


Liu et al. (2010) published a gene-based test for genome-wide association studies called VEGAS (versatile gene-based association study). The approach provides a versatile framework to test genes/genomic regions for genetic association, only requiring summary statistics from single-variant analysis. The approach does not require genotype data. It accounts for linkage disequilibrium (LD) by using simulations from a multivariate normal distribution in which the correlation matrix is defined by the local LD structure. The test is implemented in the software package VEGAS, and the LD information is extracted from the HapMap reference panel (International HapMap 3 Consortium, 2010). Recently, Mishra and MacGregor (Reference Mishra and MacGregor2015) extended and improved VEGAS by incorporating the 1,000 Genomes data as the reference panel (1,000 Genomes Project Consortium, 2012) for the local LD structure (VEGAS2).
The new software package VEGAS2 builds on the VEGAS methodology. Both tools provide the option that only a specific percentage of the most promising single nucleotide polymorphisms (SNPs), that is, the SNPs with the strongest association test signals, are included in the gene-based test. In this case, the observed gene-based statistic in VEGAS is the sum of the squared single-variant association tests for the most promising SNPs. To calculate the empirical/permutation-based p value of this test statistic, in each replicate of the simulations, the sum of the top percentage of the simulated squared z scores is computed and compared to the observed statistic.
However, in the software implementation of VEGAS as well as VEGAS2, the top percentage of the signed z scores is determined and the sum is taken over the squared values. The corresponding R function in the code of VEGAS is
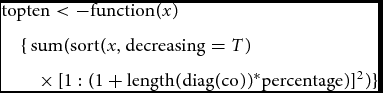 $$\begin{eqnarray}
&& \hspace*{-18pt} {\rm{topten}} < - {\rm{function}}(x) \nonumber\\
&& \hspace*{-18pt} \quad \{ \, {\rm{sum}}({\rm{sort}}(x,{\rm{decreasing}} = T) \nonumber\\
&& \hspace*{-18pt} \qquad \times\, {[1:(1 + {\rm{length}}({\rm{diag}}({\rm{co}}))^{*}{\rm{percentage}})]^2}) \}
\end{eqnarray}$$
$$\begin{eqnarray}
&& \hspace*{-18pt} {\rm{topten}} < - {\rm{function}}(x) \nonumber\\
&& \hspace*{-18pt} \quad \{ \, {\rm{sum}}({\rm{sort}}(x,{\rm{decreasing}} = T) \nonumber\\
&& \hspace*{-18pt} \qquad \times\, {[1:(1 + {\rm{length}}({\rm{diag}}({\rm{co}}))^{*}{\rm{percentage}})]^2}) \}
\end{eqnarray}$$
In order to compute the correct test statistic in each replicate, the function should be defined as
 $$\begin{eqnarray}
&& \hspace*{-18pt} {\rm{topten}} < - {\rm{function}}(x) \nonumber \\
&& \hspace*{-18pt} \quad \{ \, {\rm{sum}}({\rm{sort}}({x^ \wedge }2,{\rm{decreasing}} = T) \nonumber\\
&& \hspace*{-18pt} \qquad \times\, [1:(1 + {\rm{length}}({\rm{diag}}({\rm{co}}))^{*}{\rm{percentage}})]) \}
\end{eqnarray}$$
$$\begin{eqnarray}
&& \hspace*{-18pt} {\rm{topten}} < - {\rm{function}}(x) \nonumber \\
&& \hspace*{-18pt} \quad \{ \, {\rm{sum}}({\rm{sort}}({x^ \wedge }2,{\rm{decreasing}} = T) \nonumber\\
&& \hspace*{-18pt} \qquad \times\, [1:(1 + {\rm{length}}({\rm{diag}}({\rm{co}}))^{*}{\rm{percentage}})]) \}
\end{eqnarray}$$
The current implementation (1) therefore provides incorrect p values for the test statistic, as replicates that consists of large z scores with different signs contribute only their largest, positive z scores to the computation of the empirical p value. The correct implementation (2) squares the z scores and then determines the top percentage of the squared statistics. The empirical p values provided by VEGAS are therefore often smaller/anti-conservative than the correct, permutation-based p values. The implementation of VEGAS2 also uses definition (1) instead of (2).
Since the decision to perform the top percentage test is very reasonable and it is frequently used in substantive papers, we strongly believe that a correction of this software bug is very valuable for applied research. This is especially true given the high number of citations for VEGAS/VEGAS2 in data analysis papers. On January 27, 2016, we informed Dr Stuart MacGregor about this mistake by email. Dr MacGregor replied that it was a bug indeed and assured us that his group would fix this mistake by the next day. However, to the best of our knowledge, there have not been any software updates of the VEGAS/VEGAS 2 packages since we sent the email to Dr MacGregor, which was about 1 year ago. We think that this is unfortunate since, as we outline below, our VEGAS2 results for the Bipolar Disorder Meta-Analysis of the PGC clearly show that the current implementation (1) of the VEGAS test statistic can lead to false-positive findings, as VEGAS provides p values that are substantially anti-conservative. In all our analyses, we used the most recent versions of VEGAS/VEGAS2 (12/15/2016).
To illustrate the impact of this software error, we performed a genome-wide, gene-based analysis using the results of the Bipolar Disorder Meta-Analysis of the Psychiatric GWAS Consortium Bipolar Disorder Working Group (2011). We performed the analysis with the offline version of VEGAS2, as this is the latest version. We used the whole gene list provided by VEGAS2, a flanking sequence of +/- 10kb and considered the top 10% SNP test. For all other parameters, the default option was used.
We performed the tests with the original offline version of VEGAS2 software and a modified version of VEGAS2 in which we corrected the software bug as described above. The analysis resulted in gene-based p values for 23,158 genes.
With the exception of 24 genes, the p value of the original VEGAS2 implementation (1) was smaller than the correct p value based on implementation (2). The average deflation factor between both implementations for the p value computations was ~1.80. For 218 genes, the factor was greater than 3 and the greatest observed factor was 69.93. Clearly, the factor is expected to be greater if the number of SNPs mapped to the specific gene increases. For CSMD1, the gene with the highest factor of 69.93, the number of mapped SNPs within the flanking sequence was 5,559. For an overall significance level of 5%, we assessed the number of significantly associated genes after Bonferroni correction for n = 23,158 genes. While 15 genes achieved genome-wide significance with incorrect implementation (1), only nine genes were genome-wide significant with the modified/correct implementation (2).
To emphasize the impact of this software bug on the validity of the test results, we analyzed the example input file, which is available for download on the VEGAS homepage with the web-based version of VEGAS and the latest offline version of VEGAS. We computed the top 9%, top 20%, and the best SNP test with the HapMap CEU (Utah residents with Northern and Western European ancestry from the CEPH collection) information. Using the example input file, VEGAS maps four variants to the RABL4 and 11 variants to the FOXRED2 gene. Given the number of variants in each gene, the top 20% procedure should correspond to the best SNP test for RABL4 and the top 9% test to the best SNP test for FOXRED2. In Table 1, we provide the empirical p values for our analyses. The results of the modified version, using (2) instead of (1), are in the last column. All results are based on 105 simulations, suggesting sufficient number of replicates in order to obtain stable estimate for the p values.
TABLE 1 Comparison of Gene-Based Test Association Results for RABL4 and FOXRED2 Using Different Implementations of VEGAS

The results of ‘VEGAS web-based top X%’ and ‘VEGAS offline top X%’ match very well, suggesting the same implementation in the web-based version and in the offline version. However, the results are substantially different from the results obtained by ‘VEGAS web-based best SNP’, although theoretically they should be identical. The VEGAS web-based best SNP results agree well with the results of VEGAS offline modified top X%. In conclusion, the results of Table 1 prove that the p values provided by VEGAS are too small, and that the software bug still exists in the web-based and offline version of VEGAS.
In this communication, we described why the software implementation of the popular gene-based test frameworks VEGAS and VEGAS2 for the top percentage option is incorrect. Since the top percentage test is frequently applied, we examined the impact of the incorrect computation of the empirical p value in the top 10% gene-based test statistic by application to the PGC meta-analysis results for bipolar disorder. As the p values were substantially deflated by the incorrect implementation, VEGAS2 provided false-positive results for six genes. Furthermore, we demonstrated inconsistencies between the best SNP and the top percentage test that also arise from the incorrect implementation. Given the consequences and/or implications of false-positive results, we strongly believe that user of VEGAS/VEGAS2 should replace (1) in their software copy by the implementation that we provided in (2) so that in the literature correct p values can be reported.
Acknowledgements
We would like to thank Nan M. Laird and Michael H. Cho for helpful discussions.
Drs MacGregor and Mishra comment
We thank Dr Lange and colleagues for reporting the bug in VEGAS. The bug was fixed on the VEGAS2 web-based version in January 2016 but unfortunately not until January 2017 on the VEGAS2 offline version. Users who ran the top % test (not the default option) using VEGAS2 prior to these dates (or using the now retired VEGAS1) should re-run their analysis using VEGAS2.
Brisbane, February 20, 2017.

