



Introduction
The family Allocreadiidae contains trematodes commonly found, as adults, in the intestine of freshwater fishes, and occasionally in snakes, salamanders and frogs (Caira and Bogéa, Reference Caira, Bogéa, Jones, Bray and Gibson2005). Adult allocreadiids typically possess an unspined tegument, a well-developed cirrus-sac, gonads in tandem and relatively large eggs. Allocreadiid metacercariae encyst in the haemocoel of aquatic arthropods; the cercariae are ophthalmoxiphidocercariae and develop in sphaeriid clams rather than in gastropods (Caira and Bogéa, Reference Caira, Bogéa, Jones, Bray and Gibson2005; Choudhury et al., Reference Choudhury, Rosas-Valdez, Johnson and Pérez-Ponce de León2007). Allocreadium is the type-genus and the most speciose among allocreadiids. The somewhat complex taxonomic history of the family was discussed in detail by Caira and Bogéa (Reference Caira, Bogéa, Jones, Bray and Gibson2005). These authors also revised the genus composition within the family and, after discussing the doubtful validity of 13 of them, concluded at the time that Allocreadiidae comprised 14 genera. The phylogenetic position of the family within the trematode order Plagiorchiida was first tested using molecular data by Curran et al. (Reference Curran, Tkach and Overstreet2006) and Choudhury et al. (Reference Choudhury, Rosas-Valdez, Johnson and Pérez-Ponce de León2007) using 28S rDNA sequences or a combined analysis of 28S and 18S rDNA, respectively. In both analyses, allocreadiids formed a highly supported monophyletic clade with species of Callodistomidae and Gorgoderidae, which, in turn appeared as sister taxa to a clade containing representative species of Encyclometridae, Brachycoeliidae, Dicrocoelidae and Orchipedidae.
Traditional approaches based on morphology have not been useful to assess the interrelationships among members of the family, although a significant progress has been made unravelling the species delimitation, evolutionary history, classification and historical biogeography of this group of trematodes by using DNA sequences. For instance, 2 genera validated by Caira and Bogéa (Reference Caira, Bogéa, Jones, Bray and Gibson2005), Paracreptotrematina and Polylekithum, were proved not to belong in Allocreadiidae (Curran et al., Reference Curran, Tkach and Overstreet2006, Reference Curran, Tkach and Overstreet2011; Choudhury et al., Reference Choudhury, Rosas-Valdez, Johnson and Pérez-Ponce de León2007); the genera Margotrema and Wallinia were validated as allocreadiids and not macroderoidids (Pérez-Ponce de León et al., Reference Pérez-Ponce de León, Choudhury, Rosas-Valdez and Mejía-Madrid2007); furthermore, 3 of the genera considered invalid by Caira and Bogéa (Reference Caira, Bogéa, Jones, Bray and Gibson2005) have been resurrected, i.e., Acrolichanus, Stephanophiala and Megalogonia (Atopkin et al., Reference Atopkin, Sokolov, Vainutis, Voropaeva, Shedko and Choudhury2020; Vainutis et al., Reference Vainutis, Voronova and Urabe2021); finally, 4 new genera have been described (Paracreptotrema, Paracreptotrematoides, Pseudoparacreptotrema and Mesoamericatrema) (Choudhury et al., Reference Choudhury, Pérez-Ponce de León, Brooks and Daverdin2006; Pérez-Ponce de León et al., Reference Pérez-Ponce de León, Pinacho-Pinacho, Mendoza-Garfias, Choudhury and García-Varela2016, Reference Pérez-Ponce de León, Sereno-Uribe, García-Varela, Mendoza-Garfias, Hernández-Mena, Pinacho-Pinacho and Choudhury2020; Mendoza-Garfias et al., Reference Mendoza-Garfias, García-Teh, Caspeta-Mandujano, Vidal-Martínez and Hernández-Mena2022). Our own account on the taxonomic composition of Allocreadiidae shows that the family currently contains 15 genera and approximately 130 species. This provides but a glimpse on the progress made on the classification scheme and species composition within the family Allocreadiidae after the revisionary work by Caira and Bogéa (Reference Caira, Bogéa, Jones, Bray and Gibson2005).
The application of molecular tools has provided useful information that expands our knowledge about the species diversity of the family and their interrelationships (e.g., Faltýnková et al., Reference Faltýnková, Pantoja, Skírnisson and Kudlai2020; Pérez-Ponce de León et al., Reference Pérez-Ponce de León, Sereno-Uribe, García-Varela, Mendoza-Garfias, Hernández-Mena, Pinacho-Pinacho and Choudhury2020; Franceschini et al., Reference Franceschini, Aguiar, Zago, de Oliveira, Yamada, Bertholdi and da Silva2021; Petkevičiūtė et al., Reference Petkevičiūtė, Stunžėnas and Stanevičiūtė2023; Sokolov et al., Reference Sokolov, Khasanov and Lebedeva2023; Vainutis et al., Reference Vainutis, Voronova, Urabe and Kazarin2023). Since the year 2000, c. 40 new species have been described following an integrative taxonomy approach, and molecular phylogenetic hypotheses have addressed aspects of the classification scheme of the group, providing more robust species delimitation criteria upon which biogeographical interpretations are made and nomenclatural changes proposed (see Atopkin et al., Reference Atopkin, Sokolov, Vainutis, Voropaeva, Shedko and Choudhury2020; Franceschini et al., Reference Franceschini, Aguiar, Zago, de Oliveira, Yamada, Bertholdi and da Silva2021; Vainutis et al., Reference Vainutis, Voronova and Urabe2021). In the same period, about 30 papers generated information to enhance the genetic library of ribosomal and mitochondrial genes for allocreadiid species, particularly the 28S rDNA and the cytochrome oxidase subunit 1, cox1.
Complete mitochondrial genomes (mt genomes) obtained through high throughput sequencing methods have been used in the last 20 years for reconstructing the phylogenetic history of trematodes and to test the higher-level classification scheme. The use of mt genomes is based on the premise that the number of sequenced base pairs, gene order and genome arrangement may provide evidence of shared ancestry (see Littlewood et al., Reference Littlewood, Lockyer, Webster, Johnston and Le2006), although their power to resolve deep levels of phylogenetic trees remains controversial (Pérez-Ponce de León and Hernández-Mena, Reference Pérez-Ponce de León and Hernández-Mena2019). Still, mt genomes have not been generated for species in the family Allocreadiidae to test their interrelationships with other trematodes. Thus, the main objective of this paper was to sequence the mt genome of 3 species of allocreadiids, namely Allocreadium lobatum, Creptotrematina aguirrepequenoi and Wallinia mexicana, parasites of freshwater fishes, to determine the gene content, arrangement, and composition, and to assess the phylogenetic relationships with other trematode species belonging in the order Plagiorchiida.
Materials and methods
Sample collection, morphological identification, and DNA extraction
Specimens of allocreadiids were sampled from the intestines of their host as follows: Specimens of C. aguirrepequenoi, W. mexicana, and A. lobatum were isolated from Astyanax mexicanus (Jalpan, Queretaro, Mexico), Astyanax aeneus (Matías Romero, Oaxaca, Mexico) and Luxilus cornutus/Semotilus atromaculatus (West Twin River, Wisconsin, USA), respectively. Some specimens were killed and relaxed with nearly boiling tap water and stored in vials with 96% ethanol for further processing (staining and mounting) and morphological identification. Trematodes were identified to species level following previous bibliographic accounts. Some other specimens were thoroughly washed in physiological saline solution and fixed in 100% ethanol for molecular analysis. To confirm the taxonomic identity, DNA was extracted from 1 specimen of each morphologically identified species and a fragment of the 28S ribosomal RNA gene was amplified as described in previous studies, using the primers 502 (5′ – CAAGTACCGTGAGGGAAAGTTGC- 3′) and 536 (5′ – CAGCTATCCTGAGGGAAAC-3′) (García-Varela and Nadler, Reference García-Varela and Nadler2005). PCR products were sequenced at the LANBIO-IBUNAM, Mexico City and a BLAST-N search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was performed.
For mt genome sequencing, genomic DNA was extracted from a pool of 5–9 individuals of each trematode species using QIAamp Fast DNA Tissue kit (Qiagen) according to the manufacturer's protocol. The dosage of the obtained DNA was determined in a microvolume spectrophotometer (NanoDrop_ND-1000).
High throughput sequencing and assembly
Extracted genomic DNA of each trematode species was shotgun sequenced on an Illumina HiSeq 4000 with a 100x depth, and 150-bp paired-end libraries were built with Nextera adaptors at Azenta GENEWIZ (NJ, USA). Bioinformatic analysis and de-novo genome assembly of the obtained reads were also conducted by Azenta. SOAPDeNovo2 (Luo et al., Reference Luo, Liu, Xie, Li, Huang, Yuan and Wang2012) was used on each of the samples with a minimum contig length of 1000 bp. EMBOSS tools GetOrf was then used to find the open reading frames (ORF) within the de novo assembled genome. The protein sequences from ORF were then annotated using Diamond BLASTp. To extract the mt genome, scaffolds were mapped onto a barcode sequence of the cox1 gene from each allocreadiid species in this study using the software Geneious Prime v.11 with the highest sensitivity and up to 5 iterations. This initial assembly was then used to seed iterative extensions until even and thorough coverage was obtained. The final assembly was annotated with MITOS Webserver (refseq = 63, translation table = 5) (Bernt et al., Reference Bernt, Donath, Jühling, Externbrink, Florentz, Fritzsch, Pütz, Middendorf and Stadler2013). The ORF of each protein gene was verified using Geneious, employing the invertebrate mitochondrial genetic code. Putative tRNA genes and their secondary structure were identified using ARWEN (Laslett and Canbäck, Reference Laslett and Canbäck2008) and MITOS. Circular mitochondrial genome maps were drawn using GenomeVx tool (Conant and Wolfe, Reference Conant and Wolfe2008).
Comparative analysis among allocreadiid mt genomes.
Contents of A + T and G + C were calculated in Geneious. AT-skew and GC-skew values were calculated using the equations AT-skew = (A- T)/(A + T) and GC-skew = (G- C)/(G + C) in each of the 12 protein-coding genes (PCGs) and the non-coding regions (NCRs). The PCG sequences were translated into their corresponding amino acid sequences using MEGA X (Kumar et al., Reference Kumar, Stecher, Li, Knyaz and Tamura2018) and the invertebrate mitochondrial genetic code. Amino acid composition and relative synonymous codon usage (RSCU) was also estimated in MEGA X. Mutation rate (non-synonymous/synonymous, dN/dS) ratios among the 12 PCGs of the 3 newly sequenced allocreadiid mt genomes were calculated using DnaSP v.6.12 (Rozas et al., Reference Rozas, Ferrer-Mata, Sánchez-Del Barrio, Guirao-Rico, Librado, Ramos-Onsins and Sánchez-Gracia2017). To estimate nucleotide diversity (π) across genes, a sliding window analysis was implemented in DnaSP using a window size of 300 bp and a step size of 30 bp, for the 12 PCGs plus the 2 rRNAs (rrnS and rrnL); values of nucleotide diversity were then plotted according to the midpoint position of each window. Nucleotide and amino acid differences were calculated using MEGA X
Phylogenetic analyses
The newly sequenced mt genomes were used along with those of other plagiorchiid trematodes downloaded from the GenBank database to reconstruct phylogenetic trees. Analyses included species allocated in the suborders Echinostomata, Opisthorchiata and Xiphidiata. Based on previous molecular phylogenetic analyses (Pérez-Ponce de León and Hernández-Mena, Reference Pérez-Ponce de León and Hernández-Mena2019), Fasciola hepatica (AF216697) was used as an outgroup. A concatenated matrix of the 12 PCGs was built in Geneious. Previously, each gene was independently aligned in ClustalW through the GenomeNet web interface (www.genome.jp). Phylogenetic relationships were inferred using 2 datasets: the first one with the nucleotide alignment of 12 PCGs plus the 2 ribosomal genes (rrnS and rrnL), and the second with the amino acid alignment. Phylogenetic trees were constructed using the maximum likelihood (ML) method in RaxML (Stamatakis, Reference Stamatakis2014). Empirical substitution models GTR + CAT and PROTGAMMA were employed for nucleotide and amino acid data sets, respectively; bootstrap support values for nodes were estimated with 1000 replicates.
Results and discussion
General features of allocreadiid mitogenomes
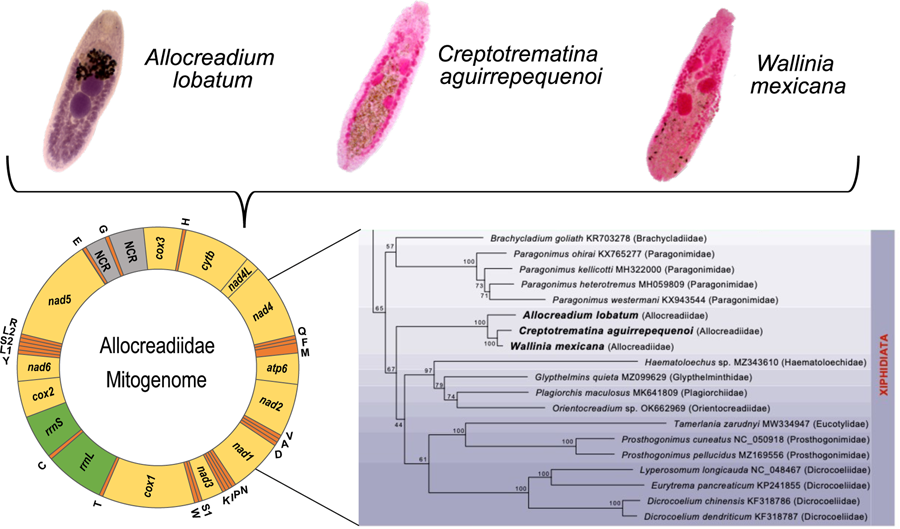
The complete circular mt genomes of A. lobatum, C. aguirrepequenoi and W. mexicana were 14,427, 13,798 and 13 926 pb long, respectively (GenBank accession numbers: OR987847, OR987848, OR987849, respectively). These mt genomes were composed of 12 PCGs (cox1 – 3, nad 1–6, cytb and atp6; atp8 gene is lacking), 2 rRNA genes (rrnS and rrnL), 22tRNAs (1 for each amino acid and 2 each for leucine and serine), and 2 non-coding regions (NCR) (Fig. 1 and Table 1). All genes were transcribed in the same direction. Total length and gene order of all 3 newly sequenced allocreadiid mt genomes corresponds to the typical arrangement observed in other species allocated in the order Plagiorchiida (Liu et al., Reference Liu, Yan, Otranto, Wang, Zhao, Jia and Zhu2014; Briscoe et al., Reference Briscoe, Bray, Brabec and Littlewood2016; Chang et al., Reference Chang, Liu, Gao, Zheng, Zhang, Duan, Yue, Fu, Su, Gao and Wang2016; Qian et al., Reference Qian, Zhou, Li, Wang, Miao and Hu2018; Le et al., Reference Le, Nguyen, Nguyen, Doan, Agatsuma and Blair2019; Suleman et al., Reference Suleman, Khan, Tkach, Muhammad, Zhang and Zhu2019, Reference Suleman, Tkach, Muhammad, Zhang, Zhu and Ma2020, Reference Suleman, Khan, Tkach, Ullah, Ehsan, Ma and Zhu2021; Guo et al., Reference Guo, Li, Gao, Qiu, Jin, Gao, Zhang, An, Chang, Gao and Wang2022; Gao et al., Reference Gao, Zhang, Wei, Jia, Zhang, Li, Chen, Sun, Hou, Liu, Wang, Zhang and Wang2023). The order of the 12PCGs seems to be highly conserved among trematodes, with the location of tRNAs as variable among species (Suleman et al., Reference Suleman, Khan, Tkach, Muhammad, Zhang and Zhu2019; Gao et al., Reference Gao, Zhang, Wei, Jia, Zhang, Li, Chen, Sun, Hou, Liu, Wang, Zhang and Wang2023).

Figure 1. Gen map of the 3 allocreadiid mitochondrial genomes, Allocreadium lobatum, Creptotrematina aguirrepequenoi, Wallinia mexicana. NCR, non-coding regions.
Table 1. Comparison of the mitochondrial genome arrangement of Allocreadium lobatum (Al), Creptotrematina aguirrepequenoi (Cp.) and Wallinia mexicana (Wa)

Similarity values are expressed in percentage. NN, nucleotides; AA, amino acids.
Intergenic spacers ranged from 1–34 bp in A. lobatum and C. aguirrepequenoi, and from 1–63 bp in W. mexicana. A 40 bp overlap between nad4L and nad4 was present in the 3 mt genomes. This is consistent with some species of xiphidiatan trematodes such as Paragonimus heterotremus, P. ohirai, Prosthogonimus spp., Tamerlania zarudnyi and Plagiorchis maculosus (Qian et al., Reference Qian, Zhou, Li, Wang, Miao and Hu2018; Le et al., Reference Le, Nguyen, Nguyen, Doan, Agatsuma and Blair2019; Suleman et al., Reference Suleman, Khan, Tkach, Muhammad, Zhang and Zhu2019, Reference Suleman, Khan, Tkach, Ullah, Ehsan, Ma and Zhu2021; Guo et al., Reference Guo, Li, Gao, Qiu, Jin, Gao, Zhang, An, Chang, Gao and Wang2022). However, other xiphidiatans (e.g., Dicrocoelium dendriticum, D. chinensis and Paragonimus westermani) show an overlap of just 1– 14 bp (Biswal et al., Reference Biswal, Chatterjee, Bhattacharya and Tandon2014; Liu et al., Reference Liu, Yan, Otranto, Wang, Zhao, Jia and Zhu2014).
The rrnL gene was located between trnT and trnC, while the rrnS gene was located between trnC and cox2 in the 3 allocreadiid species. The length of the 22 tRNA genes was 1403 bp in A. lobatum, 1404 bp in C. aguirrepequenoi, and 1,427 in W. mexicana, whereas individual tRNA gene lengths ranged from 54 to 72 bp (Table 1). The secondary structures predicted for the tRNA genes were similar among the 3 allocreadiids. Twenty of the tRNAs in C. aguirrepequenoi and W. mexicana, along with 18 tRNAs in A. lobatum can be folded into the conventional cloverleaf secondary structure. The D-arm is absent in the tRNA for Serine 1 (trnS1) in the 3 species, in the trnS2 only in A. lobatum, and in the tRNA for Cysteine in C. aguirrepequenoi and W. mexicana (Fig S1). The tRNAs for Proline (trnP) and Lysine (trnK) are lacking the T-arm in A. lobatum (Fig S1). The lack of D-arm in the Serine 1 tRNAs is consistent with previous reports on mt genomes of other trematodes (Chang et al., Reference Chang, Liu, Gao, Zheng, Zhang, Duan, Yue, Fu, Su, Gao and Wang2016; Le et al., Reference Le, Nguyen, Nguyen, Doan, Agatsuma and Blair2019; Suleman et al., Reference Suleman, Khan, Tkach, Ullah, Ehsan, Ma and Zhu2021; Guo et al., Reference Guo, Li, Gao, Qiu, Jin, Gao, Zhang, An, Chang, Gao and Wang2022). Likewise, tRNAs with non-canonical structures have been observed in different organisms including mammals, nematodes and arthropods, where either T-arm, D-arm or both could be missing (Krahn et al., Reference Krahn, Fischer and Söll2020).
The 3 species contain 2 NCRs between trnE and cox3, separated by trnG (Fig. 1). The shorter NCR was found between trnE and trnG and varies in size from almost 500 bp in A. lobatum to 167 bp in W. mexicana; the longer NCR was located between trnG and cox3 and ranged from 300 bp in C. aguirrepequenoi to 580 bp in A. lobatum. The pattern of 2 NCRs, one shorter than the other, has also been observed in other plagiorchiida including Lyperosomum longicauda, T. zarudnyi, Paragonimus ohirai; however, these are located between trnG and cox3, separated by trnE (Le et al., Reference Le, Nguyen, Nguyen, Doan, Agatsuma and Blair2019; Suleman et al., Reference Suleman, Tkach, Muhammad, Zhang, Zhu and Ma2020, Reference Suleman, Khan, Tkach, Ullah, Ehsan, Ma and Zhu2021). This arrangement differs in Prosthogonimus spp. and Plagiorchis maculosus since both species present only 1 long NCR (Suleman et al., Reference Suleman, Khan, Tkach, Muhammad, Zhang and Zhu2019; Guo et al., Reference Guo, Li, Gao, Qiu, Jin, Gao, Zhang, An, Chang, Gao and Wang2022), indicating that the length, arrangement and composition of the NCRs are not ubiquitous traits in Plagiorchiida mt genomes.
Comparative analysis of allocreadiid mitogenomes
Gene order of the mt genome was the same among the 3 species of allocreadiids (Fig. 1); likewise, the size of each gene was similar. The rank order of the 12 PCGs by length was: nad5 > cox1> nad4 > cytb > nad1 > nad2 > cox3 > cox2 > atp6 > nad6 > nad3 > nad4L (Table 1). Nucleotide composition of the 3 mt genomes was biased towards A and T, with and overall AT content of 63.5% in A. lobatum, 64.7% in C. aguirrepequenoi, and 65.1% in W. mexicana (Fig. 2), as observed in most trematodes (Gao et al., Reference Gao, Zhang, Wei, Jia, Zhang, Li, Chen, Sun, Hou, Liu, Wang, Zhang and Wang2023). A + T contents of cytb, nad4L, nad1, nad3, nad6 and cox1 were the lowest in A. lobatum, but the highest in W. mexicana; the opposite was true for cox3 and cox2, with the highest A + T content in A. lobatum. The genes atp6 and nad2 showed highest A + T content in C. aguirrepequenoi and the lowest values in W. mexicana (Fig. 2). The AT-skew values in the A. lobatum mitogenome ranged from −0.16 (cox2) to −0.50 (nad6), the GC-skew values ranged from 0.23 (cox1 and cox2) to 0.67 (nad3). In C. aguirrepequenoi the AT-skew values ranged from −0.25 (cox2) to −0.50 (nad2), the GC-skew values ranged from 0.20 (nad5) to 0.31 (nad3, nad4L, nad6). Finally, for W. mexicana AT-skew values ranged from −0.30 (cox2) to −0.48 (nad3), and GC-skew values ranged from 0.15 (atp6) to 0.46 (nad3) (Fig. 2).

Figure 2. The nucleotide skewness (A) and AT content (B) of 3 species of Allocreadiids mitochondrial genomes (Allocreadium lobatum, Creptotrematina aguirrepequenoi Wallinia mexicana).
The most common start codon among the 12 PCGs of the 3 allocreadiid mt genomes was ATG, with a frequency of 10/12 in A. lobatum and C. aguirrepequenoi, and 8/12 in W. mexicana. The TAG was the predominant stop codon in A. lobatum (8/12), as well as in C. aguirrepequenoi and W. mexicana (7/12) (Table 1). There were 3341, 3342 and 3,328 amino acids in 12 PCGs of A. lobatum, C. aguirrepequenoi and W. mexicana, respectively; the RSCU is shown in Fig. 3. In the 3 mt genomes, the most frequent amino acid was Leu (16.2–16.8%), followed by Phe (11.5–11.6%), Ser (11.1–11.4) and Val (9.2–9.6%). The least frequent amino acid was Gln (0.8–0.9%).

Figure 3. Relative synonymous codon usage (RSCU) of 12 protein coding genes of the 3 allocreadiid mt genomes: a) Allocreadium lobatum, c) Creptotrematina aguirrepequenoi; w) Wallinia mexicana.
Overall nucleotide similarity for the 12 PCGs among the 3 allocreadiid mt genomes was 75.5%, whereas amino acids showed 61.6% overall similarity. Genes with high nucleotide diversity among allocreadiids were atp6 > nad3 > nad2> rrnS > cox1, which is consistent with previous studies using sliding window analysis in members of Plagiorchiida (Table 1). To determine the highly conserved and variable mitochondrial genes among the 3 allocreadiid trematodes, a sliding window analysis was conducted by the concatenated nucleotide sequence of 12 PCGs plus the 2 rrn genes. Nucleotide diversity (π) among the 3 mt genomes ranged from 0.004 to 0.29; atp6 (π = 0.241) was the most variable gene, while cytb (π = 0.10) and nad1 (π = 0.09), showed low sequence variation (Fig. 4). Cox 1, rrnS and rrnL show intermediate molecular variation (π = 0.19, 0.21 and 0.15, respectively).

Figure 4. Nucleotide variation across Alocreadium lobatum, Creptotrematina aguirrepequenoi and Wallinia mexicana mt genomes. (A) Sliding window analysis of the 12 PCGs plus rrnS and rrnL. The black line represents nucleotide variation in a window of 300 bp (step size = 10 bp, with the value inserted at its mid-point). (B) Ratios of non-synonymous to synonymous (dN/dS) substitution rates calculated from individual protein-coding genes of 3 newly allocreadiid mt genomes.
Analysis of mutation rates among the 12 PCGs of the 3 allocreadiid mt genomes showed that all PCGs were under purifying selection (dN/dS < 1) (Fig. 4); this coincides with previous estimations for other trematode species (Suleman et al., Reference Suleman, Tkach, Muhammad, Zhang, Zhu and Ma2020; An et al., Reference An, Qiu, Lou, Jiang, Qiu, Zhang, Li, Zhang, Wei, Chen, Gao and Wang2022). The highest non-synonymous substitution rate (dN) was observed in nad4L and nad1, while cox1 gene showed the lowest dN/dS value, followed by atp6, indicating a higher synonymous variation in these genes among allocreadiids.
These results plus the distribution of nucleotide diversity among genes indicate cox1 and atp6 as useful genetic markers for genetic lineage determination and species identification studies in the family Allocreadiidae. It has been observed in other trematode mt genomes that atp6 and nad2 genes evolve faster than cox1, suggesting that these genes may also serve as genetic markers to capture genetic variation among closely related trematode species or even populations (Suleman et al., Reference Suleman, Tkach, Muhammad, Zhang, Zhu and Ma2020).
Phylogenetic relationships of Allocreadiidae
The ML phylogenetic trees using either nucleotides or amino acids recovered Allocreadiidae as a highly supported monophyletic group (Figs 5, 6), with C. aguirrepequenoi clustered together with W. mexicana, and these 2 with A. lobatum. In both analyses, allocreadiids appeared as sister taxa of other species included in the suborder Xiphidiata; this is consistent with results obtained through phylogenetic analyses based on 18S and 28S rRNA genes (Pérez-Ponce de León and Hernández-Mena, Reference Pérez-Ponce de León and Hernández-Mena2019), in which Allocreadiidae was recovered as a monophyletic group within the suborder Xiphidiata. Figure 5 depicts the phylogenetic tree inferred from the concatenated nucleotide sequences of 12 PCGs and 2 rrn for the same dataset; here the family Allocreadiidae was recovered as the sister group of a clade containing Haematoloechidae, Glypthelminthidae, Plagiorchiidae and Orientocreadiidae, plus a clade comprising Eucotylidae, Prosthogonimidae and Dicrocoeliidae. In the phylogenetic tree inferred with amino acids (Fig. 6), Allocreadiidae appeared as an early divergent clade with respect to a clade containing paragonimids plus brachycladiids (both members of Xiphidiata in the current classification, see Pérez-Ponce de León and Hernández-Mena, Reference Pérez-Ponce de León and Hernández-Mena2019), and all of them as sister taxa of 2 families of Opisthorchiata (Fig. 6), although these relationships are poorly supported by bootstrap values.

Figure 5. Maximum likelihood phylogenetic tree obtained with the nucleotide dataset of 12 protein-coding genes and 2 regions of the ribosomal RNA. Species in the suborder Xiphidiata are framed in purple shade. Species in Opisthorchiata are framed in green shade. The name of the species is followed by the mt genome accession number in the Genbank dataset, and in parentheses the family to which it belongs. The species sequenced in this study are highlighted in bold. Numbers near the nodes are the Bootstrap support values.

Figure 6. Maximum likelihood phylogenetic tree obtained the amino acids translated of 12 PCG's. Species in the suborder Xiphidiata are framed in purple shade. Species in Opisthorchiata are framed in green shade. The name of the species is followed by the mt genome accession number in the Genbank dataset, and in parentheses the family to which it belongs. The species sequenced in this study are highlighted in bold. Numbers near the nodes are the Bootstrap support values.
In the current study, phylogenetic analyses of mt genomes of 28 species of trematodes allocated in 12 families using either nucleotides or amino acids, yielded contrasting results about the monophyly of Xiphidiata, since the nucleotide tree yielded the suborder as monophyletic, whereas the amino acids tree yielded Xiphidiata paraphyletic. Our results are in contrast with Suleman et al. (Reference Suleman, Khan, Tkach, Ullah, Ehsan, Ma and Zhu2021) who found consistent results of the paraphyly of Xiphidiata using either nucleotide or amino acid sequences; Suleman et al. (Reference Suleman, Khan, Tkach, Ullah, Ehsan, Ma and Zhu2021) used the mt genomes of 30 species of trematodes allocated in 14 families. Likewise, Pérez-Ponce de León and Hernández-Mena (Reference Pérez-Ponce de León and Hernández-Mena2019) also found Xiphidiata as paraphyletic, and the suborder Haploporata was proposed for keeping the former as monophyletic. Other mt genome phylogenetic analyses using amino acids for building the trees have consistently shown the paraphyly of Xiphidiata (Guo et al., Reference Guo, Li, Gao, Qiu, Jin, Gao, Zhang, An, Chang, Gao and Wang2022; Gao et al., Reference Gao, Zhang, Wei, Jia, Zhang, Li, Chen, Sun, Hou, Liu, Wang, Zhang and Wang2023). In the most recently published study, Atopkin et al. (Reference Atopkin, Semenchenko, Solodovnik and Ivashko2023) used a comprehensive dataset of 61 mitogenomes of trematodes and corroborated Xiphidiata as paraphyletic, although their dataset was not analysed using nucleotides. Apparently, interrelationships between paragonimids and dicrocoeliids as members of the superfamily Gorgoderoidea is problematic even though both have been considered as members of Xiphidiata. Interestingly, Suleman et al. (Reference Suleman, Khan, Tkach, Ullah, Ehsan, Ma and Zhu2021) and Atopkin et al. (Reference Atopkin, Semenchenko, Solodovnik and Ivashko2023) found that Paragonimidae plus Brachycladiidae are nested with Opisthorchiidae and Heterophyidae. The amino acid phylogenetic tree in our study uncovered the same relationships, although Allocreadiidae appeared as the sister clade to these 4 families. The number and selection of taxa representative of different families, and the molecular data employed (nucleotides or amino acids), seem to exert an effect when assessing interrelationships of trematodes using mt genomes. As shown in our study, a shorter number of terminals, and a different selection of taxa may yield contrasting results.
There is no doubt that the discordant phylogenies of mt genomes obtained when using nucleotide or amino acid data, or through different inference methods, and the constant changes in the interrelationships of higher taxa when adding newly sequenced mt genomes is posing new challenges to trematode classification. In our opinion, 3 aspects must be considered to fully assess the value of mt genomes in reconstructing the phylogenetic relationships, and in elucidating higher-level interrelationships of this group of parasitic platyhelminthes. These aspects include the number of taxa sampled, outgroup selection, and use of nucleotide or amino acids. These remain key elements that need to be assessed to produce the most natural classification scheme of digenean trematodes using modern high throughput sequencing technologies.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182024000064.
Data availability statement
Mitogenome sequence data is available on the NCBI GenBank database.
Acknowledgements
We sincerely thank Laura Márquez and Nelly López from the Laboratorio Nacional de Biodiversidad (LaNabio, Biology Institute, UNAM) for their help with DNA sequencing processing.
Authors’ contributions
GPPL, BSG and DHM designed the study; GPPL, DHM and AC conducted field work, and processed the specimens for morphological identification. BSG conducted the experiments to obtain the mitogenomes. BSG and DHM analysed the molecular data and submitted the mitogenome sequences to GenBank. All authors discussed the results. GPPL and BSG drafted the manuscript which was reviewed and greatly improved by a DHM and AC.
Financial support
This research was supported by a grant from the Consejo Nacional de Ciencia y Tecnología (CONACYT A1-S-21694), and partially funded by the Programa de Apoyo a Proyectos de Investigación e Inovación Tecnológica to GPPL (PAPIIT-UNAM IN212621).
Competing interests
The authors declare they have no competing financial interests or personal relationships that could have influenced the work reported in this manuscript.
Ethical standards
All applicable institutional, national and international guidelines for the care and use of animals were followed.









 Open access
Open access