



Introduction
The complement system plays a key role in innate immune surveillance, including initiating a defense response to pathogens and maintaining host homeostasis through clearance of toxic materials. Over 30 component proteins, receptors, and regulators in the complement system’s intricate signaling network are tightly controlled to ensure response to threats is initiated and appropriately amplified, while avoiding host tissue damage from overactivity. Reference Merle, Church, Fremeaux-Bacchi and Roumenina1,Reference Chamberlain, Huda, Whittam, Matiello, Morgan and Jacob2 Unsurprisingly, there is a growing list of inflammatory, autoimmune, and degenerative diseases in which abnormal complement system function or activation contributes to tissue damage. Reference Morgan and Harris3 In many of these diseases, complement dysfunction is not the primary cause of disease, but is secondary to other mechanisms of inflammation or tissue injury. However, some diseases are now recognized to have complement dysfunction at the core of pathogenesis, providing a strong rationale for therapeutic targeting of complement components. Reference Morgan and Harris3 Among these are two autoantibody-mediated neurological conditions: myasthenia gravis (MG) and neuromyelitis optica spectrum disorders (NMOSD). The purpose of this paper is to describe the role of complement activation in the pathogenesis of MG and NMOSD, to discuss the rationale and evidence for complement inhibition as a method to manage these diseases, and to provide a Canadian perspective on the use of complement inhibition therapy in real-world cases of MG and NMOSD.
Background
Complement Activation Pathways
At any time, components of the complement system can be found throughout bodily fluids and tissues in an inactive state; however, they can be activated upon sensing a danger signal. Reference Janeway, Travers, Walport and Shlomchik4 Complement activation can occur through three different pathways: (1) the classical pathway, which is triggered by binding of an antigen-antibody complex to the C1q complement component, (2) the mannose-binding lectin pathway, which is triggered by binding of the mannan-binding lectin recognition molecule to mannose residues found on bacterial cell surfaces, and (3) the alternative pathway, which is triggered by the binding of spontaneously activated complement components to pathogen surfaces. All these pathways lead to the formation of the C3 convertase, and subsequently, the C5 convertase, both of which amplify a series of enzymatic reactions (Figure 1). The products of these reactions ultimately lead to host protection by stimulating opsonization (marking pathogens for phagocytic engulfment), recruiting additional inflammatory cells, and directly killing pathogens through the formation of the membrane attack complex (MAC). Reference Janeway, Travers, Walport and Shlomchik4

Figure 1: Complement activation through multiple pathways. The complement system is activated through three main pathways which all lead to the formation of C3 and C5 convertases (dashed red boxes). These trigger a series of enzymatic reactions necessary to form the membrane attack complex (MAC, red steps 7/8). The classical pathway is initiated upon binding of an antigen-antibody complex to the C1q complement component, leading to its auto-activation (blue steps 1–2). The mannose-binding lectin pathway is initiated upon binding of the mannan-binding lectin (MBL) recognition molecule to mannose residues found on bacterial cell surfaces, leading to its auto-activation (green steps 1 and 2). Both the activated C1q and MBL molecules are then able to cleave complement components C2 and C4 (gold step 3). The cleavage products of these reactions, C4b and C2a, can subsequently bind, forming the C3 convertase (gold step 4). The alternative complement pathway is activated through spontaneous hydrolysis of the C3b complement component at the cell surface (orange step 1). C3b subsequently binds to factor Bb, a product of the proteolysis of Factor B, to form a C3 convertase (orange steps 1–2). C3 convertases from all pathways cleave C3, forming C3b which interacts with the C3 convertase to form the C5 convertase (gold steps 5–6, orange steps 4–5). The C5 convertase cleaves component C5, producing C5b which is important for recruitment and formation of the MAC complex (gold step 7, orange step 6). Other products of the C3 and C5 cleavage reactions, C3a and C5a, play a role in chemotaxis and other inflammation-promoting immune cell signaling.
Myasthenia Gravis: Complement-Mediated Pathogenesis and Treatment
MG is a rare neuromuscular autoimmune disorder occurring at a rate of 0.25–2 people per 100,000 per year. Reference Chamberlain, Huda, Whittam, Matiello, Morgan and Jacob2 It is a T-cell dependent-B-cell mediated disease, requiring the activation of CD4+T cells to initiate the autoimmune process of T regulatory cell, cytokine secretion, and B-cell activation impairment. Reference Chamberlain, Huda, Whittam, Matiello, Morgan and Jacob2 The disease primarily manifests as muscle weakness upon repeated muscle use and most commonly affects the oculobulbar muscles, often asymmetrically. Approximately 15% of patients only present with ocular symptoms, whereas 85% have generalized MG which affects proximal muscles of the extremities and trunk. Reference Gilhus, Tzartos, Evoli, Palace, Burns and Verschuuren5
Roughly, 80%–85% of MG cases are linked to the presence of antibodies against the nicotinic acetylcholine receptor (AChR; primarily of the immunoglobulin [Ig] G1 and Ig G3 subclasses), which is expressed on the postsynaptic muscle endplate of the neuromuscular junction (NMJ). Reference Zisimopoulou, Brenner, Trakas and Tzartos6 In these cases, the pathogenesis of MG can be partly attributed to the AChR antigen-antibody complex triggering classical complement activation, which ends in MAC-mediated lysis of AChR and disturbance of the postsynaptic membrane. Reference Chamberlain, Huda, Whittam, Matiello, Morgan and Jacob2 This in turn leads to faulty synaptic transmission across the NMJ and contributes to the progressive muscle weakness and fatigability in patients with MG. Other autoantibodies have also been identified in patients with MG, including anti-MuSK antibodies, which are detected in approximately 5% of patients with MG. Reference Zisimopoulou, Brenner, Trakas and Tzartos6 The pathogenesis for anti-MuSK-positive MG differs from anti-AChR-positive MG as anti-MuSK antibodies belong to the IgG4 subclass which cannot activate the complement cascade. Reference Gilhus, Tzartos, Evoli, Palace, Burns and Verschuuren5 The mechanism of pathogenesis of MG with anti-MuSK and other autoantibodies is described elsewhere. Reference Howard7
Although in most cases MG can be effectively managed with a combination of acetylcholinesterase inhibitors and immunosuppressants, 10%–15% of patients require alternative therapeutic options as they have incomplete disease control, are treatment-refractory, or have become intolerant to immunosuppressive therapy. Reference Levine8 Therapy for these patients may include azathioprine, methotrexate, mycophenolate mofetil, tacrolimus, cyclosporine, corticosteroids, intravenous immunoglobulin (IVIG), plasma exchange (PLEX), or rituximab. Reference Narayanaswami, Sanders and Wolfe9,Reference Farmakidis, Pasnoor, Dimachkie and Barohn10 Although these therapies do not have Health Canada-approved indications for generalized MG, their use is supported by case series, years of clinician experience, and, in some cases, small randomized controlled trials. Given the strong evidence for the role of complement-mediated destruction in the pathogenesis of anti-AChR-positive MG, Reference Howard7 therapies directly targeting the complement system have been evaluated in these patients.
Eculizumab, a drug targeting the complement component C5, is one such therapy that has received Health Canada approval for the treatment of generalized MG. 11 It is the first approved antibody therapy targeting the complement system and is also approved for the treatment of other disorders where complement activation contributes pathologically, including paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome. Regulatory approval of eculizumab was based on a phase III, randomized, double-blind trial (REGAIN) which evaluated the efficacy of eculizumab versus placebo in 125 patients with anti-AChR antibody-positive, refractory generalized MG. Reference Howard, Utsugisawa and Benatar12 The study failed to meet the primary endpoint, which intended to demonstrate a statistically significant increase in change in Myasthenia Gravis Activities of Daily Living (MG-ADL) score at 26 weeks for eculizumab versus placebo, using the prespecified worst rank analytical approach. However, post hoc sensitivity analyses of changes in MG-ADL, Quantitative MG, MG Composite, and 15-item MG Quality of Life scores did show significant improvements with eculizumab compared with placebo. Additionally, longer follow-up (up to 2.5 years) in the open-label extension study showed the rate of MG exacerbation was reduced by 75% from the year before REGAIN (p < 0.0001), and over 50% of patients achieved a postintervention status of minimal manifestations or better after 1 year of eculizumab treatment. Reference Muppidi, Utsugisawa and Benatar13,Reference Mantegazza, Wolfe and Muppidi14 Improvements in activities of daily living, muscle strength, functional ability, and quality of life were also maintained through 3 years of eculizumab treatment. Reference Muppidi, Utsugisawa and Benatar13
As activation of the complement system is critical for mounting a host defense against Neisseria meningitidis infection, an increased risk for meningococcal infection has been documented in patients on eculizumab therapy. Reference Ladhani, Campbell and Lucidarme15 This led to the requirement for vaccination against N. meningitidis at least 2 weeks before starting treatment in the REGAIN study. Fortunately, no cases of meningococcal infection occurred in either the placebo-controlled study or open-label extension. Reference Howard, Utsugisawa and Benatar12 The most common adverse events (AEs) occurring in both the eculizumab and placebo arms were headache and upper respiratory tract infection (each occurring in 16% of patients in the eculizumab arm and 19% of patients in the placebo arm). With longer follow-up in the open-label extension study, the safety profile of eculizumab remained consistent with the REGAIN trial. Reference Muppidi, Utsugisawa and Benatar13,Reference Mantegazza, Wolfe and Muppidi14
Based on the above data, the International Consensus Guidelines 2020 Update, developed by a task force of the MG Foundation of America, recommends eculizumab for the treatment of severe, refractory, AChR antibody-positive generalized MG, with its role in the treatment of MG likely to evolve over time. Reference Narayanaswami, Sanders and Wolfe9 Other therapies targeting the C5 complement component are also being studied in clinical trials for patients with MG, including zilucoplan and ravulizumab. Notably, ravulizumab has been designed to have enhanced plasma terminal half-life and thus need not be given as often as eculizumab (which is dosed bi-weekly). Reference Menon, Barnett and Bril16
Neuromyelitis Optica Spectrum Disorder: Complement-Mediated Pathogenesis and Treatment
NMOSD is a rare, autoimmune disease that ranges in prevalence from 0.5−4/100,000 worldwide, with prevalence reaching up to 10/100,000 in certain racial groups. Reference Hor, Asgari and Nakashima17 It attacks the central nervous system (CNS), with the potential to cause blindness and paralysis, among other CNS symptoms. Although historically NMOSD was considered a variant of multiple sclerosis and known as Devic’s disease, the identification of the pathogenic IgG against aquaporin-4 (AQP4) that is present in 70%–80% of clinically defined cases has solidified NMOSD as a distinct condition. Reference Chamberlain, Huda, Whittam, Matiello, Morgan and Jacob2 AQP4 is a water channel protein that is abundantly expressed throughout the body but in the CNS is mainly on astrocytic foot processes at fluid-parenchymal surfaces. It plays an essential role in supporting neuroexcitation and glutamate reuptake following synaptic transmission. Binding of AQP4-IgG to AQP4 on the surface of astrocytic endfeet in patients with NMOSD presents several vulnerabilities to complement-mediated damage. Firstly, AQP4 tetramers cluster together to form orthogonal arrays of particles on astrocytic surfaces. This aggregation of antigen allows the potential to trigger a large stimulus for classical complement activation. Reference Chamberlain, Huda, Whittam, Matiello, Morgan and Jacob2 Secondly, a study by Saadoun and Papadopoulos observed a lack of complement system regulators at the astrocytic endfeet in normal brain tissue, Reference Saadoun and Papadopoulos18 explaining at least partially why disease is centered on the CNS and not on any other organ system.
Indeed, there is an abundance of evidence to support the significant role of complement-mediated attack against AQP4 in NMOSD pathogenesis, which is described in detail elsewhere. Reference Chamberlain, Huda, Whittam, Matiello, Morgan and Jacob2 Briefly, in NMOSD, the classical complement pathway is activated upon binding of the AQP4-IgG:AQP4 complex to complement component C1q. The amplification products of this complement signaling cascade promote the recruitment of other inflammatory cells and the terminal complement molecules promote astrocyte cell death through MAC formation. This intense tissue inflammation initially triggered through complement activation can invoke collateral damage to nearby oligodendrocytes and neurons, ultimately leading to myelin loss and necrosis in both gray and white matter. Reference Chamberlain, Huda, Whittam, Matiello, Morgan and Jacob2,Reference Lucchinetti19
As NMOSD is a relapsing disease, resulting in cumulative neurological damage and disability upon repeated immune attacks, the main goals of therapy are to suppress acute inflammation that leads to relapses thus preventing relapse related progression. Reference Kessler, Mealy and Levy20 Some of the traditional therapies for management of NMOSD are all aimed at reducing inflammation and include high-dose corticosteroids, azathioprine, mycophenolate mofetil, rituximab, PLEX, and IVIG. Reference Chamberlain, Huda, Whittam, Matiello, Morgan and Jacob2 Like in generalized MG, the use of these standard therapies is largely supported by case series, retrospective analyses, and clinical experience.
Like in MG, eculizumab has demonstrated excellent efficacy in NMOSD, owing to the large role of complement-mediated destruction in its pathogenesis. It is also currently approved in Canada for the treatment of adults with AQP4-positive NMOSD, based on results from the PREVENT trial. In the phase III PREVENT trial, a double-blind study of 143 adults with relapsing AQP4-IgG-positive NMOSD, eculizumab demonstrated a significantly lower risk in adjudicated relapses versus placebo (94% reduction in relapses; hazard ratio 0.06). Reference Pittock, Berthele and Fujihara21 Analyses of the open-label extension to the PREVENT study demonstrated a sustained ability of eculizumab to prevent relapse, with 94.4% of patients remaining adjudicated relapse-free at 3.7 years. Reference Wingerchuk, Fujihara and Palace22 Safety analyses showed that in the eculizumab group, more frequent upper respiratory tract infections and headaches were reported. One death from pulmonary empyema occurred in the eculizumab group, and no cases of meningococcal infection were reported at a median follow-up of 133.3 weeks (362.3 patient-years). Reference Wingerchuk, Fujihara and Palace22
Other complement-directed therapies under investigation for NMOSD include a C1-esterase inhibitor, which inactivates parts of the C1 recognition complex, and the use of statins to upregulate the CD55 complement regulator on astrocytes. Novel therapies targeting other aspects of immune attack in NMOSD include inebilizumab, an anti-CD19 antibody inducing B-cell depletion, Reference Frampton23 and satralizumab, an anti-interleukin-6 receptor antibody targeting T- and B-cell maturation, Reference Heo24 the latter of which has been approved by Health Canada. 25
Canadian Experience
Illustrative Cases of Eculizumab in Myasthenia Gravis
Eculizumab has been authorized for use in Canada for adult patients with generalized MG since August 2018 11 ; however, access to the drug is variable across the country. This was based on the study of eculizumab in patients with treatment-refractory anti-AChR antibody-positive generalized MG where treatment refractoriness was defined as failure of treatment with two or more immunosuppressive therapies either in combination or as monotherapy, or failure of at least one immunosuppressive therapy and requiring chronic PLEX or IVIG to control symptoms. The Canadian Agency for Drugs and Technology in Health (CADTH) has recommended eculizumab be reimbursed for patients with generalized MG as per the Health Canada indication if several conditions are met, including a reduction in drug price and a 6-month assessment of disease improvement required for therapy continuation. 26 As of June 2021, eculizumab is not yet funded publicly in any provinces; however, it may be available to patients through private insurance or can be accessed through a compassionate use program.
With the availability of eculizumab, Canadian neurologists are beginning to gain real-world experience in treating MG patients with this drug. Table 1 illustrates seven retrospectively and sequentially selected patients with treatment-refractory, generalized MG who were receiving eculizumab treatment at Toronto Western Hospital or the Montreal Neurological Institute and Hospital under the care of Dr D. Dodig or Dr A. Genge, from August 2018 to February 2021. Patients received eculizumab at 900 mg/week for 4 weeks, followed by 1200 mg every 2 weeks. All cases were anti-AChR antibody-positive and included five female and two male patients with a mean age of 54 years. The mean time since diagnosis was 18 years, with two patients having longstanding disease beyond 35 years. Four patients had a prior thymectomy. Patient 2 had been incidentally diagnosed with thymoma and treated by thymectomy 2 years prior to onset of MG with no evidence of reoccurrence of thymoma. The appearance of MG after thymectomy has previously been reported in the literature. Reference Kang, Lee, Choi and Kang27,Reference Beckers, Mercelis and Heyman28
Table 1: Myasthenia gravis patient cases treated with eculizumab
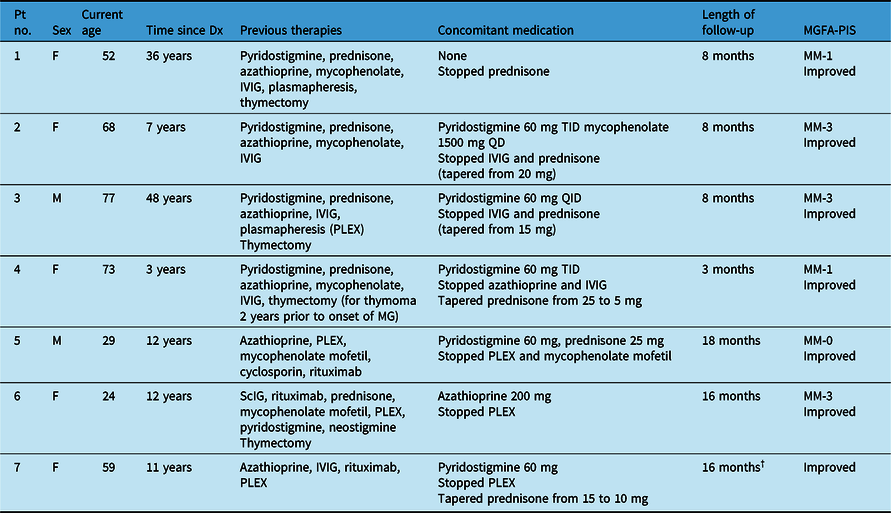
F, female; IVIG, intravenous immunoglobulin; M, male; MG, myasthenia gravis; MGFA-PIS, Myasthenia Gravis Foundation of America Postintervention Status; MM, minimal manifestations; PLEX, plasma exchange; QD, once-daily; ScIG, subcutaneous immunoglobulin; TID, twice-daily.
† Patient discontinued eculizumab at this time point.
Eculizumab was chosen in the seven patients described because their disease could not be adequately controlled despite being on chronic IVIG (three patients) or chronic PLEX (three patients) or the patient was experiencing negative side effects due to long-term, high-dose steroid use (one patient, 20 mg prednisone for 30 years). These cases align with the Health Canada indication for eculizumab in generalized MG and with the population in the REGAIN trial. 11,Reference Howard, Utsugisawa and Benatar12 For each patient, MG-ADL and QMG were collected during follow-up visits. Myasthenia Gravis Composite Scores (MGCS) were also collected at follow-up in the majority of cases but were collected retrospectively for a few visits conducted via telemedicine due to COVID-19 pandemic-related restrictions.
At 3–12 months after starting eculizumab, a clinically meaningful improvement in MGCS (three or more points from baseline) was observed in all seven patients (Figure 2). In addition, five of seven patients had a clinically meaningful improvement in MG-ADL (score decrease of three or more from baseline) and four of seven patients had a clinically meaningful improvement in QMG score (score decrease of five or more from baseline). An MG-ADL improvement greater than 8 and QMG improvement greater than 10 were observed in two patients each. This improvement in MG disease scores is similar to the REGAIN trial which reported clinically meaningful improvements in MG-ADL and QMG scores from baseline in 60% and 45% of patients, respectively (vs. 40% and 19% in the placebo arm) at 26 weeks. Reference Howard, Utsugisawa and Benatar12 A particularly encouraging improvement after eculizumab treatment was noted in patient 3 who had regained bilateral power in his hip flexors, which had persistently been weak prior to therapy (grade 3 on Medical Research Council scale). Based on clinical observations of the disease, it has been deemed that in the ultimate stage of "burned-out" disease, untreated weakness may become fixed in association with muscle atrophy. Reference Juel and Massey29 Therefore, improvement of longstanding weakness supports the hypothesis that complement inhibition can facilitate restructuring of chronic damage to the NMJ architecture. MG exacerbations did not occur in four patients, they were decreased in two patients, and no serious treatment-related AEs were observed during eculizumab therapy.
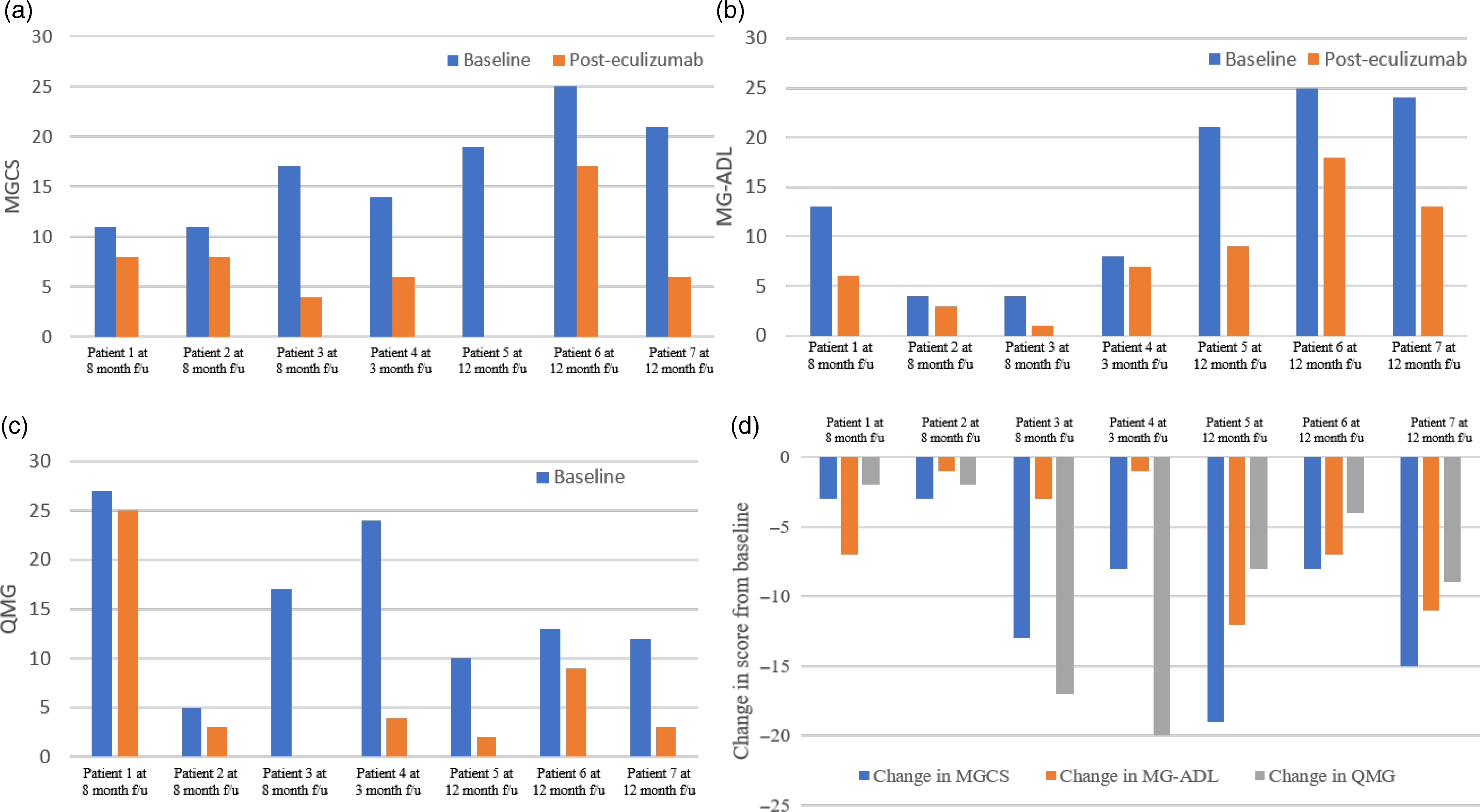
Figure 2: Change in MGCS, MG-ADL, and QMG scores from baseline. (A) MGCS at baseline and posteculizumab treatment. (B) MG-ADL at baseline and posteculizumab treatment. (C) QMG at baseline and posteculizumab treatment. (D) Change in MG scores from baseline. f/u, follow-up; MG-ADL, Myasthenia Gravis Activities of Daily Living; MGCS, Myasthenia Gravis Composite Score; QMG, Quantitative Myasthenia Gravis score.
The cases presented provide additional support for the use of agents that target complement activity, such as eculizumab, for the treatment of AChR antibody-positive MG and are in line with what was observed in the REGAIN trial. However, we recognize the limitations to the conclusions that can be drawn from both REGAIN and these real-world cases, as well as practical challenges in having widespread access to eculizumab in Canada.
Firstly, the case studies presented, and the population studied in the REGAIN trial may not reflect the general population of patients with AChR antibody-positive refractory generalized MG. In the cases presented, there is a risk for selection bias based on individual practices of the clinicians, individual province-based access to off-label use medications, and ability of the patient to afford/access eculizumab. Similarly, the population enrolled in the REGAIN trial may not be reflective of the refractory generalized MG population seen in Canadian practice. For example, in REGAIN, less than 50% of patients had previously received mycophenolate mofetil and only 11% of patients had received prior rituximab therapy, Reference Howard, Utsugisawa and Benatar12 compared with approximately 70% and 40% of patients, respectively, in the small sample of presented MG cases.
Secondly, the actual degree of improvement with eculizumab remains unclear, as there were no specific analyses to determine this. This may also be partly attributed to the failure of meeting the designated primary endpoint in the REGAIN trial (although this may have also been influenced by a less than optimal primary outcome choice). In addition, although the analysis of secondary outcomes consistently demonstrated a benefit for eculizumab over placebo, there was a large variation in the level of response among patients receiving eculizumab, Reference Gilhus30 which was also observed in the presented cases. Without the presence of a control, the extent to which a placebo effect has impacted the outcomes in the cases presented is also unclear. As observed in other clinical trials in neurological disorders, including the REGAIN trial, patients in the placebo arm can report meaningful improvements in disease measures. Reference Howard, Utsugisawa and Benatar12 This could be attributed to the presence of concomitant medications or due to patients being more closely monitored, which may influence reported outcomes.
Lastly, longer follow-up is needed to understand how eculizumab can maintain symptom improvement in generalized MG. Thus far, with a relatively short follow-up, only patient 7 discontinued eculizumab treatment at 16 months due to both declining drug effects and the opportunity to begin a clinical trial. The open-label extension of the REGAIN trial has followed patients receiving eculizumab up to approximately 2.5 years and has demonstrated sustained achievement in Myasthenia Gravis Foundation of America postintervention status of minimal manifestations; Reference Mantegazza, Wolfe and Muppidi14 however, given the chronic nature of MG and the lack of data to understand the optimal length of treatment, additional follow-up is needed.
These caveats tied to eculizumab become increasingly important given the significant cost of eculizumab compared to other standard of care treatments in generalized MG (over $700,000 CAD annually as submitted by manufacturer, although price negotiations are ongoing). 31 In a reanalysis of the sponsor’s economic model performed by the CADTH, the incremental cost-effectiveness ratio for eculizumab plus standard of care compared to standard of care alone in patients with refractory AChR antibody-positive generalized MG was $1,505,712 per quality-adjusted life-year (QALY). 31 This is significantly beyond the willingness to pay threshold set at $50,000 per QALY. Albeit, the cost-effectiveness thresholds leveraged by CADTH in the Common Drug Review are consistent across all therapeutic areas and do not take different patient populations into consideration (i.e., common chronic diseases vs. orphan diseases). Reference Black32 Although there were significant uncertainties within the parameters of this economic model that affect the precision of the cost-effectiveness projection, sustainable access to eculizumab may nevertheless be challenging in the Canadian healthcare system at its current cost.
Eculizumab therapy is also associated with an increased risk of meningococcal infection, with a rate of 0.25 infections per 100 patient-years reported in a registration study of PNH. Reference Socié, Caby-Tosi and Marantz33 Infection risk appears to persist among patients who received meningitis vaccination, as there is evidence to suggest attenuated vaccine response and infection postvaccination in some patients receiving eculizumab. Reference McNamara, Topaz, Wang, Hariri, Fox and MacNeil34 For this reason, some countries recommend antimicrobial prophylaxis for the duration of eculizumab treatment; 35,36 however, the added benefit of long-term prophylaxis in this setting has not yet been elucidated. Reference McNamara, Topaz, Wang, Hariri, Fox and MacNeil34,Reference Bozio, Isenhour and McNamara37,Reference Patriquin, Kulasekararaj and Peffault de Latour38 There are currently no guidelines in Canada that recommend continuous antibiotic prophylaxis for patients receiving eculizumab. In all presented cases, as per product monograph guidance, patients received meningitis vaccination at least 2 weeks prior to the start of eculizumab without the use of prophylactic antibiotics and thus far no meningococcal infections were reported.
Despite the aforementioned caveats, eculizumab’s specificity in targeting disease pathogenesis and favorable safety profile make it an important therapeutic option for Canadian patients with generalized MG who fail to respond to or cannot tolerate traditional immunosuppressive therapies. It is a particularly important therapy that can address some limitations with current treatment options for generalized MG. For example, chronic treatment with IVIG or PLEX has some disadvantages, particularly in the setting of the COVID-19 pandemic. This includes the frequent shortages of Ig product occurring globally over the last few years which has been predicted to be exacerbated by the COVID-19 pandemic. This is due to a potential decrease in source plasma collection and an increase in the investigational use of plasma products for the treatment of COVID-19 itself. Reference Hartmann and Klein39 With many provinces initiating programs to ration IVIG use, this may not reflect a sustainable long-term therapy for patients with MG. The main disadvantages of PLEX are the invasiveness of placing an IV port for patients receiving frequent treatment and the long timeframe required to receive a full course of therapy in a hospital setting, which puts pressure on an already taxed healthcare system during the pandemic and increases the potential for the patient to be exposed to SARS-CoV-2.
Rituximab is another biologic therapy that may be effective in treatment-refractory AChR antibody-positive gMG, although a larger clinical benefit has been reported in patients who are positive for anti-MuSK antibodies. Reference Banerjee and Adcock40 It functions by targeting the CD20 antigen on B cells, leading to direct complement-mediated and antibody-dependent cytotoxicity. Reference Menon, Barnett and Bril16 An international consensus guidance for the treatment of MG recommends rituximab be considered as an early therapeutic option in patients with MuSK antibody-positive MG who have an unsatisfactory response to initial immunotherapy and notes that its efficacy in AChR antibody-positive disease is uncertain and therefore, rituximab may be an option if patients fail or do not tolerate other immunosuppressive agents. Reference Narayanaswami, Sanders and Wolfe9 Although approved for the treatment of other autoimmune disease, rituximab is not approved for the treatment of MG in Canada. For this reason, it is difficult to gain off-label access to rituximab for MG in most provinces. Also, it is of note that use of B-cell-depleting agents such as rituximab have been cautioned during the COVID-19 pandemic as some reports suggest a higher risk of infection and severe COVID-19 outcomes for patients receiving rituximab therapy, as well as a potential decrease in immune response to SARS-CoV-2 vaccines. Reference Avouac, Drumez and Hachulla41,Reference Baker, Roberts and Pryce42
Finally, while the patients with MG may be maintained on long-term high doses of prednisone to manage their disease, these can also be associated with unfavorable side effects such as weight gain, bruising, osteoporosis, cataracts, glaucoma, onset or worsening of diabetes, and psychological disorders. Reference Sanders and Evoli43 Eculizumab may serve as a steroid sparing-agent, which has been demonstrated in the REGAIN and open-label extension trials, where the mean dose of prednisone (as well as azathioprine and mycophenolate) was significantly decreased from baseline to last assessment. Reference Nowak, Muppidi, Beydoun, O'Brien, Yountz and Howard44 The benefit of eculizumab in reducing the use of prednisone was also observed in the cases described, with five patients being able to stop or decrease prednisone dose after treatment initiation.
Although the complement pathway is a rational target for anti-AChR antibody-positive MG, the role for agents inhibiting complement is less clear in patients positive for other autoantibodies such as anti-MuSK. Therefore, patients with anti-AChR antibody-negative refractory MG are in need of therapies with novel mechanisms of action to target other aspects that drive disease. Fortunately, many other therapeutic avenues are being explored in MG that target different aspects of disease pathophysiology, including neonatal Fc receptor inhibitors, B-cell-depleting agents, proteosome inhibitors, T-cell- and cytokine-based therapies, and autologous stem cell transplantation. Reference Menon, Barnett and Bril16
Illustrative Cases of Eculizumab in Neuromyelitis Optica Spectrum Disorder
Eculizumab has been authorized for use in Canada for adult patients with anti-AQP4 antibody-positive NMOSD since September 2019; however, it is not intended for acute treatment of an NMOSD relapse. Similar to MG, access for this indication is variable across the country. The CADTH has recommended eculizumab be reimbursed for patients with anti-AQP4-positive NMOSD with several initiation criteria. These include the patient requiring a history of at least two relapses during the previous 12 months or three relapses during the last 2 years (with at least one of these relapses occurring within the last 12 months) despite adequate trial of other preventative NMOSD medications; the patient having an expanded disability status scale (EDSS) score of 7 or less; initiation of eculizumab not be given during a relapse episode; and a maximum 12-month duration of initial authorization. 45 A reduction in price is also noted as a conditional requirement for public reimbursement. Currently, eculizumab may be available to patients with NMOSD through private insurance or can be accessed through a compassionate use program.
Table 2 illustrates retrospectively and sequentially selected patients with anti-AQP4-positive NMOSD who were receiving eculizumab treatment at St. Michael’s Hospital, University of Toronto or the Ottawa Hospital under the care of Dr D. Selchen or Dr M.S. Freedman from September 2019 to February 2021. Five female patients aged 26–49 were included. Only patient 1 fit the criteria of the PREVENT trial of having two relapses within the last 12 months before starting eculizumab treatment. In the two prior relapse episodes, the patient presented with left optic neuritis, one of which accompanied other symptoms (severe bilateral visual impairment, avascular necrosis of the hip, glaucoma) and led to hospitalization, the other occurring while on treatment with rituximab. This patient has an additional diagnosis of systemic lupus erythematosus. Of the remaining patients, patient 3 and 4 received eculizumab as a first-line therapy based on the distressing nature of the first episodes and the ability to access the therapy through private insurance. Patients 2 and 5 received eculizumab after first relapse on azathioprine and rituximab, respectively, with patient 2 requesting the most effective therapy they could access and patient 5 having a particularly severe first relapse involving a spinal event (EDSS 8).
Table 2: Neuromyelitis optica spectrum disorder patient cases treated with eculizumab

EDSS, expanded disability status scale; F, female; NMO, neuromyelitis optica; PLEX, plasma exchange; SLE, systemic lupus erythematosus.
The length of follow-up at last assessment for the presented cases is 6–12 months. At this early follow-up, all patients had stable disease and no relapses or hospitalizations have occurred; however, given the median time to relapse in the placebo arm of the PREVENT trial was almost 2 years, Reference Pittock, Berthele and Fujihara21 a longer follow-up is needed to assess the efficacy of eculizumab in these patients. Of note, four patients are currently being maintained on eculizumab with either no prednisone use or are tapering prednisone. Similar to the PREVENT trial, eculizumab treatment had minimal effect on EDSS scores, with patient 2 having a 2.5 point score reduction which can be attributed to rescue therapy at relapse (baseline EDSS score was taken at relapse) and patient 5 having a score reduction of 1 point. However, given EDSS is a non-validated assessment for disability in NMOSD, the conclusions that can be drawn from these data are limited.
NMOSD is a devastating and unpredictable disease that progresses at each relapse; however, access to treatment options that effectively prevent relapse is limited. The cases presented reflect the need for access to effective treatments, such as eculizumab, to prevent relapse in patients with anti-AQP4-positive NMOSD. The fast onset of action and long-term efficacy of eculizumab observed in the PREVENT study, along with its ability to specifically target the innate immune system without affecting acquired immunity, make eculizumab an excellent treatment option in NMOSD. However, drug cost is a major deterrent to its use, as well as the frequent dosing required, and variable access across Canada. Other potential treatments in preventing NMOSD relapse include rituximab and satralizumab. There is evidence to support the use of rituximab in reducing relapse rates and disability for patients which NMOSD, Reference Damato, Evoli and Iorio46 and it was used in 2 of the presented cases, though relapses did occur while these patients were on rituximab therapy. As the use of rituximab in NMOSD is off-label in Canada, access to this therapy may still be limited for some patients. Satralizumab is another option that may be available to patients with NMOSD through private insurance or compassionate use. It has received Health Canada approval as monotherapy or in combination with immunosuppressive therapies for the treatment of adult and adolescent patients with NMOSD who are anti-AQP4 seropositive. Like eculizumab, satralizumab is not intended for acute treatment of an NMOSD relpase. 25 It has also received a positive recommendation for reimbursement from CADTH for anti-AQP4-positive NMOSD patients who have had at least 1 relapse in the 12 months before initiation despite an adequate trial of other accessible/tolerable preventive treatments for NMOSD, on the condition of an 80%–89% price reduction. The ease of administration (subcutaneous) and less frequent dosing (every 2 weeks for first 3 weeks, followed by every 4 weeks thereafter) for satralizumab are important considerations for treatment decisions.
Conclusion
Since the complement system has been implicated in the pathogenesis of MG and NMOSD, agents targeting this signaling pathway continue to be investigated for therapeutic potential. Eculizumab, a humanized monoclonal antibody targeting the C5 component of the complement pathway, has demonstrated excellent efficacy and tolerability in clinical trials of patients with MG and NMOSD. The benefits of eculizumab in these patient groups have been observed in our clinical experience thus far and provide additional support for the utility of this drug and other potential drugs targeting the complement system in selected patients with MG and NMOSD. However, cost will need to be addressed for broad applicability of eculizumab in patients with MG and NMOSD in the Canadian healthcare system.
Acknowledgements
Medical writing support was provided by Anna Christofides and Sarah Doucette of IMPACT Medicom Inc., funded by Alexion Pharmaceuticals Inc. Alexion Pharmaceuticals provided a medical accuracy review of the final draft.
Disclosures
AG has received honoraria from Alexion Pharmaceuticals Inc. DS has received speaking fees and has participated on an advisory board for Alexion Pharmaceuticals Inc. MF has received research or educational grants from Sanofi-Genzyme Canada; has received honoraria or consultation fees from Actelion (Janssen/J&J), Alexion, Bayer Healthcare, BiogenIdec, Celgene (BMS), EMD Inc., Sanofi-Genzyme, Hoffman La-Roche, Merck Serono, Novartis, and Teva Canada Innovation; has participated as a member of an advisory board or board of directors for Actelion (Janssen/J&J), Alexion, Atara Biotherapeutics, Bayer Healthcare, BiogenIdec, Celgene (BMS), Clene Nanomedicine, Hoffman La-Roche, Magenta Therapeutics, Merck Serono, Novartis, Sanofi-Genzyme, and Teva Canada Innovation; and has participated in a company-sponsored speaker’s bureau for Sanofi-Genzyme and. EMD Serono.
Statement of Authorship
Authors contributed to the development of this paper as follows: Conception and design (D.D., A.G., D.S., and M.S.F.); acquisition of data for cases (D.D., A.G., D.S., and M.S.F.); interpretation of data (D.D. and M.S.F.); drafting of the manuscript (D.D.); critical review of the manuscript (D.D., M.S.F.); approval of final version of the manuscript (D.D., A.G., D.S., and M.S.F.).





 Open access
Open access