an upper limit for nitrate in food in 1962, which was based on studies showing that daily doses of 500 mg nitrate per kg body weight had no adverse effects on rats and dogs. This value was divided by 100 to obtain an ADI for humans of 5 mg sodium nitrate or 3·7 mg nitrate per kg body weight(Reference Katan26). However, food choices within a dietary pattern such as the Dietary Approaches to Stop Hypertension (DASH) diet(Reference Svetkey, Sacks and Obarzanek27) can yield differences from 2·8 to 19·7 mmol nitrate, exceeding the ADI by 550 % for a 60 kg adult(Reference Hord, Tang and Bryan28). Therefore, the variability in nitrate content between vegetables and within species has a large influence on meeting the ADI for nitrate.
Metabolism of dietary nitrate
The metabolism of dietary nitrate is shown in Fig. 1. In humans the majority of ingested nitrate is readily absorbed in the upper gastrointestinal tract(Reference Wagner, Schultz and Deen29) into the circulation where it mixes with endogenously generated nitrate, which is present as a result of the oxidation of NO. Plasma nitrate levels peak 1·5–1·8 h after the ingestion of nitrate-rich foods and remain elevated for up to 5–6 h postprandially(Reference Van Velzen, Sips and Schothorst30). Although, the majority (65–70 %) of ingested nitrate is excreted in urine via the kidneys, approximately 25 % of the nitrate is actively reabsorbed and concentrated by a factor of ten from plasma into the saliva, and re-secreted into the upper intestinal tract(Reference Duncan, Dougall and Johnston31–Reference Tannenbaum, Fett and Young33). In the oral cavity salivary nitrate is reduced to nitrite by facultative anaerobic bacteria, mainly Actinomyces, Rothia and Staphylococcus epidermis that reside on the dorsal surface of the tongue and use nitrate as an alternative electron accepter to oxygen during respiration(Reference Duncan, Dougall and Johnston31, Reference Doel, Benjamin and Hector34). When the nitrite-rich saliva is swallowed and reaches the acidic environment of the stomach, nitrite is reduced to nitrous acid, which spontaneously decomposes to NO and other bioactive nitrogen oxides(Reference Lundberg, Weitzberg and Lundberg35). Furthermore, additional reactions occur in the presence of reducing compounds such as ascorbic acid and polyphenolic compounds, which cause rapid decomposition, ultimately forming NO(Reference Gago, Lundberg and Barbosa36–Reference Rocha, Gago and Barbosa38). In the gastric milieu nitrate–nitrite-derived NO increases host defence(Reference Lundberg, Weitzberg and Lundberg35, Reference Benjamin, O'Driscoll and Dougall39), blood flow and mucus production(Reference Björne, Petersson and Phillipson40, Reference Petersson, Carlström and Schreiber41). Some of the nitrite formed following the reduction of nitrate in the oral cavity is absorbed in the stomach and enters the systemic circulation(Reference Lundberg, Weitzberg and Cole7), where plasma concentrations peak after approximately 3 h. In the circulation and tissues such as the heart, nitrite is metabolised to NO by deoxyhaemoglobin(Reference Cosby, Partovi and Crawford42), deoxymyoglobin(Reference Shiva, Huang and Grubina43), cytoglobin(Reference Petersen, Dewilde and Fago44), neuroglobin(Reference Tiso, Tejero and Basu45), xanthine oxidoreductase(Reference Webb, Milsom and Rathod46), aldehyde oxidase(Reference Li, Cui and Kundu47), endothelial NOS (eNOS)(Reference Gautier, van Faassen and Mikula48–Reference Vanin, Bevers and Slama-Schwok50) and cytochrome P450(Reference Li, Liu and Cui51). The reduction of nitrite to NO in the circulation has been shown to have a number of beneficial effects on the cardiovascular system, which will be detailed below.
Endothelial function
The function of the vascular endothelium is essential for the maintenance of vascular homeostasis. It provides a cellular layer to all blood vessels in the circulatory system, and forms a structural barrier between circulating blood and the smooth muscle cells(Reference Furchgott52, Reference Rubanyi53). This monolayer of cells is essential in the regulation of vascular tone, which is maintained by the synthesis of vasoconstrictors (thromboxane A2, PGH2 and endothelin 1) and vasodilators (NO, endothelium-derived hyperpolarising factor and prostacyclin), released in response to local physical stimuli(Reference Moncada, Palmer and Higgs54, Reference Naseem55). The endothelium is also involved in the inhibition of smooth muscle cell proliferation, inflammation, vascular permeability, leucocyte adhesion, platelet aggregation and maintenance of the balance between prothrombotic and profibrinolytic activity. Indeed, many of the functions of the endothelium are mediated by NO(Reference Hoak, Czervionke and Fry56).
Role of nitric oxide
The principal role of NO is in the maintenance of vascular homeostasis. Endothelium-derived NO diffuses into underlying vascular smooth muscle cells where the classical NO–cyclic guanylate monophosphate (cGMP)–protein kinase G signalling pathway causes vascular relaxation(Reference Palmer, Ferrige and Moncada57). NO also plays a critical role in the inhibition of thrombosis by inhibiting platelet adhesion, activation and agonist-induced secretion, and has been shown to promote platelet disintegration and prevent the binding of fibrinogen(Reference Loscalzo58). This is thought to be partly through a cGMP-mediated mechanism(Reference Loscalzo58). However, endothelium-derived NO has effects beyond that of the vasculature; NO inhibits leucocyte adhesion and inhibits injury-induced intimal proliferation by inhibiting the proliferation and migration of smooth muscle cells. Conversely, when the bioavailability of NO is compromised, these beneficial effects are lost and endothelial dysfunction predominates(Reference Münzel, Daiber and Ullrich59).
Clinical assessment of endothelial function
There are a number of ways in which endothelial function can be assessed in vivo (Reference Donald, Charakida and Cole60). In the coronary circulation endothelial function can be measured by quantitative angiography, which examines change in diameter in response to intracoronary infusions of acetylcholine. In healthy blood vessels acetylcholine increases the production of NO and hence induces endothelium-dependent vasodilation(Reference Ludmer, Selwyn and Shook61). However, in individuals with endothelial dysfunction this process is blunted(Reference Ludmer, Selwyn and Shook61). Due to the invasive nature, expense and inaccessibility of the methodology described above a number of non-invasive measurements of endothelial function have been developed. For example, flow-mediated dilatation (FMD) measures the diameter of the brachial artery by non-invasive ultrasound before and after increasing shear stress by reactive hyperaemia, with the degree of dilatation reflecting arterial endothelial NO release(Reference Celermajer, Sorensen and Gooch62). FMD has been shown to correlate with measures of coronary endothelial function(Reference Anderson, Uehata and Gerhard63). This technique has been widely employed; however, there are questions regarding reproducibility and repeatability, since it is very technically demanding and specific training is needed(Reference Anderson, Uehata and Gerhard63). Furthermore, changes in forearm blood flow can be measured by strain-gauge venous occlusion plethysmography. The principle behind this technique is to detect the changes in forearm blood flow in response to intra-arterial administration of agonists that stimulate NO production(Reference Wilkinson and Webb64). This technique had been widely used in acute intervention studies when a number of repeated measurements are needed(Reference Wilkinson and Webb64). The major limitations of this method are reproducibility and its invasive nature. However, finger plethysmography employs a similar principle to venous occlusion plethysmography, but non-invasively measures changes in finger blood flow before and after reactive hyperaemia. Another technique is laser Doppler imaging with iontophoresis, which measures the endothelial function of the peripheral microcirculation. The degree of endothelial dysfunction occurring in the microcirculation has been shown to be proportional to that occurring in the coronary arteries(Reference Stehouwer65). This technique measures the response of cutaneous blood vessels to transdermal delivery of two contrasting vasoactive agents: acetylcholine (endothelium-dependent vasodilator) and sodium nitroprusside (endothelium-independent vasodilator) by iontophoresis, and has the advantage of being less technically demanding compared with FMD. Thus, a reduced local vasodilatory response to acetylcholine is associated with endothelial dysfunction(Reference Ramsay, Ferrell and Greer66). A number of studies have used pulse wave analysis and/or pulse wave velocity as a measure of endothelial function(Reference Hayward, Kraidly and Webb67, Reference Wilkinson, Hall and MacCallum68). Pulse wave analysis detects waveform traces of the peripheral waveform from the radial artery and generates the corresponding aortic waveform from which the augmentation index, which is a composite measure of global stiffness, can be calculated. Pulse wave velocity is a direct measure of arterial stiffness and is calculated from sequential recordings of pressure waveforms from the carotid and femoral arteries using applanation tonometry. Arterial stiffness has been shown to relate to FMD of the brachial artery(Reference Nigam, Mitchell and Lambert69), suggesting a role of arterial stiffness in the measurement of endothelial function. Although endothelial function and arterial stiffness represent different aspects of CVD, it is likely that the pathophysiological processes are interlinked.
Endothelial dysfunction
Endothelial dysfunction is broadly characterised as an imbalance between the release of vasoconstrictors and vasodilators, which is initiated by damage or impairment of the endothelial cell layer(Reference Drexler and Hornig70). A number of studies have shown endothelial dysfunction to be highly related to atherosclerosis and the development of CVD(Reference Anderson, Uehata and Gerhard63, Reference Schächinger, Britten and Zeiher71). Indeed, endothelial dysfunction, as defined by decreased production or activity of NO, may be one of the earliest signs of atherosclerosis and is associated with the development of a number of cardiovascular disorders such as hypertension, coronary artery disease, chronic heart failure and peripheral artery disease(Reference Cannon6). Therefore, increasing and/or improving NO bioavailability in the vasculature are likely to have considerable implications for the development of cardiovascular disorders. There is increasing evidence to suggest that dietary nitrate may improve NO bioavailability, increase vasodilation, inhibit platelet aggregation and thus improve cardiovascular health(Reference Lidder and Webb72). Research into the emerging area of dietary nitrate and endothelial function will be the focus of the remainder of the review.
Dietary nitrate and blood pressure
A number of epidemiological studies suggest that increased consumption of vegetables is associated with a reduced risk of CVD(Reference Bazzano, He and Ogden73–Reference Ness and Powles75). Indeed, the greatest protection against CHD and stroke was associated with the consumption of green leafy vegetables(Reference Joshipura, Ascherio and Manson76, Reference Joshipura, Hu and Manson77). Green leafy vegetables are amongst the highest nitrate-accumulating vegetables and have recently been postulated to be responsible for the cardioprotective effects observed from dietary patterns such as the Mediterranean and traditional Japanese(Reference Raat, Noguchi and Liu78, Reference Sobko, Marcus and Govoni79). In addition, evidence from prospective controlled intervention studies further strengthens this association(Reference Appel, Moore and Obarzanek80).
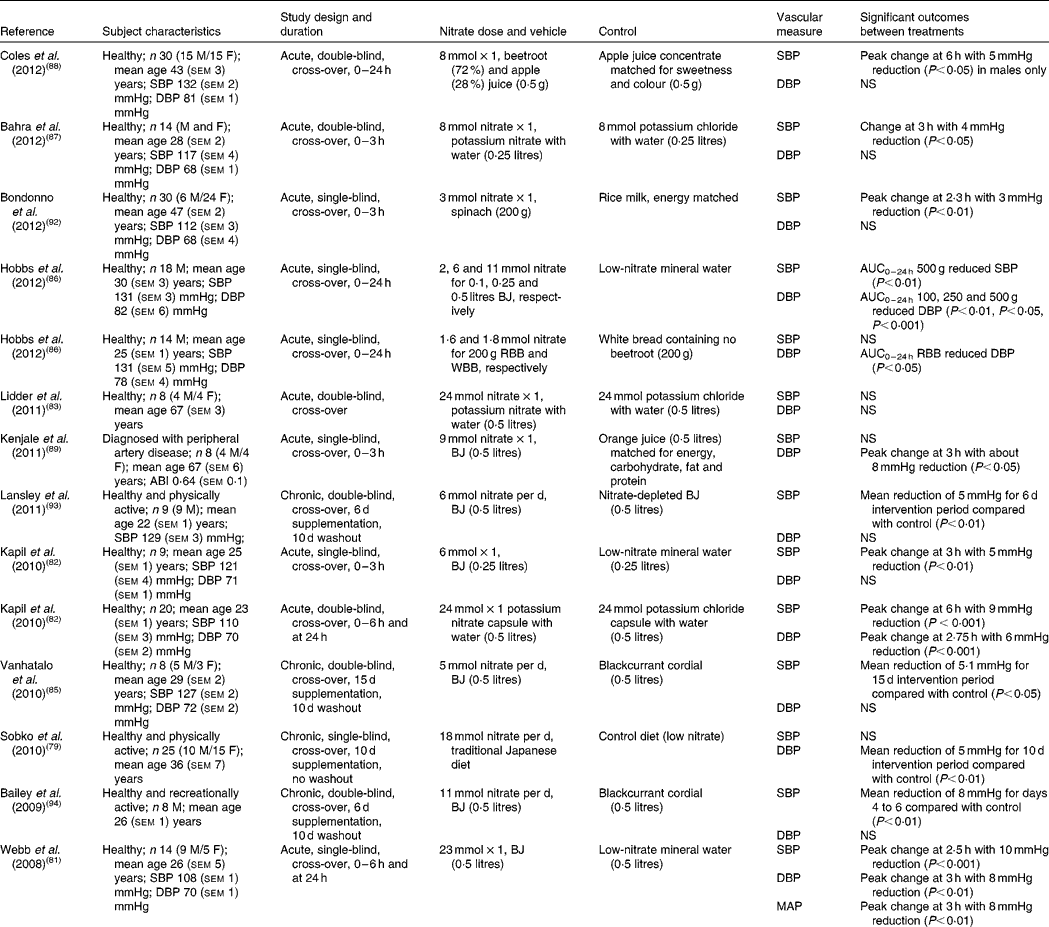
A number of human intervention studies have been conducted to assess the effects of dietary nitrate ingestion on BP either by the use of inorganic salts such as potassium nitrate or sodium nitrate or with dietary sources of nitrate such as beetroot juice (Table 2). In 2008, Webb et al. (Reference Webb, Patel and Loukogeorgakis81) showed that ingestion of a single dose of beetroot juice (500 ml; 23 mmol nitrate) by fourteen healthy subjects reduced systolic BP (SBP) and DBP by 10 and 8 mmHg, respectively. The peak effect in BP occurred at 3 h after ingestion and in association with an increase in plasma nitrite concentration, supporting the hypothesis that the delayed appearance of nitrite is due to the sequential bioconversion of nitrate to nitrite and further to NO in vivo. In a later study, the same group showed a 5 mmHg reduction in SBP in nine healthy subjects, using a lower dose of beetroot juice (250 ml; 5 mmol nitrate)(Reference Kapil, Milsom and Okorie82). Furthermore, these effects coincided with an increase in plasma cGMP, which lends itself to the possibility of soluble guanylyl cyclase (sGC)–cGMP-mediated vasodilation. In contrast to the studies detailed above, Lidder et al. (Reference Lidder, Hunt and Omar83) did not find a significant reduction in BP with potassium nitrate (24 mmol nitrate) compared with the equivalent potassium chloride as control. However, a 75 g glucose tolerance test to examine the potential effect of nitrate on glucose handling was performed in conjunction with the BP measurements. It may be that ingestion of a large dose of glucose caused vasoconstriction of the blood vessels to an extent that was not reversed by the provision of nitrate–nitrite-derived NO, thus affecting the BP response. This hypothesis is supported by a study by Zhang et al. (Reference Zhang, Chu and Yu84), which found that rats assigned to glucose of 15, 25 or 35 g/kg per d, orally, for 3 months showed glucose-induced hypertension. The authors postulated that the reason for this was due to the inhibition of the transcription of both 11β-hydroxysteroid dehydrogenase 2 (11β-HSD2) and aldosterone synthase (CYP11B2) genes in the vasculature by glucose, leading to lower aldosterone and higher corticosterone production in the blood vessels. This resulted in increased vasoconstrictor responses to noradrenaline, a well-known vasopressor medication used to increase BP in patients with critically low BP(Reference Zhang, Chu and Yu84). Although the study was performed in rats and was carried out over a 3-month period, it suggests a detrimental impact of glucose on BP control.
Table 2 Acute and chronic randomised placebo-controlled trials investigating the effects of dietary nitrate on blood pressure
M, male; F, female; SBP, systolic blood pressure; DBP, diastolic blood pressure; BJ, beetroot juice; RBB, red beetroot bread; WBB, white beetroot bread; ABI, ratio of BP in the lower legs to BP in the arms; MAP, mean arterial pressure.
The majority of human studies to date have investigated the effects of dietary nitrate on BP in healthy young subjects(Reference Kapil, Milsom and Okorie82, Reference Vanhatalo, Bailey and Blackwell85–Reference Coles and Clifton88), but similar effects have also been reported in older subjects diagnosed with peripheral artery disease(Reference Kenjale, Ham and Stabler89) and hypertensives(Reference Ghosh, Kapil and Fuentes-Calvo90). In the study by Kenjale et al. (Reference Kenjale, Ham and Stabler89) the consumption of beetroot juice (500 ml; 9 mmol nitrate) resulted in a DBP reduction of 8 mmHg, an increase in the onset of claudication pain and prolonged peak walking time(Reference Kenjale, Ham and Stabler89). Taken together, this suggests that dietary nitrate may have a role in the regulation of BP, even when the endothelial-derived NO synthesis is compromised, such as in the case of peripheral artery disease. In line with this, Ghosh et al. (Reference Ghosh, Kapil and Fuentes-Calvo90) investigated the effects of elevation of circulating nitrite levels in fifteen drug-naive grade 1 hypertensives using 3·5 mmol nitrate. The elevation of plasma nitrite levels by approximately 1·5-fold coincided with a substantial reduction in SBP by 12 mmHg(Reference Ghosh, Kapil and Fuentes-Calvo90). The BP-lowering effects observed in this study are comparable with those achieved by a single antihypertensive drug. These findings therefore show a promising role for dietary nitrate in the management of hypertension.
When evaluating the studies investigating the acute effects of dietary nitrate from beetroot juice or inorganic supplements on BP a significant inverse relationship between dose of nitrate and reduction in SBP (R 2 − 0·45; P= 0·033; Fig. 2(a)) was observed, although this did not reach significance for DBP (R 2 0·233; P= 0·27; Fig. 2(b)). The study by Hobbs et al. (Reference Hobbs, Kaffa and George86) demonstrated that the ingestion of three doses of beetroot juice (100, 250 and 500 g containing 2, 6 and 11 mmol nitrate, respectively) dose-dependently reduced SBP over a period of 24 h, with dose-dependent effects being less apparent for DBP. In addition, lower doses of dietary nitrate (100 g, 2 mmol) were shown to reduce DBP(Reference Hobbs, Kaffa and George86), suggesting that even relatively small doses, that are easily achievable in the diet, have BP-lowering effects.

Fig. 2 Acute dose-dependent effects of dietary nitrate from beetroot juice and inorganic salts on peak change in systolic (a) and diastolic (b) blood pressure (BP) in healthy normotensive subjects. Data have been extracted from studies by Hobbs et al. (Reference Hobbs, Kaffa and George86), Kapil et al. (Reference Kapil, Milsom and Okorie82), Bahra et al. (Reference Bahra, Kapil and Pearl87), Coles et al. (Reference Coles and Clifton88) and Webb et al. (Reference Webb, Patel and Loukogeorgakis81). For systolic BP, R 2 0·45 (P= 0·033); for diastolic BP, R 2 0·27 (P= 0·233).
In addition, foods that are rich in dietary nitrate are often also a good source of other components that may have additive or synergistic effects on BP. Betalains, a group of N-containing colour compounds responsible for the red/purple colour of beetroot, have been shown to affect the body's redox balance, decrease damage to lipids and increase antioxidant status compared with vitamin C in healthy subjects after a 2-week supplementation with 250 g cactus pear fruit containing 10·5 mg betalains per 100 g fruit pulp or 75 g vitamin C, suggesting a role for betalains in age-related degenerative diseases(Reference Tesoriere, Butera and Pintaudi91). In order to test the potential effect of betalains on BP response to dietary nitrate, Hobbs et al. (Reference Hobbs, Kaffa and George86) assessed the effects of white beetroot (betacyanin-deficient variety) or red beetroot-enriched bread on BP in fourteen healthy subjects. The results of the study showed that there were no significant differences in BP reduction between bread enriched with 100 g red or white beetroot (both 2 mmol nitrate)(Reference Hobbs, Kaffa and George86), suggesting that dietary nitrate present in the beetroot was responsible for the BP-lowering effects, whereas betalains had minimal effect. Other sources of nitrate-rich vegetables have been investigated and their potential interaction with flavonoid-rich foods studies. In the study by Bondonno et al. (Reference Bondonno, Yang and Croft92) the combined effects of nitrate-rich spinach and epicatechin-rich apples on BP and FMD in thirty healthy subjects were investigated. The study showed a significant reduction in SBP with 200 g spinach containing 3 mmol nitrate, but not when spinach and apple interventions were given simultaneously(Reference Bondonno, Yang and Croft92). There were no effects of any of the interventions on DBP. The authors suggest that a possible explanation for the reduced, but still significant, effects is that administering the flavonoid-rich apples and nitrate-rich spinach simultaneously resulted in augmentation of NO production from nitrite in the acidic environment of the stomach with less nitrite being available for absorption into the circulation(Reference Bondonno, Yang and Croft92). The observed interaction and less-than-additive effects of the combination intervention on plasma nitrite concentrations are consistent with this suggestion.
In addition to the aforementioned acute studies, a few studies have investigated the long-term effects of dietary nitrate on BP, mainly in healthy young subjects(Reference Sobko, Marcus and Govoni79, Reference Vanhatalo, Bailey and Blackwell85, Reference Lansley, Winyard and Fulford93–Reference Larsen, Ekblom and Sahlin95). In 2006, Larsen et al. (Reference Larsen, Ekblom and Sahlin95) showed an increase in plasma nitrite and DBP reduction of 4 mmHg after 3 d of supplementation with sodium nitrate (0·1 mmol/kg per d) in seventeen physically active subjects. Bailey et al. (Reference Bailey, Winyard and Vanhatalo94) used beetroot juice (500 ml; 11 mmol nitrate) daily for 6 d in recreationally active male subjects and reported a reduction in SBP of 8 mmHg. In addition, Sobko et al. (Reference Sobko, Marcus and Govoni79) found mean significant reductions in DBP of 5 mmHg in twenty-five healthy and physically active subjects with a traditional Japanese diet (18 mmol nitrate) compared with a control diet for 10 d, which were hypothesised to be due to the dose of nitrate rather than due to other components in the traditional Japanese diet.
In summary, human intervention studies to date suggest that dietary nitrate acutely lowers BP in healthy humans. Further long-term studies are needed to establish if the BP-lowering effects of dietary nitrate can be replicated, particularly in subjects with hypertension.
Dietary nitrate and endothelial function
There are a limited number of human intervention studies investigating the effects of dietary nitrate on endothelial function(Reference Webb, Patel and Loukogeorgakis81, Reference Bahra, Kapil and Pearl87, Reference Bondonno, Yang and Croft92, Reference Heiss, Meyer and Totzeck96, Reference Gilchrist, Winyard and Aizawa97) (Table 3). Webb et al. (Reference Webb, Patel and Loukogeorgakis81) showed that beetroot juice (500 ml; 23 mmol nitrate) completely reduced endothelial dysfunction induced by ischaemic–reperfusion injury to the forearm and preserved the FMD response, whereas the FMD response was reduced by 60 % in the control subjects. However, Bahra et al. (Reference Bahra, Kapil and Pearl87) did not find a significant effect of 8 mmol potassium nitrate on FMD, but reported a significant reduction (0·3 m/s) in pulse wave velocity and SBP (4 mmHg) at 3 h compared with the potassium chloride control in fourteen healthy subjects. This suggests that although inorganic nitrate is not able to alter endothelial function, it does appear to increase blood flow in combination with reductions in BP.
Table 3 Randomised placebo-controlled trials investigating the effects of dietary nitrate on endothelial function

M, male; F, female; SBP, systolic blood pressure; DBP, diastolic blood pressure; BJ, beetroot juice; FMD, flow-mediated dilatation; LDI, laser Doppler imaging; aPWV, aortic pulse wave velocity.
Overall, whilst the current studies provide some promising evidence on the beneficial effects of dietary nitrate on endothelial function, there is need for replication of the positive findings using a wider range of in vivo measures to assess endothelial function.
Potential mechanisms by which dietary nitrate may reduce blood pressure and increase endothelial function
The cardioprotective effects of nitrate interventions may be due to the promotion of vasodilation and inhibition of vasoconstriction, inhibition of platelet aggregation and the production of free radicals. In most of the studies examined, peak reduction in BP compared with baseline occurred at 2–3 h after the consumption of dietary nitrate, which is the same time at which peak plasma nitrite concentrations have been observed(Reference Webb, Patel and Loukogeorgakis81, Reference Kapil, Milsom and Okorie82, Reference Hobbs, Kaffa and George86, Reference Bahra, Kapil and Pearl87, Reference Kenjale, Ham and Stabler89, Reference Bondonno, Yang and Croft92), suggesting that dietary nitrate and its sequential reduction to nitrite and NO in the circulation are responsible for the cardioprotective effects observed.
Acting as a substrate to endothelial nitric oxide synthase
Under normal conditions NO is synthesised by eNOS from the amino acid l-arginine in the presence of oxygen. Therefore, when the oxygen supply becomes compromised such as in myocardial ischaemia, generation of NO by the endothelium is reduced. However, studies suggest that nitrate and nitrite may act as a substrate for eNOS-mediated NO generation, even when oxygen levels are limited. For example, Gautier et al. (Reference Gautier, van Faassen and Mikula48) showed that eNOS could reduce nitrite to NO under total oxygen depletion at a physiological pH using recombinant eNOS holoenzyme and its oxygenase domain. Compared with the conventional and oxygen-dependent l-arginine pathway, the nitrite reduction produced up to 6-fold more NO(Reference Gautier, van Faassen and Mikula48). In addition, immortalised murine endothelial cells were used by Vanin et al. (Reference Vanin, Bevers and Slama-Schwok50) to show that a significant amount of NO was produced under total oxygen depletion conditions and confirmed that the NO released was from nitrite reduction by eNOS. Cao et al. (Reference Cao, Bell and Mohanty98) demonstrated that under hypoxic conditions nitrite promotes ATP synthesis and release by erythrocytes, which in turn stimulated eNOS to produce NO.
Inhibitor of mitochondrial reactive oxygen species
There is speculation on whether the effects of dietary nitrate and nitrite are due to their ability to inhibit mitochondrial production of reactive oxygen species (ROS), since oxidative stress and the associated pro-inflammatory response are atherogenic and attributed to reduced endothelial function. ROS, in particular superoxide anions, rapidly react with NO to form peroxynitrite, which, among other effects, can oxidise LDL or up-regulate pro-inflammatory cytokines and adhesion molecules. This ultimately results in reduced NO bioavailability and impaired endothelial function. As well as being the major source of cellular energy, mitochondria are also a source of ROS overproduction in a number of cardiovascular diseases(Reference Ballinger99). Nitrite has been shown to inhibit mitochondrial ROS over production by S-nitrosation of mitochondrial respiratory chain complex I enzyme(Reference Shiva, Huang and Grubina43, Reference Shiva and Gladwin100). Furthermore, the inhibition of ROS by nitrite may also lead to the protection of NO generated by the classical l-arginine pathway, because ROS can oxidise tetrahydrobiopterin (BH4), (essential cofactor for eNOS) into dihydrobiopterin, resulting in reduced NO production(Reference Landmesser, Dikalov and Price101). This is also supported by Stokes et al. (Reference Stokes, Dugas and Tang102), who found that mice that were fed a high-cholesterol diet and were supplemented with sodium nitrite (50 mg/l) in drinking water exhibited an increase in BH4 levels compared with control. Therefore, reduced mitochondrial ROS production and increased bioavailability of NO by nitrite may help to prevent vascular dysfunction.
Increasing vasodilation
The mechanisms underlying the vasodilatory effects of dietary nitrate and nitrite remain unclear. Laustiola et al. (Reference Laustiola, Vuorinen and Pörsti103) showed that sodium nitrite administration resulted in vascular smooth muscle relaxation in pre-contracted rat mesenteric arteries via cGMP production, an effect that was amplified in the presence of exogenous guanosine triphosphate. It is likely that the effects of dietary nitrate and nitrite on smooth muscle relaxation are due to their reduction to NO and therefore the activation of sGC in smooth muscle cells. Indeed, earlier studies by Arnold et al. (Reference Arnold, Mittal and Katsuki104) and Gruetter et al. (Reference Gruetter, Kadowitz and Ignarro105) showed that nitrite concentrations above physiological levels activated sGC and vasodilated vascular smooth muscle preparations, thus suggesting a role of nitrite in vasodilation.
Inhibition of platelet aggregation
During acute damage to the endothelium, platelets are activated by contact with exposed collagen and aggregate together at the wound site to initiate clotting and stop bleeding(Reference Loscalzo58). The principal role of platelets is to form a physical plug to seal a haemorrhaging vessel(Reference Loscalzo58). However, milder injury to the endothelium, perhaps as a result of prolonged raised BP, raised plasma cholesterol, or smoking, also causes platelets to adhere to the internal walls of arteries(Reference Loscalzo58). The rapid adhesion and activation of platelets initiates an inflammatory response of the vessel wall, which predisposes the endothelium to vascular complications such as thrombosis, premature heart disease, myocardial infarction or stroke, and diabetes. Therefore, it is essential that during normal vascular homeostasis platelet activation is tightly controlled. Indeed, both platelets and endothelial cells produce and secrete chemicals that directly inhibit platelet aggregation, for example NO.
A few studies have found inhibitory effects of dietary nitrate on platelet aggregation, suggesting that inhibition of abnormal platelet aggregation may be partly responsible for the cardiovascular benefits of nitrate supplementation. For example, Richardson et al. (Reference Richardson, Hicks and O'Byrne106) demonstrated that a single 2 mmol bolus of potassium nitrate inhibited platelet aggregation by up to 31 % in healthy individuals. In a study by Webb et al. (Reference Webb, Patel and Loukogeorgakis81) supplementation of healthy individuals with beetroot juice (550 ml; 23 mmol nitrate) also had an inhibitory effect on ex vivo platelet aggregation in response to collagen and adenosine diphosphate. The exact mechanism by which dietary nitrate attenuates platelet aggregation is still unknown, but it is likely that NO formed by the sequential reduction of nitrate activates sGC by binding to the haem moiety of the enzyme(Reference Craven and DeRubertis107). This results in increased cGMP concentrations in platelets, and this is thought to be the mechanism by which platelet function is inhibited(Reference Moro, Russel and Cellek108, Reference Radomski, Palmer and Moncada109).
Conclusion
There is a modest but convincing body of evidence from acute and chronic human intervention studies that show BP-lowering effects of dietary nitrate, with limited but promising evidence for potential beneficial effects on endothelial function. In addition, animal studies have provided insight into the potential physiological and molecular mechanisms of action. Preliminary studies suggest that dietary nitrate, at doses as low as 2 mmol, may have a role in increasing peripheral vasodilation and therefore lowering BP, appearing to be dependent on dose. In vitro studies show that the beneficial effects of dietary and inorganic nitrate on BP and vascular reactivity may be mediated by a pathway that is independent of the endothelium and relies initially on the reduction of nitrate to nitrite by oral bacteria and further to NO in blood vessels. There are currently a few studies underway examining the long-term effects of dietary nitrate on BP and endothelial function (clinicaltrials.gov: no. NCT01405898, controlled-trials.com: no. ISRCTN03012467, clinicaltrials.gov: no. NCT01681810 and controlled-trials.com: no. ISRCTN25003627). However, further long-term and well-designed human intervention studies are required, particularly in individuals at risk of CVD and in those with hypertension.
Acknowledgements
This research received no specific grant from any funding agency in the public, commercial or non-profit sectors.
D. A. H. conducted the review of the literature and drafted the manuscript. T. W. G. and J. A. L. contributed to the drafting of the manuscript. All authors made a critical review of the draft.
There are no conflicts of interest.