



LEARNING OBJECTIVES
After reading this article you will be able to:
• understand current genetic testing techniques in their historical context
• understand how the pathogenicity of genetic variants is determined and the results in genetic testing reports
• broadly outline the current understanding of polygenic mental disorders.
The decoding of the human genome led to great excitement in the scientific community about future genetic research and personalised medicine (Collins Reference Collins, Green and Guttmacher2003). Rapid development in DNA sample analysis has led to an exponential rise in genetic testing in many areas of medicine since then, including aspects of psychiatry. However, it has become clear that for the majority of mental disorders, the genetics are complex owing to the role of multiple genes, interactions between different genes and environmental influences. There can be common genetic variants each with a small effect as well as rare variants with a large effect. Not only can this be daunting for psychiatrists, it is a challenge for geneticists! However, it is recommended that psychiatrists should understand the role of genes in psychiatric disorders and be able to discuss the clinical implications with patients (Nurnberger Reference Nurnberger, Austin and Berrettine2018). The Royal College of Psychiatrists’ Membership examination (MRCPsych) syllabus underlines the importance of the link between neuroscience and psychiatry by requiring that ‘the trainee shall demonstrate knowledge of the neuroscience that underpins the practice of clinical psychiatry’ (Royal College of Psychiatrists 2013: p. 5). This includes more detailed understanding of genetics and methods used in genetic testing than in previous syllabuses because of the growing relevance of genetics in clinical settings.
This narrative aims to cut through the jargon to give a clear overview of the relevance of genetics to psychiatric conditions.
DNA changes
As a brief recap, coils of DNA form the 46 chromosomes carried in human cells. These are paired, with 23 inherited from each parent. The chromosomes determining sex are X and Y, with XX denoting a female and XY a male. The double-stranded molecule of DNA itself is characterised by two phosphate–deoxyribose strands bound together by four DNA bases – adenine (A), cytosine (C), guanine (G) and thymine (T). Base A always pairs with T, and C pairs with G. Genes are portions of code along the DNA molecule that act as a blueprint for the production of proteins. There are various ways in which the DNA sequence can mutate (Fig. 1). These may be grouped as follows:
• structural variants:
◦ copy number variations (CNVs), where there are deletions, insertions or duplications in the DNA of 1000 or more bases compared with a reference sample
◦ smaller deletions, insertions or duplications
◦ inversions, where the orientation of portions of DNA is reversed
◦ translocations, where a part of a chromosome breaks off and attaches to another
• single nucleotide variations (SNVs) and single nucleotide polymorphisms (SNPs), where individual DNA base pairs are changed
• expanding trinucleotide repeats, where three base pairs repeatedly duplicate.
When the DNA blueprint is used to produce proteins, the gene code is read in groups of three bases. Each corresponds to 1 of the 20 amino acids that can be joined together to form a protein. Therefore if a DNA mutation disrupts or ‘shifts’ this triplet ‘reading frame’ then the DNA sequence from that point onwards will be read incorrectly. This is denoted a ‘frameshift’ mutation and very often results in an abnormally short protein or no protein at all. The function of DNA can also be altered by processes that do not involve a change to the DNA sequence itself; this is known as epigenetics. The changes can involve processes such as methylation (the addition of methyl groups to the DNA) and altered expression of promoters or repressors of gene function. It is beyond the scope of this article to expand on these.

FIG 1 Types of DNA variant. delin*, deletion–insertion.
History of genetic testing
In crude terms, the study of changes in DNA structure large enough to affect the architecture of chromosomes is known as ‘cytogenetics’ and the examination of smaller changes in the DNA sequence that can not be seen under the microscope is termed ‘molecular genetics’. The techniques most relevant to psychiatry are described below. Although some are gradually being superseded by more advanced methods, they are included to reflect the updated MRCPsych curriculum.
Cytogenetics
Karyotyping
This is the isolation, staining and visual examination of chromosomes under a microscope (Sinclair Reference Sinclair2002). Chromosomal studies in the 1950s began to identify disorders that alter chromosome number and cause conditions associated with mental disorder (specifically intellectual disability). These included Down syndrome (trisomy 21), Klinefelter syndrome (XXY), Turner syndrome (XO) and Fragile-X syndrome (constriction on the long arm of the X chromosome).
In situ hybridisation and fluorescence in situ hybridisation
In the late 1960s, ‘in situ hybridisation’ was developed to analyse chromosomal structure using portions of DNA (known as probes) that were complementary to a DNA region of interest and hence would bind to it. Various ways of locating the bound probes were developed, first by labelling with radioactive material (Pardue Reference Pardue and Gall1969) and soon after with a stain (dye) called Giemsa (Bhasin Reference Bhasin and Foerster1972). Later, fluorescence in situ hybridisation (FISH) was developed, in which antibodies with fluorescent molecules attached bind to modified DNA probes so that the probes can be located by the fluorescent markers (Fig. 2). The major advantage of FISH was the ability to use multiple colours of fluorescent molecule in the same preparation to study different regions of DNA.

FIG 2 Schematic diagram of fluorescence in situ hybridisation. (a) The double-stranded DNA is separated by DNA helicase enzyme and a region of interest identified. (b) A DNA probe that is complementary to the region of interest is constructed. (c) The probe is fluorescently labelled so that the portion of DNA can be located with a fluorescence detector.
This work revealed chromosome bands, transverse light and dark ‘stripes’ unique to each chromosome. Alterations such as relatively large deletions, duplications, translocations and inversions could now be seen and not just changes in the gross chromosomal structure or number (Fig. 3). This was useful for decades to come, for example to identify the 15q11-13 deletion on chromosome 15, which is the most frequent cause of both Angelman syndrome and Prader–Willi syndrome (Hawkey Reference Hawkey and Smithies1976; Ledbetter Reference Ledbetter, Riccardi and Airhart1981; Pembrey Reference Pembrey, Fennell and van den Berghe1989).

FIG 3 Schematic diagram of a normal male human karyotype (not to scale). Chromosomes are arranged from largest to smallest and each has its own banding pattern.
Molecular genetics
Molecular genetic techniques identify smaller DNA changes and various techniques exist to ‘read’ the DNA sequence.
Sanger sequencing
Sanger sequencing (Sanger Reference Sanger, Nicklen and Coulson1977) uses single strands of DNA, one of which acts as a template in the process of ‘building’ a complementary DNA strand. The DNA sample is first amplified into very large amounts using the polymerase chain reaction or PCR (Mullis Reference Mullis, Faloona and Scharf1986). It is then separated into its two single strands and mixed with:
• a primer – a short section of DNA designed to bind at the DNA area of interest, which acts as the ‘starting point’ of the sequencing reaction
• A, C, G and T DNA bases (also known as ‘nucleotides’)
• DNA polymerase enzyme, which joins each nucleotide to the previous one
• modified nucleotides, which prevent further elongation of the DNA strand when they bind in place of an ‘unedited’ form of the nucleotide. Each is fluorescently labelled with a different colour.
Therefore each reaction starts from the same primer site, but terminates wherever a chemically modified DNA base binds instead of the unedited form of that base. This results in different sized fragments, the modified end of which can be identified by an automated sensor that detects the four different fluorescent dyes (Fig. 4).

FIG 4 Normal DNA replication and Sanger sequencing. (a) Normal DNA replication: an existing DNA strand is a template for the new ‘complementary’ strand; the DNA polymerase enzyme joins DNA bases to elongate the complementary strand. (b) Sanger sequencing: a modified form of each base is added to the reaction; elongation starts from where the primer (a short portion of manufactured DNA) binds; either the normal base or the modified base can join the strand; the modified form stops elongation when it binds, hence different sized fragments are made. With each method, the fragments are separated by size and electrical charge by capillary electrophoresis and the fragment code is read by laser detection of fluorescent probes attached to the modified base pairs.
Sanger sequencing is still used for the screening of small genes or portions of DNA and is more cost-effective for these than some of the ‘next-generation’ techniques described below. It is also used to validate the findings of some of the more sophisticated methods, because although they have a much higher throughput of samples, Sanger sequencing is still valued for its level of accuracy.
Restriction fragment length polymorphism (RFLP)
Restriction fragment length polymorphisms (RFLPs) enabled the location of the Huntington's disease gene to be narrowed down to chromosome 4 (Gusella Reference Gusella, Wexler and Conneally1983). RFLP relies on bacterial enzymes that can cut DNA at specific DNA sequences. These so-called ‘restriction enzymes’ are used in RFLP to compare fragment lengths in patients and their unaffected family members (Botstein Reference Botstein, White and Skolnick1980); this analysis can be done chromosome by chromosome. As Huntington's disease is a trinucleotide repeat disorder, the mutated gene is longer than normal. RFLP revealed that the locus for the difference between the DNA of patients when compared with their relatives was on chromosome 4. It was not until later, when other more complex methods were employed, that the precise gene locus for Huntington's disease on chromosome 4 was identified (MacDonald Reference MacDonald1993). Of note, the RFLP method was also used to narrow down the locus for tuberous sclerosis (Sampson Reference Sampson, Yates and Pirrit1989). It is still used today for some specific clinical purposes, but has largely been superseded.
Comparative genome hybridisation (CGH)
Comparative genome hybridisation (CGH) was initially used in cancer genetics (Kallioniemi Reference Kallioniemi, Kallioniemi and Sudar1992). It compares chromosomal DNA from a clinical population with an unaffected reference population of cells. The affected and reference DNA samples are labelled with different coloured fluorescent probes hybridised (bound) to DNA probes. The relative abundance of the fluorescence of each colour at regions of interest can then be measured. Duplication or deletion of whole chromosomes or relatively large chromosomal portions is evident through calculation of the ratio of one colour to the other. However, as it can only detect differences in DNA abundance, CGH will miss any variants that do not result in changes in the amount of DNA: so-called ‘balanced variants’.
Modern sequencing techniques
So we come to the techniques most widely used today. The first is array CGH, also known as chromosomal microarray (CMA), which was developed in the 1990s. This is a refinement of CGH in which reference samples of areas of interest rather than all the DNA in a cell are added to DNA probes immobilised on a silicon microchip. As with CGH, the patient's DNA is compared with a reference DNA sample and in this case the patient's sample is fluorescently labelled green and the reference red (Fig. 5); sophisticated computer software then calculates the ratio of the two colours. It is now sensitive enough to detect single nucleotide variants, as even with only one base pair alteration, the DNA binds less efficiently to the probe so the fluorescence signal reaching the detector is lower.

FIG 5 Schematic diagram of chromosomal microarray. Patient and reference DNA are attached to different coloured fluorescent molecules and added to a solid surface (now a silicon microchip) on which there are many DNA probes. Differences between the patient and reference DNA are quantified by computer software.
Different microarrays targeted towards different clinical conditions are used to tailor the approach, for example by using a panel of reference genes in which variants are known to be associated with intellectual disability. The development of CMA was particularly important to the field of intellectual disability, as the ability to identify a genetic cause increased dramatically. In 2010, the International Standards for Cytogenomic Arrays (ISCA) Consortium found that CMA had a diagnostic yield of 15–20%, compared with approximately 3% with Giemsa staining (Miller Reference Miller, Adam and Aradhya2010).
Although targeting genetic testing to certain clinical conditions by using specific gene panels in this way is more cost-effective, the downside is that only genes known to be associated with that condition are analysed and the genes may vary depending on which array is being used. It is therefore possible that variants in other areas of the genome may account for the clinical presentation.
Next-generation sequencing
Next-generation sequencing is defined as technology allowing one to determine in a single experiment the sequence of DNA molecule(s) with total size significantly larger than 1 million base pairs (Płoski Reference Płoski, Demkow and Płoski2016). This is achieved by sophisticated automated pipelines with the ability to run ‘massively parallel sequencing’ in which huge numbers of DNA fragments are examined in the same reaction. Analysis of how they overlap follows and therefore the overall sequence can be pieced together. There are various ways of exploring the genome for potential causes of disease using next-generation sequencing, including whole exome sequencing and whole genome sequencing.
Exons are the protein-coding portions of DNA and although they only make up around 1–1.5% of the DNA genetic code in humans, they harbour around 85% of the variants that cause single-gene (monogenic) disorders (Choi Reference Choi, Scholl and Ji2009; Płoski Reference Płoski, Demkow and Płoski2016). Therefore, whole exome sequencing (DNA sequencing that targets only the exons) is much more cost- and time-efficient than examining the whole genome, but it still yields good results in comparison to CMA or whole genome sequencing (Clark Reference Clark, Stark, Farnaes and Tan2018). However, this technique will not pick up trinucleotide repeat conditions (e.g. fragile-X syndrome) and additional tests will be warranted if these conditions are to be investigated.
Whole genome sequencing examines all non-coding portions of the DNA in addition to the exons. The importance of non-coding regions is increasingly recognised and the burgeoning use of whole genome sequencing can only serve to further our understanding of their function. However, examination of the whole genome is not yet routinely used clinically as it is costly, not only in terms of the laboratory sequencing, but because of the huge amounts of data generated. There must be sufficient capacity to store the data and computational power to analyse it. Moreover, the complexity of the identified variants and the time needed to interpret their potential pathogenicity are significantly more than with other techniques. There is little doubt though that these challenges will gradually be overcome to make whole genome sequencing more affordable and accessible in the future.
The Human Genome Project
A momentous achievement in genetics was the draft sequencing of the human genome in 2001 (International Human Genome Sequencing Consortium 2001; Venter Reference Venter, Adams and Myers2001), as it provided the first complete human DNA reference sequence. The concept was first mooted at a meeting in 1984. Discussions on its feasibility, given the available technology, began more formally in 1986, by which time the polymerase chain reaction was in use (Mullis Reference Mullis, Faloona and Scharf1986). It made the project significantly more achievable, as it abolished the need to grow millions of human cells to gather enough DNA to analyse the sequence.
The Human Genome Project was formally launched in 1990 and began the slow and laborious process of piecing together the human genetic sequence by hundreds of scientists in an international collaborative project. One scientist, Craig Venter, left the Human Genome Sequencing Consortium and, via a private company, set about separate efforts to sequence the human genome. His strategy was to use ‘shotgun sequencing’, which involves breaking up the DNA into millions of fragments and piecing together overlapping fragments to read the genetic sequence. The downside of shotgun sequencing is that there is potential for gaps if fragments do not overlap. The Consortium took a more systematic approach, working carefully through the DNA base by base. During this time, several animal genomes were also sequenced, including worms, flies and mice. This revealed for the first time that there was considerable genetic similarity between species, something that is now utilised by scientists the world over to study different species to model illnesses and disorders affecting people.
The publishing of the human genome was felt to hold great promise for elucidating the genetic basis of major mental illnesses (Cowan Reference Cowan, Kopnisky and Hyman2002). Having the genome sequence allowed researchers to start identifying SNPs and also, in time, surveying the whole genome for associations with psychiatric disorders (Kelsoe Reference Kelsoe2004).
Clinical interpretation of genetic testing results
As no human genome is identical, deciding whether a variant is deleterious is complex. Work is continuing to ensure that catalogues of normal genetic variation accurately represent human diversity, as there is some concern that there is no global catalogue of genetic variation and there is potential for racial and ethnic differences from the reference sequence too (Manrai Reference Manrai, Funke and Rehm2016).
Hence, since the advent of CMA, various guidelines have been drawn up to try to standardise practice. They have come to a consensus that identified DNA variants should be classified as ‘pathogenic’, ‘likely pathogenic’, ‘uncertain significance’, ‘likely benign’ or ‘benign’. Evidence supporting the allocation to each of the above criteria is reviewed using knowledge of the following (Richards Reference Richards, Aziz and Bale2015; Hoffman-Andrews Reference Hoffman-Andrews2017; Ellard Reference Ellard, Baple and Callaway2020):
• the likely structural effect of a variant on the gene and therefore the protein it codes for – there are various computerised algorithms to model this
• the likely disease mechanism, for example the likely physiological effect of a change in a protein's structure secondary to a genetic variant: sometimes this can result in a gain of protein function (a new function) or loss of function, both of which might be deleterious
• whether the variant has been previously identified in individuals with the condition of interest
• clinical history and examination findings and detail about the patient's family history that are consistent with the condition of interest
• whether the particular variant or one very similar has been published in the medical and scientific literature
• information in large databases that record genetic variants.
Variant databases include different cohorts, for example the general population or a disease cohort. They can also vary in factors such as the age of people involved and the numbers from one family included, adding to the complexity of comparing variants against them (Richards Reference Richards, Aziz and Bale2015). The Genome Aggregation Database (gnomAD – gnomad.broadinstitute.org) is a widely used database of the general population. Hence, with certain caveats, if a variant is found in this database it is not likely to be pathological as it is carried by unaffected persons. Databases such as the Database of Genomic Variation and Phenotype in Humans using Ensembl Resources (DECIPHER – decipher.sanger.ac.uk) and ClinVar (www.ncbi.nlm.nih.gov/clinvar) are repositories for variants found in affected individuals. As per the classification system above, they do still contain ‘variants of uncertain significance’, ‘likely benign’ and ‘benign’ variants, because a definitive genetic diagnosis cannot be made in all affected individuals. Clinical laboratories can not only cross-check variants they identify, but also contribute to the available evidence by uploading information to these types of database.
To optimise the understanding of the phenotype (clinical presentation) of variants, a ‘language’ called the Human Phenotype Ontology is used (Köhler Reference Köhler, Doelken and Mungall2014). This consists of brief and standardised terms to allow for comparison of each patient's signs, symptoms and diagnoses while reducing the likelihood of identification of individuals included in the database. Psychiatric terms used include ‘psychomotor retardation’, ‘hallucinations’ and ‘suicidal ideation’. This cross-referencing against other people's variants underlines the importance of including as much information as possible about a patient's presentation when requesting genetic testing.
Even once all the above factors have been considered and databases searched, many variants do not slot into one of the diagnostic classification categories because their likelihood of being pathogenic is unknown. These are called ‘variants of uncertain clinical significance’. In some cases their impact can be further assessed with tests such as the following, and these are known as actionable variants:
• co-segregation tests – testing of family members to see whether the variant is only carried by people who are affected by the condition of interest
• clinical investigations such as assays to determine enzyme activity or brain imaging (these are less useful in psychiatry than in other medical disciplines)
• parental testing to see whether the variant has been inherited or is new in that individual (‘de novo’).
For many variants, it is not possible to further assess their impact and so guidelines recommend that these changes are not used or communicated in clinical diagnosis, to prevent confusion or misinterpretation of their importance or relevance by clinicians and patients. However, with information sharing via databases and scientific literature, the pathogenicity of such variants may be reclassified in the future as the evidence of their link to clinical presentations evolves. This is one reason why retention of DNA samples in genetic services is commonplace – another is so they can be re-tested down the line with more sensitive techniques.
Discussing clinical genetic test results with patients
Clinicians are increasingly being presented with genetic test results, mostly at present from people with single-gene disorders; this can be daunting. However, it is unreasonable for anyone to expect a psychiatrist (or a geneticist for that matter) to be familiar with every genetic condition relevant to psychiatry. The patient or family may in fact be able to provide information if they have already researched it themselves!
For a clinical psychiatrist, the most important categories to be aware of in a genetic testing report are probably:
• the gene name
• inheritance – as this may have direct implications for the family
• classification of pathogenicity
• implications for treatment (less common in psychiatry than in other disciplines to date)
• recommended action – this is not likely to be something the psychiatrist personally needs to take forward
• limitations of the test – a negative test can mean that no relevant variant has been found, but it can also mean simply that the laboratory or the type of testing used did not identify a relevant variant owing to the sensitivity of the methods used
• recurrence risk.
It is important to note that even de novo variants can recur in rare circumstances. This is due to ‘germline mosaicism’, a condition in which the DNA of some of the germline cells (ova or sperm) carry mutations whereas others do not. These variants are only present in the germline cells, hence testing of DNA from blood or saliva will not identify the mutations and so they will appear to be new (de novo) in the affected offspring. Germline mutations cannot be tested for and so only become apparent if an individual with a genetic diagnosis has a sibling with the same condition but the parents are neither affected by nor carriers of the relevant DNA variant.
Table 1 gives more explanation about the nomenclature that may be in genetic reports. There are various internet resources that can then be searched to better understand diagnosed conditions. Good starting places are databases such as DECIPHER and ClinVar, which have a lot of detail on each known variant. Others to be aware of are the Online Mendelian Inheritance in Man database (OMIM – omim.org) and GeneReviews, an inherited conditions resource in journal-style format (www.ncbi.nlm.nih.gov/books/NBK1116). Resources such as the US Library of Medicine's Genetics Home Reference (which is now part of MedlinePlus – medlineplus.gov/genetics) distil the information so it is more accessible to clinicians.
TABLE 1 Glossary of some commonly used genetic test result notation
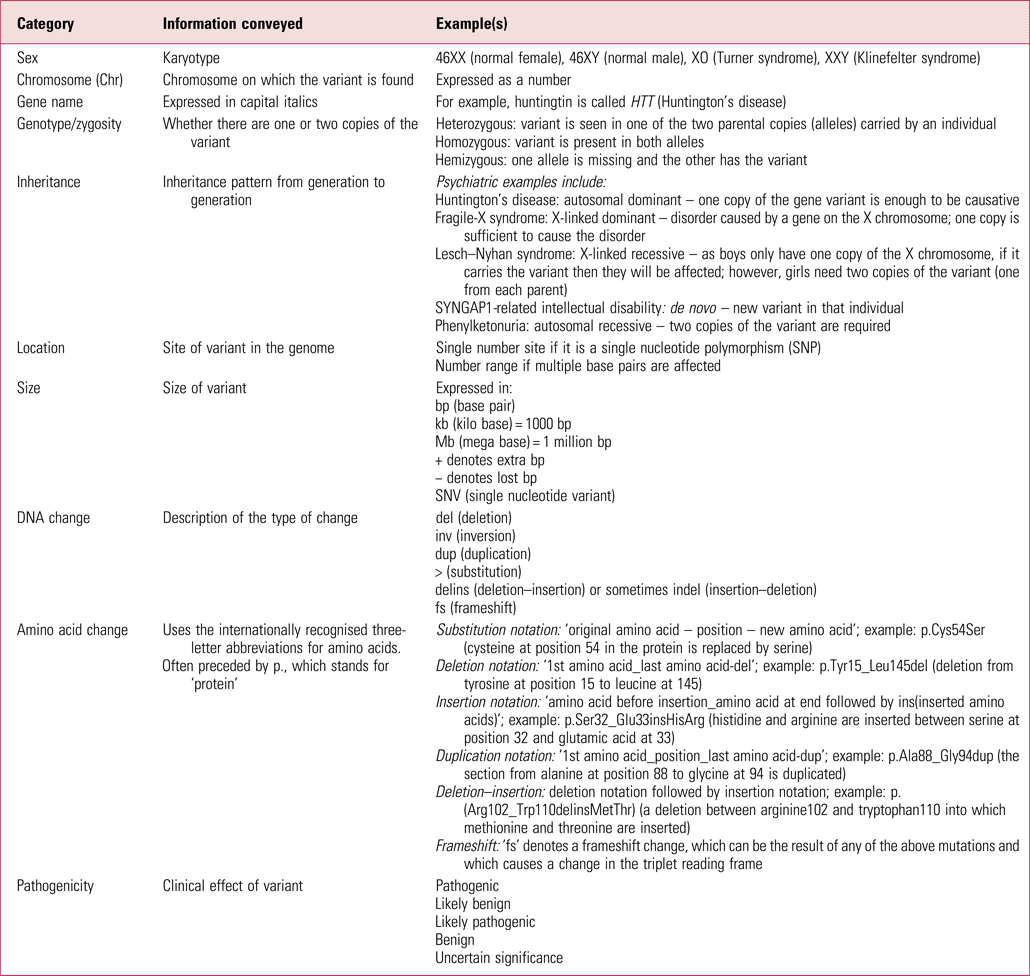
For many people, having a genetic diagnosis that explains or partially explains their condition is of psychological benefit. Therefore, patients may not look for anything more from their psychiatrist than to recognise this. However, other people may be keen to have more information, sources of which include:
• clinical genetics services
• support groups specific to that condition (typically easily found on the internet)
• more general support groups such as Rare Disease UK (www.raredisease.org.uk) or Unique (www.rarechromo.org)
• a literature search for publications about the condition of interest: often these include clinically relevant information and a search also facilitates the identification of clinicians and researchers with an interest in that condition, who may be able to provide expert advice.
Pharmacogenetic testing in psychiatry
Aside from testing for genetic variants that directly link to a mental disorder, research into pharmacogenetics will have increasing relevance to psychiatry in coming years. Associations have already been made between variants of cytochrome P450 (CYP450) liver enzymes and human leucocyte antigen (HLA) gene variants and metabolism of psychotropic medication. For example, the US Food and Drug Administration (FDA) recommends that patients of Asian descent are screened for variants that put them at higher risk of Stevens–Johnson syndrome when taking carbamazepine. The FDA also recommends that people with certain variants in the CYP450 liver enzymes that reduce the rate of drug metabolism should not take more than 20 mg of citalopram daily (Nurnberger Reference Nurnberger, Austin and Berrettine2018). The Individualized Medicine: Pharmacogenetic Assessment & Clinical Treatment (IMPACT) study in Canada has recruited thousands of people with the aim of identifying response to various psychiatric medications and experience of side-effects across various mental illnesses, such as generalised anxiety disorder, depression, schizophrenia and bipolar disorder (Herbert Reference Herbert, Neves-Pereira and Baidya2018).
More work is needed so a consensus can be reached regarding types of pharmacogenetic tests to use and their implications before they become relevant in daily psychiatric practice.
Understanding polygenic psychiatric conditions
It has long been recognised that most major psychiatric disorders are highly heritable and polygenic, meaning that multiple genetic variants contribute to the disorder. In the past, twin and adoption studies were the mainstay of research into heritability and were based on the assumption that, if a disorder is caused by genetic factors, then individuals who are genetically related should share similar risks for the disorder. However, the ability of such studies to identify specific genetic risk factors was extremely limited.
Some of the relevant genetic variants that can now be identified are of small effect and found in more than 1% of the population, others are rare (found in less than 1% of the population) and of large effect. Unlike in some monogenic disorders, an individual does not directly inherit a polygenic disorder: they are at risk of developing it and the degree of risk is dependent on the particular mix of variants in their DNA. To further complicate matters, polygenic disorders (and single-gene disorders too) are modulated by environmental effects such as age, gender, diet, season, time of day, drug and medication use, and exposure to a wide variety of environmental stimuli that can modify the expression of genes: a phenomenon known as ‘epigenetics’ (Kular Reference Kular and Kular2018; Greener Reference Greener2019).
Genome-wide association studies (GWAS) and polygenic risk scores (PRS) are now widely used to identify the location (loci) of genetic risk variants in major mental illnesses. In GWAS, cohorts of patients are compared with unaffected persons and usually tens or hundreds of thousands of cases and controls are needed to identify variants of relevance. This is because the effect of each individual variant can be so small that it takes huge numbers to give sufficient statistical power to identify them. Hence, various large consortia have been formed within psychiatric research to pool data-sets, including the Psychiatric Genomics Consortium (www.med.unc.edu/pgc) and the PsychENCODE Consortium (www.nimhgenetics.org/resources/psychencode). A statistically significant finding in a GWAS means that, at a population level, persons with the condition of interest are more likely to carry the variant(s) than controls.
There have now been many GWAS studies identifying variants at particular genetic loci that increase the risk of a variety of mental disorders, some of which are identified in multiple disorders (Wu Reference Wu, Cao and Baranova2020). Most of the data identifies SNPs that individually confer negligible risk of developing a disorder, but in combination can increase the risk. However, GWAS data have also helped to reveal the role of some rare copy number variants (CNVs) of large effect in psychiatric disorders, particularly in the case of schizophrenia (Marshall Reference Marshall, Howrigan and Merico2017), but in other conditions too (Cross-Disorder Group of the Psychiatric Genomics Consortium 2019). GWAS results can be presented in a Manhattan plot, which charts each individual variant and its chromosomal location (Fig. 6). Many of the genes affected by CNVs code for proteins known to be involved in brain functions such as neurogenesis, glutamate pathways and synaptic transmission. Knowing this not only supports some of the current theories of psychiatric pathophysiology, but also helps identify potential targets for new pharmacotherapies that will in time be available in the clinic.

FIG 6 Schematic diagram of a Manhattan plot. Each dot represents an identified single nucleotide polymorphism or copy number variant arranged by chromosome. Those above the horizontal dashed line have surpassed the stringent statistical analysis to identify them as being associated with the condition in question.
Polygenic risk scores use the results from GWAS to quantify the relative risk of having a condition on a population basis by calculating the additive effect of risk variants. The details of PRS are complex and beyond the scope of this article, but some key points are as follows. PRS are usually based on SNP data and so the analysis is complex, given the millions of SNPs in a genome, most of which do not relate to the condition of interest. Nevertheless, these scores are showing significant promise in the prediction of risk in neuropsychiatric conditions, as data collated by the Polygenic Score (PGS) Catalog shows (pgscatalog.org/). This is an open database of published PRS developed by a collaboration between the University of Cambridge, Health Data Research UK, the Baker Heart and Diabetes Institute and the European Molecular Biology Laboratory's European Bioinformatics Institute (EMBL-EBI). It provides detailed information about how each score was calculated and its predictive value. This is vital as there is growing recognition that the planned utility of the test (e.g. public, personal, etc.) and how it is devised and analysed is critical in evaluating its results (PHG Foundation 2021). To date, PRS in Alzheimer's disease, schizophrenia and depressive disorders are the most abundant psychiatric conditions in the PGS Catalog. PRS have also revealed that the interplay between polygenic conditions and monogenic disorders is perhaps more complex than previously thought, with evidence that in developmental disorders, the clinical presentation of an individual with a monogenic disorder can be influenced by having parents with a higher PRS for neuropsychiatric disorders (Niemi Reference Niemi, Martin and Rice2018).
At present, PRS are informative when comparing cohorts of people, for example those with a condition and those without, but their clinical utility for an individual continues to be debated. They are also limited by:
• any unmodelled environmental factors (Lewis Reference Lewis and Vassos2020) – this is a considerable concern in psychiatry, given the very significant influence of environment and life events
• the population from which the data were derived – currently almost always people of European ancestry, although this is gradually being addressed.
It is hoped that PRS will in time enable categorisation of patients on the basis of the genetic cause of their symptoms, not just on their clinical presentation (Zheutlin Reference Zheutlin and Ross2018). They could also lead to stratification of patient groups, for example by prognosis, treatment resistance, optimal mood stabiliser selection, suicidality and the risk of physical health conditions that can be exacerbated or precipitated by psychotropic medication (Fullerton Reference Fullerton and Nurnberger2019). There is no doubt that PRS will continue to evolve and there will be progression in their clinical utility in coming years.
Direct-to-consumer genetic testing kits
In addition to testing from clinical genetics services, it is important for psychiatrists in the clinic to be aware of direct-to-consumer genetic testing kits, which have become increasingly popular in recent years. These are used not just for health purposes, but also for things like ancestry analysis. One of the most well-known is 23andMe, but there are many on the market and it is increasingly likely that clinicians will be faced with the challenge of patients presenting them with their results. The main issues to be aware of are as follows (these are expanded on in: Wilde Reference Wilde, Meiser and Mitchell2010; Horton Reference Horton, Crawford and Freeman2019; Dinulos Reference Dinulos and Vallee2020; Majumder Reference Majumder, Guerrini and McGuire2021):
• these kits vary significantly in the methods they use and their accuracy, so there is a real risk of false-positive or false-negative results
• companies selling these kits often do not offer access to a genetic counsellor, but even when they do, many people choose not to contact them
• there are concerns about privacy of the data and potential discrimination by insurance companies or employers on the basis of the results
• there are various third-party companies to which people can upload the raw data for further analysis; these companies are unregulated and there is often a lack of transparency about the way they come to their conclusions or their clinical relevance
• there is controversy about collaborations between some companies that make such kits and pharmaceutical companies.
Hence, although these kits can be popular and there is some suggestion that they might help motivate people to change relevant lifestyle factors, they risk causing significant psychological harm to patients who do not understand the implications of the results and have not sought advice from a reputable genetic counsellor or medical practitioner. Many of these tests are now reporting polygenic risk scores related to mental health conditions despite the complexity of interpreting PRS data. The potential negative impact is tangible, as a study of simulated genetic testing in people with mild depression showed that a result indicating a putative higher risk of developing a depressive illness became a self-fulfilling prophecy, as the participants felt less able to cope with their symptoms (Lebowitz Reference Lebowitz and Ahn2018).
Therefore, if faced with questions about or results from direct-to-consumer testing in the clinic, such limitations and concerns need to be openly discussed with patients and consideration given as to whether there is a clinical indication for referral to the local National Health Service (NHS) genetics service. In many cases there will not be, particularly when the results are a PRS and careful explanation of this will be required.
Conclusions
Our understanding of genetics has developed apace over recent decades and, although psychiatry may lag somewhat behind other medical disciplines, the utility of genetic assessments is increasing. To date, their role in our specialty has largely related to rare individual variants with very significant effects on clinical presentation. However, there is great promise that, as scientific understanding and computational power increase, polygenic information will be increasingly useful in psychiatry. The general public, and therefore patients, are becoming more aware of aspects of genetics and the potential relevance to mental disorder. They also have easy access to testing through direct-to-consumer kits and hence psychiatrists need to have an understanding of genetic testing, its clinical relevance and its potential risks and benefits.
Acknowledgements
I am grateful to Dr Morad Ansari, MRC Human Genetics Unit, Institute of Genetics and Molecular Medicine, University of Edinburgh, for his assistance with the technical detail of the manuscript. I also thank Professor Andrew McIntosh, Professor of Biological Psychiatry, and Dr Andrew Stanfield, Senior Clinical Research Fellow, both at the University of Edinburgh, for reviewing the manuscript.
Author contributions
L.A.M.M. conceived of and prepared the manuscript.
Funding
L.A.M.M. is currently employed and funded by the Patrick Wild Centre, University of Edinburgh, to carry out clinical research into genetic causes of intellectual disability in adults.
Declaration of interest
None.
MCQs
Select the single best option for each question stem
1 The genetics of which of the following disorders was not discovered with karyotyping?
a Down syndrome
b Huntington's disease
c Fragile-X syndrome
d Klinefelter syndrome
e Turner syndrome.
2 Which of the following techniques is least useful for identifying single-gene polymorphisms (SNPs)?
a whole genome sequencing
b fluorescence in situ hybridisation
c Sanger sequencing
d chromosomal microarray
e whole exome sequencing.
3 Which of the following is not one of the recommended classification terms for genetic variants?
a pathogenic
b benign
c probably pathogenic
d likely benign
e uncertain significance.
4 Which of the following is a good source of clinical information about disease-causing genetic variants?
a gnomAD
b DECIPHER
c HPO
d PolyPhen-2
e FISH.
5 GWAS data have been particularly fruitful in identifying copy number variants (CNVs) that significantly raise the risk of which of the following?
a attention-deficit hyperactivity disorder
b panic disorder
c generalised anxiety disorder
d Tourette syndrome
e schizophrenia.
MCQ answers
1 b 2 b 3 c 4 b 5 e








eLetters
No eLetters have been published for this article.