



The aetiology of CHD is multifactorial and involves genetics, embryology, and environmental exposures. Reference Fahed, Gelb and Seidman1 Some forms of CHD are known to have a specific genetic cause Reference Zaidi and Brueckner2,Reference Wang, Li and Chen3 and most, if not all, patients with moderate-to-severe CHD will undergo some form of genetic testing. As genetic technologies have evolved, so has the detection of genetic variants associated with CHD. Karyotyping was one of the first genetic testing modalities used clinically, it revealed chromosomal aberrations in 8–13% of neonates with CHD. Reference Aksglaede, Jensen, Carlsen, Pham, Neil and Patel4 This era was followed by combined testing with karyotyping and fluorescence in situ hybridisation analysis. The combination was able to diagnose aneuploidies and more subtle genetic abnormalities such as deletions and duplications. Reference Bachman, De Ward, Chrysostomou, Munoz and Madan-Khetarpal5 Our previous institutional review identified chromosomal abnormalities in 17.8% of neonates with structural heart disease by karyotyping and fluorescence in situ hybridisation analysis. Reference Dykes, Al-Mousily and Abuchaibe6 Chromosomal microarray analysis is a valuable clinical tool for the identification of submicroscopic chromosomal aberrations that cannot be detected by conventional cytogenetic methods. Reference Blue, Kirk and Giannoulatou7
In this study, we sought to compare the diagnostic yield of two different eras of genetic testing at our institution: chromosomal microarray, conducted between 2012 and 2017 and karyotyping and fluorescence in situ hybridisation analysis, conducted between 2004 and 2010 as previously reported. Reference Dykes, Al-Mousily and Abuchaibe6 In both eras, patients with CHD routinely underwent genetic testing upon admission to the CICU, with neonates admitted between 2004 and 2010 undergoing karyotyping and fluorescence in situ hybridisation analysis and neonates admitted between 2012 and 2017 undergoing chromosomal microarray testing.
Materials and methods
The Institutional Review Board at Nicklaus Children’s Hospital approved this retrospective comparative study. Karyotyping and fluorescence in situ hybridisation analysis data on 482 neonates less than 30 days of age with CHD admitted to the cardiac intensive care unit at Nicklaus Children’s Hospital between 2004 and 2010 were reported in our previous study. Reference Dykes, Al-Mousily and Abuchaibe6
Inclusion criteria for chromosomal microarray analysis were all patients less than 30 days of age with CHD admitted to the CICU between 2012 and 2017. Demographic data including gestational age, birth weight, gender, and race were collected. The presence of dysmorphic features was confirmed by physical examination by consulting board-certified geneticists. Cardiac lesions were evaluated and diagnosed by transthoracic echocardiography. Genetic testing via chromosomal microarray is currently standard of care for all neonates admitted with CHD to the CICU at Nicklaus Children’s Hospital.
Chromosomal microarray testing was performed using the Affymetrix CytoScan HD Array, which features 2.6 million genetic markers including ∼750,000 single-nucleotide polymorphisms and 1.9 million non-polymorphic probes for the detection of copy number variations, loss of heterozygosity, uniparental isodisomy, regions indicated by descent, and measurements of low-level mosaicism and heterogeneity.
All patients with genetic results not reported as “normal chromosomal microarray” were categorised as an anomalous chromosomal microarray result. Anomalous chromosomal microarray results included aneuploidies, deletions, duplications, loss of heterozygosity, and variants of uncertain significance. Chromosomal microarray results were also subdivided into four subcategories: abnormal results with a known association with CHD; abnormal results with no known association with CHD; variants of uncertain significance; and benign familial variants. Abnormal results with known associations with CHD consisted of abnormal genetic findings with well-known associations with cardiac lesions such as deletions involving TBX1, GATA4, CREBBP, ELN. 8 Abnormal results with no known association with CHD consisted of abnormal, but well-described genetic variants without documented associations with CHD. Variants of uncertain significance represent a chromosomal anomaly (i.e. deletion or duplication) for which there are insufficient data, thus, the impact on health and disease risk is uncertain. Reference Blue, Kirk and Giannoulatou7 Benign familial variants are a chromosomal anomaly with no association with a specific condition. Reference Richards, Aziz, Bale, Bick, Das and Gastier-Foster9
Descriptive data are presented as mean ± standard deviation for normally distributed continuous data or median (25–75% interquartile range) otherwise and as n (%) for categorical data. Chi-square tests were used to compare differences in proportions between the categorical outcome groups, and the Mann–Whitney U-test or Student’s t-test were used to test for differences between groups for continuous variable, depending on the distribution. All statistical tests were two-tailed and a p-value <0.05 was considered significant. Analyses were performed with SPSS version 23.0 (IBM Statistics, Chicago, IL, USA).
Results
Eight-hundred and forty patients underwent genetic testing in 2 different eras of genetic testing at our institution; there were no statistically significant differences in gestational age or gender in patients tested between 2012 and 2017 and between 2004 and 2010 (Table 1).
Table 1. Comparison of demographics and cardiac lesions between the karyotype and FISH testing group versus the CMA group and between normal and abnormal CMA results

Values expressed as n (%).
CMA = chromosomal microarray; FISH = fluorescence in situ hybridisation analysis.
Three-hundred and sixty-seven neonates admitted to the CICU at Nicklaus Children’s Hospital between 2012 and 2017 met the inclusion criteria (Table 2). 135/367 (36.7%)patients had anomalous chromosomal microarray analysis results. Of the anomalous results, deletions were the most common finding, present in 52/135 (38.5%), followed by gains/duplications (41/135; 30.4%), aneuploidies (17/135; 13%), loss of heterozygosity (14/135; 10%), multiple anomalous results (9/135; 6.7%), and XX/XY mosaicism (Table 2). According to our sub-classification, abnormal results with a known genetic association with CHD were present in 43/135 (31.8%) of the abnormal results; abnormal results with no known association with CHD comprised 29/135 (21.5%) of abnormal results; variants of uncertain significance were present in 61/135 (45.2%); and benign familial variants were present in 2/135 (1.5%) patients (Table 2).
Table 2. Breakdown of abnormal chromosomal microarrays and their associations with CHD

Values expressed as n (%).
Of all patients with abnormal chromosomal microarray analysis results, 76/135 (56.3%) of patients had genetic abnormalities known to be associated with risk for neurodevelopmental disorders. In 46/76 (60.5%) of patients with neurodevelopmental disorders, the genetic abnormality would not have been identified by karyotyping and fluorescence in situ hybridisation analysis testing alone. Karyotyping and fluorescence in situ hybridisation analysis can identify aneuploidies and 22q11 deletions, so patients with trisomy 13, 18, and 21 and 22q11 deletion syndrome were excluded from the analysis of neurodevelopmental disorders. Forty-six genetic abnormalities were detected by chromosomal microarray analysis that is known to be associated with neurodevelopmental disorders such as autosomal-dominant intellectual disability, Jacobsen syndrome, and various seizure disorders. A complete list of the identified genetic abnormalities and their associated neurodevelopmental disorders and cardiac lesions are shown in Table 3.
Table 3. All genetic defects exclusively identified by chromosomal microarray associated with neurodevelopmental abnormalities
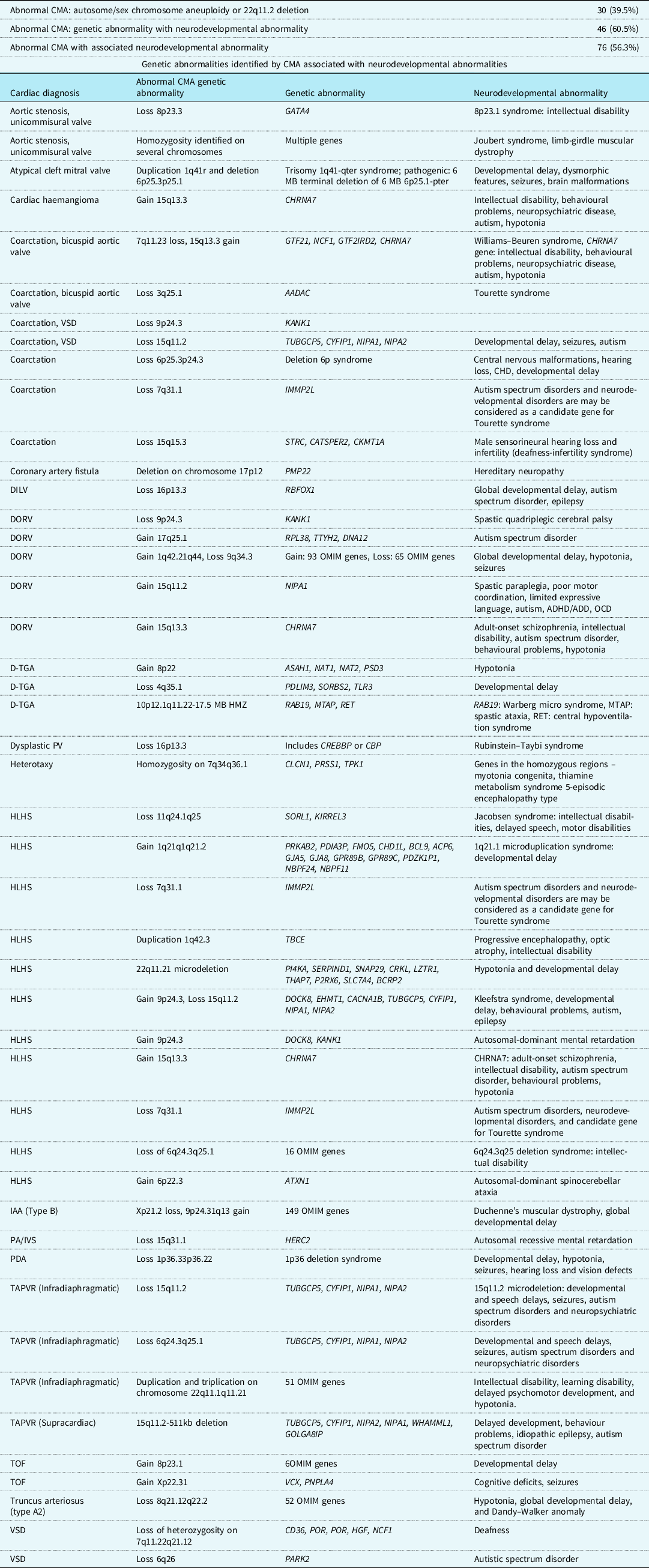
DILV = double inlet left ventricle; DORV = double outlet right ventricle; DORV w/HLV = DORV with hypoplastic left ventricle; d-TGA = d-transposition of the great arteries; PV = pulmonary valve; HLHS = hypoplastic left heart syndrome; IAA = interrupted aortic arch; PA/IVS = pulmonary atresia with intact ventricular septum; PDA = patent ductus arteriosus; TAPVR = totally anomalous pulmonary venous return; TOF = tetralogy of Fallot; VSD = ventricular septal defect.
Dysmorphic features were defined as physical examination findings that were deemed abnormal rather than a variant of normal. Dysmorphic features were identified by physical examination and documented by the same group of geneticists in the two eras of testing. In the patients that underwent chromosomal microarray analysis testing, dysmorphic features were documented in 84/367 (22.9%) patients. The most commonly described dysmorphic features were malformations of the ears, mouth, eyes, and limbs. Of the patients with dysmorphic features, 50/84 (59.5%) had abnormal chromosomal microarray analysis results (p < 0.00001) (Table 1). In our previous study, dysmorphic features were present in (68/86) 79.1% of patients with abnormal genetic results. Reference Dykes, Al-Mousily and Abuchaibe6 Of note, our previous study classified sacral dimple, cleft, and/or tuft of hair as dysmorphic features, whereas the current study did not.
Multiple cardiac lesions were identified both with and without genetic associations. Comparing our results to our previous study using karyotyping and fluorescence in situ hybridisation analysis, Reference Dykes, Al-Mousily and Abuchaibe6 chromosomal microarray analysis detected a higher number of genetic abnormalities (36.7% of patients) than karyotyping or fluorescence in situ hybridisation analysis (17.8% of patients) (Table 4). Conotruncal abnormalities were the most common cardiac diagnosis in our cohort and comprised 41/135 (30.4%) of patients with abnormal chromosomal microarray analysis results. Surprisingly, 7/54 (13.0%) patients with d-transposition of the great arteries were found to have abnormal chromosomal microarray analysis results, while karyotyping and fluorescence in situ hybridisation analysis detected no genetic abnormalities in this population.
Table 4. Cardiac lesions and their frequency of genetic abnormalities detected by different testing modalities

R=anomalous pulmonary venous return; ASD=atrial septal defect; AVC=atrioventricular canal; DILV=double inlet left ventricle; DORV=double outlet right ventricle; DORV w/HLV=DORV with hypoplastic left ventricle; d-TGA=d-transposition of the great arteries; HLHS=hypoplastic left heart syndrome; PA/IVS=pulmonary atresia with intact ventricular septum; l-TGA=l-transposition of the great arteries; TOF=tetralogy of Fallot; VSD=ventricular septal defect.
Discussion
With rapid advances in genomic technologies, various forms of genetic testing are now offered to CHD patients and their families. However, there is little consensus on the optimal test, the specific clinical indication for testing, the interpretation of test results, and their prognostic yield. At our institution, since 2004, every neonate admitted with CHD has undergone some form of genetic testing. Between 2004 and 2010, all neonates admitted to the cardiac ICU underwent genetic testing by karyotyping or karyotyping and fluorescence in situ hybridisation analysis. When we reported these data in 2016, 86/482 (17.8%) of patients had a chromosomal abnormality. Reference Dykes, Al-Mousily and Abuchaibe6 In 2010, the American College of Medical Genetics issued practice guidelines for chromosomal microarray analysis and recommended chromosomal microarray analysis as first-tier testing for postnatal patients with multiple congenital anomalies. Reference Miller, Adam, Aradhya, Biesecker, Brothman and Carter10 In 2012, our institution began to regularly use chromosomal microarray analysis as the screening genetic test in CHD patients. In our cohort, 135/367 (37%) patients had genetic abnormalities detected by chromosomal microarray analysis. As chromosomal microarray analysis offers higher resolution compared to karyotyping and/or fluorescence in situ hybridisation analysis, an increased detection rate of abnormalities is consistent with the current literature. Reference Bachman, De Ward, Chrysostomou, Munoz and Madan-Khetarpal5,Reference Miller, Adam, Aradhya, Biesecker, Brothman and Carter10–Reference Wu, Li, Fu, Pan, Han and Yang12
Of interest, 76/135 (56.3%) of patients with abnormal chromosomal microarray analysis results had chromosomal abnormality associated with the risk of neurodevelopmental disorders. Of these 76 patients, 30 had one of the following: trisomy 18, 21, or 22, sex chromosomal aneuploidy, or 22q11.2 deletion. All of which can be detected by karyotyping and fluorescence in situ hybridisation analysis. So, 46/76 (60.5%) of patients with risk of neurodevelopmental disorders had genetic abnormalities that were only detectable by chromosomal microarray analysis. Recognising neurodevelopmental problems in patients with CHD is of paramount importance in the neonatal period, since these disorders affect 10% of all patients with CHD and 50% of patients with severe heart disease. Reference Marino, Lipkin and Newberger15 Traditionally, consideration of risk factors for CHD-associated neurodevelopmental delay has focused on the role of cardiopulmonary bypass, the byproduct of abnormal physiology, and post-operative complications. Reference Marelli, Miller, Marino, Jefferson and Newburger16 It is likely that these factors contribute to some degree of neurodevelopmental delay in our patients, together with genetic abnormalities. Neurodevelopmental risk stratification remains difficult in the neonatal CHD population, but readily identifying patients at high risk due to genetic abnormalities allows early intervention with multidisciplinary care and more accurate pre-surgical prognostic counselling for families. Several studies have shown the benefit of early intervention in these patients and its effect on neurodevelopmental outcomes, such as executive function in at-risk children. Reference Diamond, Barnett and Thomas17
Dysmorphic features were identified and documented by the same group of board-certified geneticists in both studies. In our current cohort, 84/367 patients had dysmorphic features, 50/84 (59.5%) of whom had an abnormal chromosomal microarray analysis result. Two-hundred and eighty-three patients did not exhibit any dysmorphic features, and 85 (30.0%) of these had an abnormal chromosomal microarray analysis result. Of all patients with an abnormal chromosomal microarray analysis result, dysmorphic features were noted in 50/135 (37%) patients. Of note, dysmorphic features were also noted in 34/235 (15%) of patients with a normal chromosomal microarray analysis. Our previous study detected dysmorphic features in 79% of patients with abnormal cytogenetic results and in 25% of patients with normal cytogenetic results, Reference Dykes, Al-Mousily and Abuchaibe6 higher than in the current study, which may be due to the inclusion of sacral dimple and cleft as a dysmorphic feature in the previous study. Most dysmorphic patients lacked a specific history or physical examination features to suggest a specific genetic disorder. Reference Miller, Adam, Aradhya, Biesecker, Brothman and Carter10 In some institutions, genetic testing is only considered if the patient exhibits dysmorphic features. Reference Chaix14 However, our data indicate that nearly half of patients with CHD and genetic anomalies do not have any dysmorphic features on physical examination. Thus, using dysmorphic features to risk stratify candidates with CHD for genetic testing may overlook a significant number of patients with genetic abnormalities.
Conotruncal abnormalities were the most common diagnosis in both our previous study and the current chromosomal microarray analysis study. In our previous study, conotruncal defects were present in 33.2% (160/482) of patients and accounted for 30.2% (26/86) of all abnormal genetic results. In the current study, conotruncal defects were present in 36.8% (135/367) of patients and comprised 30.4% (41/135) of patients with abnormal chromosomal microarray analysis results. Of patients with d-transposition of the great arteries, 7/54 (12.9%) had abnormal chromosomal microarray analysis results, while no genetic abnormalities were detected in patients with d-transposition of the great arteries by karyotyping and fluorescence in situ hybridisation analysis. Reference Dykes, Al-Mousily and Abuchaibe6 D-transposition of the great arteries is not generally associated with genetic anomalies and has been reported to have a very low recurrence risk, Reference Digilio, Casey, Toscano, Calabrò, Pacileo and Marasini13 so our result is surprising. Of our patients with d-transposition of the great arteries and abnormal genetic results, one patient had an abnormal result with known association with CHD, two patients had an abnormal result with no known association with CHD, and four patients had variants of uncertain significance. The patient with an abnormal result with known association with CHD had a gain of 622 kb at 8p22, a region containing four Online Mendelian Inheritance in Man genes: ASAH1, NAT1, NAT2, and PSD3. Duplications of this region have been associated with coarctation of the aorta and generalised neonatal hypotonia. 8 Of the two patients with abnormal results with no known association with CHD, one had a loss of 10p12.1q11.22 and the other had a 47, XYY karyotype. 8 Four patients had variants of uncertain significance: a gain of 246 kb at 15q15.3, loss of 336 kb at 13q21.31, loss of 252 kb at 22q21.1, and a 251 kb loss at 7q34. 8 Despite the current literature reporting genetic abnormalities as rare in patients with d-transposition of the great arteries, 13% of patients with d-transposition of the great arteries and abnormal genetic test results. Of the seven patients with d-transposition of the great arteries and anomalous chromosomal microarray, one patient was at risk of developing a neurodevelopmental disorder.
As expected, we detected a much higher rate of genetic abnormalities by chromosomal microarray analysis compared to the previous era of testing with karyotyping and fluorescence in situ hybridisation analysis at our institution. Despite a large number of results being variants of uncertain significance, we detected several clinically impactful genetic abnormalities. With the current growth in cardiac genetics clinical research, variants of uncertain significance may yet become clinically significant. Reference Erdogan, Larsen, Zhang, Tümer, Tommerup and Chen18–Reference Warburton, Ronemus, Kline, Jobanputra, Williams and Anyane-Yeboa20 CHD patients who do not display dysmorphic features on clinical examination should undergo genetic evaluation and testing, as 15% of our patients with abnormal chromosomal microarray analysis results had no dysmorphic features identifiable by board-certified geneticists. We identified 60% more patients with potential neurodevelopmental abnormalities by chromosomal microarray analysis compared to karyotyping and fluorescence in situ hybridisation analysis. The ability to recognise patients who are of innately high neurological risk allows us to provide appropriate family counselling, anticipatory guidance, and early interventions. Chromosomal microarray analysis is a valuable routine tool to supplement the lifelong care of patients with CHD.
Genetic testing continues to rapidly advance, as next-generation sequencing of multiple targets or whole-genome sequencing becomes readily available and affordable. Sequencing tests detect variants within single genes and chromosomal microarray detects copy number variants. While a few studies have reported targeted use of next-generation sequencing of multiple genes and whole-genome sequencing with high diagnostic yield in CHD, Reference Hu, Qiao and Wang22,Reference Gillian, Edwin and Giannoulatou23 further studies are required for routine clinical application in paediatric CHD.
Limitations of this study include the retrospective study design, the comparison of different genetic testing modalities in two different groups rather than the same group, and a lack of long-term follow-up in patients with an identified risk of neurodevelopmental disorders. Nevertheless, despite the two groups examined undergoing different genetic testing modalities, there were no statistically significant differences between the two populations in terms of gender, gestational age, and the majority of cardiac lesion subgroups (Table 1). Birth weight could not be compared, as raw data were not available from the previous study. There was a significant difference in race, possibly attributable to the introduction of electronic medical records in 2012, as race was exclusively documented for each patient in the paper records but the more recent electronic medical records contain two fields for race or ethnicity, with ethnicity often the only documentation and race then auto-populated as “other”. The strengths of this study include the large sample size of patients who underwent two distinct genetic testing modalities with available results and clinical correlation.
Acknowledgements
None.
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guidelines on human experimentation (please name) and with the Helsinki Declaration of 1975, as revised in 2008, and has been approved by Institutional Review Board at Nicklaus Children’s Hospital.





 Open access
Open access