


Implications
Reference population size is a key factor affecting accuracy of genomic prediction. In dairy cattle, reference population usually consisted of progeny-tested bulls. Limited number of progeny-tested bulls is a limitation to the accuracy of genomic prediction in numerically small breeds. The results from this study indicate that sharing reference data and including cows in reference population can greatly increase reliability of genomic prediction for the populations where the size of domestic bull reference population is small.
Introduction
Genomic selection has been widely implemented in dairy cattle breeding. Its success depends on accurate genomic predictions. A key factor affecting accuracy of genomic prediction is the amount of information from reference population (Daetwyler et al., Reference Daetwyler, Villanueva and Woolliams2008; Goddard, Reference Goddard2009; Goddard and Hayes, Reference Goddard and Hayes2009). In dairy cattle, reference populations are usually composed of progeny-tested bulls, since they have reliable phenotypic information from a large group of daughters. However, the number of progeny-tested bulls is limited for numerically small dairy cattle populations, such as Danish Jersey.
Currently, there are about 60 000 cows in the Danish Jersey population. Until now, only about 1200 to 1400 Danish progeny-tested bulls (depending on trait) are available to be used as reference bulls. Due to the small reference population, accuracy of genomic prediction in the Danish Jersey is much lower than in the Danish Holstein and Red Cattle populations (Su et al., Reference Su, Brondum, Ma, Guldbrandtsen, Aamand and Lund2011 and Reference Su, Guldbrandtsen, Aamand, Stranden and Lund2012c; Gao et al., Reference Gao, Christensen, Madsen, Nielsen, Zhang, Lund and Su2012; Thomasen et al., Reference Thomasen, Guldbrandtsen, Su, Brondum and Lund2012). Therefore, it is important to find efficient approaches to improve accuracy of genomic prediction in this population.
Several approaches have been proposed to improve accuracy of genomic prediction for small dairy cattle populations (Lund et al., Reference Lund, Su, Janss, Guldbrandtsen and Brondurn2014). An efficient approach is to use a joint reference population that combines the reference data from different populations. A large benefit from this approach has been reported in genomic prediction for North American Holstein populations (Schenkel et al., Reference Schenkel, Sargolzaei, Kistemaker, Jansen, Sullivan, Van Doormaal, VanRaden and Wiggans2009; Muir et al., Reference Mäntysaari, Liu and VanRaden2010), European Holstein populations (Lund et al., Reference Lund, Su, Nielsen and Aamand2011), Chinese Holstein population (Zhou et al., Reference Zhou, Heringstad, Su, Guldbrandtsen, Meuwissen, Svendsen, Grove, Nielsen and Lund2013) and Brown Swiss populations (VanRaden et al., Reference VanRaden, Olson, Null, Sargolzaei, Winters and van Kaam2012). However, since accuracy of genomic prediction depends on the relationship between candidates and reference animals (Lund et al., Reference Lund, de Ross, de Vries, Druet, Ducrocq, Fritz, Guillaume, Guldbrandtsen, Liu, Reents, Schrooten, Seefried and Su2009; Habier et al., Reference Habier, Tetens, Seefried, Lichtner and Thaller2010; Clark et al., Reference Clark, Hickey, Daetwyler and van der Werf2012; Pszczola et al., Reference Pszczola, Strabel, Mulder and Calus2012), it requires that the reference populations are close enough to link with the target populations. Another approach is to genotype cows and include them in the reference population. Cow reference populations have been used to predict genomic breeding values in populations where only few progeny-tested bulls have reliable phenotypic information (Ding et al., Reference Ding, Zhang, Li, Wang, Wu, Sun, Yu, Liu, Wang, Zhang, Zhang, Zhang and Zhang2013; Li et al., Reference Li, Wang, Huang, Li, Zhang and Ding2014). A more common approach is to include genotyped cows in a bull reference population. Increasing the accuracy of genomic prediction by adding cows to a progeny-tested bull reference population has been reported in previous studies (Wiggans et al., Reference Wiggans, Cooper and VanRaden2010; Calus et al., Reference Calus, de Haas and Veerkamp2013; Cooper et al., Reference Cooper, Wiggans and VanRaden2015). Although phenotypic information is much less accurate for cows than progeny-tested bulls, the increased information may be considerable because a large number of cows are available to be used as reference animals. According to previous studies (Daetwyler et al., Reference Daetwyler, Villanueva and Woolliams2008; Goddard and Hayes, Reference Goddard and Hayes2009), the gain from additional information depends on the size of original reference population.
Both approaches can be implemented to improve accuracy of genomic prediction for Danish Jersey. On one hand, US Jersey has been contributed to the Danish Jersey population for a long time, especially during the period from 1985 to 1995. It is expected that a joint reference population combining Danish and US Jersey bulls would increase accuracy of genomic prediction considerably for both Danish and US Jersey populations. Therefore, in 2013, marker data for Danish and US Jersey bulls were exchanged to create a joint reference population for genomic prediction of Jersey cattle. On another hand, adding cows to the reference population of Danish Jersey may increase accuracy of genomic prediction considerably, because the current progeny-tested bull reference population is small. Taking this into consideration, a number of females have been genotyped since 2013 with the purpose of increasing the size of the reference population.
The objective of this study was to investigate the improvement of genomic predictions in numerically small breeds by sharing reference data and including cows in reference population. Thus, this study accessed the reliability and unbiasedness of genomic breeding values predicted using a Danish Jersey bull reference population, a joint Danish–US Jersey bull reference population, a reference population consisting of Danish Jersey bulls and cows, and a reference population consisting of Danish Jersey bulls and cows as well as US Jersey bulls. The validation was carried out on Danish Jersey bulls and cows, respectively.
Material and methods
Data
The data in the analysis included 1369 Danish Jersey bulls, 1160 US Jersey bulls and 9419 Danish Jersey cows; 98.4% of Danish bulls were born from 1988 to 2010, 99.6% of US bulls from 2000 to 2009, and 95.4% of Danish cows from 2010 to 2013. The cows were from herds with good data registration. Danish Jersey bulls were genotyped with the Illumina Bovine SNP50 chip (54 609 single nucleotide polymorphism (SNP, Illumina, Inc.)). US Jersey bulls were genotyped either with the standard Illumina Bovine SNP50 chip or with the GeneSeek Genomic Profiler chip HD (near 78 000 SNP, GeneSeek, Neogen Corporation). Danish Jersey cows were genotyped either with the standard BovineLD BeadChip (6909 SNP, Illumina, Inc.) or with a customized Illumina BovineLD which included the SNP in the standard BovineLD BeadChip and near 5000 user-selected SNP, and a few cows were genotyped with Illumina Bovine SNP50 chip. The marker data of different chips were imputed to Bovine SNP50 chip using FIMPUTE (Sargolzaei et al., Reference Sargolzaei, Chesnais and Schenkel2014). The markers which are not in the Bovine SNP50 chip were excluded. After removing markers with allele frequency <1%, 39 937 autosomal markers were used to predict genomic breeding values.
De-regressed proofs (DRP) derived from the published estimated breeding values (EBV) of the Interbull 2014-12 evaluation and Nordic 2015-02 evaluation were used as response variables in the analysis. Two set of DRP were obtained. One (DRPB) was derived from a de-regression procedure in which EBV of genotyped Danish bulls and US bulls were used. The other (DRPBC) was derived based on EBV of all genotyped animals including cows. The traits with reliable DRP for both Danish and US Bulls were milk, fat, protein, body conformation, fertility, longevity, mastitis and udder conformation. The traits with DRP available for Danish bulls, US Bulls and genotyped Danish cows were milk, fat, protein, body conformation, mastitis and udder conformation. Thus, eight traits were analyzed when using DRPB, and six traits when using DRPBC.
Validation of genomic predictions
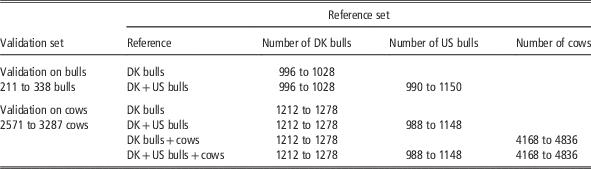
Genomic estimated breeding values (GEBV) using different data sets were validated on bulls and cows, respectively. The bull validation set comprised Danish Jersey bulls born in the years from 2005 onwards, which accounted for about 25% of the Danish Jersey bulls. This validation set was suitable for comparing GEBV from Danish bull reference and the joint Danish and US reference populations. However, it is not appropriate to use this validation set to assess genomic predictions based on the reference population including cows, since most cows in the reference population were born during 2010-12 and were the sibs or daughters of the bulls in the bull validation set. Therefore, a cow validation set was created in the following way: (1) genotyped cows born in the period from 1 July 2012 onwards were extracted; (2) these cows and their paternal female half-sibs born after 2007 were defined as the cow validation set but excluding the half-sib families with size >500 (these families were kept in the reference data in order to avoid a large reduction of reference population size). This resulted in about 3000 validation cows from 87 paternal half-sib families. When using the cow validation set, validation cows’ maternal female and male half-sibs born after 2007 were excluded from reference population. In addition, the progenies of these animals (validation cows and the sibs) were also removed from the reference population. The validation and reference populations defined this way were in order to reduce the relationship between validation and reference animals and to achieve consistency with a real life selection scenario as much as possible.
For the bull validation set, the reference populations were: (1) Danish bull reference population, and (2) the Joint Danish and US bull reference population. For the cow validation set, four reference populations were used for genomic prediction: (1) Danish bulls, (2) Danish bulls and US bulls, (3) Danish bulls and cows and (4) Danish bulls, US bulls and Danish cows. The validation scenarios, the number of animals in the reference data and the validation data are shown in Table 1. Genomic predictions using different reference data sets were evaluated by comparing GEBV and DRP for animals in the validation data. Reliability of GEBV was measured as the squared correlation between GEBV and DRP divided by the reliability of DRP (Su et al., Reference Su, Christensen, Ostersen, Henryon and Lund2012b). Unbiasedness of genomic prediction was assessed by regression of DRP on GEBV (Su et al., Reference Su, Gao and Lund2012a).
Table 1 Number of animals (dependent on traits) in different validation sets and reference sets
Prediction model
Breeding values were predicted using a genomic best linear unbiased model (GBLUP), based on different reference populations. The GBLUP model is
 $${\bf y}={\bf 1}\mu {\plus}{\bf Z}_{g} {\bf g}{\plus}{\bf e}$$
$${\bf y}={\bf 1}\mu {\plus}{\bf Z}_{g} {\bf g}{\plus}{\bf e}$$
where y is the vector of DRP, µ the overall mean, 1 a vector of ones, g the vector of additive genomic effects, Z g the incidence matrix linking g to y and e the vector of residuals.
It is assumed that g~N(0, G A σ 2 g ), and e~N(0, D σ 2 e ), where G A is a genomic relationship matrix combining marker-based relationship matrix and pedigree-based relationship matrix, σ 2 g is the additive genetic variance, D is a diagonal matrix and σ 2 e is the residual variance. Matrix D has diagonal elements d ii =(1−r 2 DRP)/r 2 DRP to account for heterogeneous residual variances due to different reliabilities of DRP (r 2 DRP).
The matrix G A was constructed in the following steps. First, an original genomic relationship matrix (G) was built according to VanRaden (Reference VanRaden2008) and Hayes et al. (Reference Hayes, Visscher and Goddard2009),
 $${\bf G}={\bf MM'}\!/\!\mathop{\sum}{2p_{i} q_{i} } $$
$${\bf G}={\bf MM'}\!/\!\mathop{\sum}{2p_{i} q_{i} } $$
where elements in column i of M are 0−2p i , 1−2p i and 2−2p i for genotypes A 1 A 1, A 1 A 2 and A 2 A 2, respectively, and q i is allele frequency of A 1 and p i is allele frequency of A 2. In this study, allele frequencies were calculated from the current marker data. G matrix used in each analysis was built using the marker data of the animals in the corresponding reference and validation data. Thus, the marker data used in construction of G matrix differed among different data sets. Second, the G matrix was adjusted to be on the same scale as the pedigree-based relationship matrix according to Christensen et al. (Reference Christensen, Madsen, Nielsen, Ostersen and Su2012). Thus, the adjusted matrix (G c ) was
 $${\bf G}_{{\bf c}} =\alpha {\plus}{\bf G}\beta $$
$${\bf G}_{{\bf c}} =\alpha {\plus}{\bf G}\beta $$
The parameter α and β were derived from the following equations,
 $$\eqalignno{ & {\rm Average}\ {\rm diagonal (}{\bf A}{\rm )}=\alpha {\plus}{\rm Average}\ {\rm diagonal}\,{\rm (}G{\rm )}\,\beta \cr & {\rm Average}\ {\rm off}{\hbox-}{\rm diagonal (}{\bf A}{\rm )}=\alpha {\plus}{\rm Average}\ {\rm off}{\hbox -}{\rm diagonal}\,{\rm (}G{\rm )}\,\beta $$
$$\eqalignno{ & {\rm Average}\ {\rm diagonal (}{\bf A}{\rm )}=\alpha {\plus}{\rm Average}\ {\rm diagonal}\,{\rm (}G{\rm )}\,\beta \cr & {\rm Average}\ {\rm off}{\hbox-}{\rm diagonal (}{\bf A}{\rm )}=\alpha {\plus}{\rm Average}\ {\rm off}{\hbox -}{\rm diagonal}\,{\rm (}G{\rm )}\,\beta $$
where A was the pedigree-based relationship matrix for the genotyped animals and was extracted from the relationship matrix built based on the whole pedigree. Furthermore, matrix G A was calculated as
 $${\bf G}_{{\bf A}} =\left( {{\rm 1}{\minus}\omega } \right){\bf G}_{{\bf c}} {\plus}\omega {\bf A}$$
$${\bf G}_{{\bf A}} =\left( {{\rm 1}{\minus}\omega } \right){\bf G}_{{\bf c}} {\plus}\omega {\bf A}$$
where ω was the relative weight on matrix A. In this study, ω=0.20 was chosen according to the previous studies in Nordic cattle populations (Su et al., Reference Su, Brondum, Ma, Guldbrandtsen, Aamand and Lund2011 and Reference Su, Guldbrandtsen, Aamand, Stranden and Lund2012c; Gao et al., Reference Gao, Christensen, Madsen, Nielsen, Zhang, Lund and Su2012). In this setting, the GBLUP model is equivalent to a GBLUP including a genomic effect and a residual polygenic effect accounting for 80% and 20% of total additive genetic variance, respectively.
Genomic predictions were performed using the DMU package (Madsen et al., Reference Madsen, Su, Labouriau and Christensen2010). Additive genetic and residual variances applied in Nordic routine genetic evaluation were used in this study to predict genomic breeding values.
Measurement of consistency in genome and genetic relationship between Danish and US Jersey populations
Gain in genomic prediction accuracy from a joint reference population depends on genetic similarity and relationship between the populations involved. In this study, the consistency of linkage disequilibrium, allele frequency and genetic relationship between Danish and US Jersey populations were investigated. The consistency of linkage disequilibrium was measured as the correlation of r values for adjacent marker pairs between the Danish Jersey bulls and US Jersey bulls, that is, Cor(r
LD(DK), r
LD(US)), where
 $r_{{{\rm LD}}} ={{f(AB){\minus}f(A)f(B)} \over {\sqrt {f(A)f(a)f(B)f(b)} }}$
for marker A (allele A and a) and marker B (allele B and b). The consistency of marker allele frequency was measured as the correlation of allele frequencies between the two populations. The genetic relationship coefficients between Danish and US Jersey bulls were calculated based on SNP markers. Following Clark et al. (Reference Clark, Hickey, Daetwyler and van der Werf2012), the maximum relationship and the mean of top 10 relationships of a Danish bull with the US bulls were used as measures of relationship between a Danish bull and the US bulls.
$r_{{{\rm LD}}} ={{f(AB){\minus}f(A)f(B)} \over {\sqrt {f(A)f(a)f(B)f(b)} }}$
for marker A (allele A and a) and marker B (allele B and b). The consistency of marker allele frequency was measured as the correlation of allele frequencies between the two populations. The genetic relationship coefficients between Danish and US Jersey bulls were calculated based on SNP markers. Following Clark et al. (Reference Clark, Hickey, Daetwyler and van der Werf2012), the maximum relationship and the mean of top 10 relationships of a Danish bull with the US bulls were used as measures of relationship between a Danish bull and the US bulls.
Results
Reliability of GEBV using Danish bull reference population
The reliabilities of GEBV using reference population comprising Danish bulls alone are shown in Table 2 for validation on bulls and in Table 3 for validation on cows. In validation on bulls, reliability of GEBV for the eight traits ranged from 0.151 (longevity) to 0.372 (milk yield) with an average of 0.262. In validation on cows, reliabilities of GEBV for the six traits ranged from 0.249 (fat) to 0.555 (mastitis) with an average of 0.394. Validation reliabilities on cows were higher than those on bulls for all the traits, except for protein in which reliability for bulls was slightly higher than that for cows. Averaged over the six traits common in the two validations, reliability in validation on cows was 11.7 percentage points higher than that in validation on bulls. There were at least two reasons for higher reliability in validation on cows than bulls. The first could be that the reference population for validation on cows was larger (>200 bulls) than that for validation on bulls. The second was that validation cows were a random sample, while validation bulls are a selected sample (selected on parent average) which would reduce the correlation between GEBV and DRP and thereby result in an underestimate of the true reliability.
Table 2 Validation reliability (%) of GEBV and regression coefficient of DRP on GEBV using Danish bull reference population (DK) and joint DK-US bull reference population (DKUS), based on validation on bulls
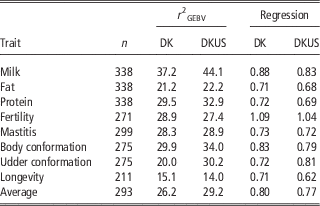
GEBV=genomic estimated breeding value; DRP=de-regressed proofs.
Table 3 Validation reliabilities (%) of GEBV using Danish bull reference population (DK), joint DK-US bull reference population (DKUS), Danish bull and cow reference population (DKCOW), DK-US bull and cow reference population (DKUSCOW), based on validation on cows
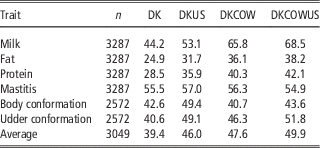
GEBV=genomic estimated breeding value.
As shown in Table 2, the regression coefficients of DRP on GEBV for validation bulls were considerably <1, except for fertility which was slightly >1. The regression coefficients were 1.09 for fertility and ranged from 0.71 to 0.88 for the other five traits. However, the regression coefficients for validation cows (Table 4) were in general slightly larger than 1, except for protein (0.93) and fat (0.80). The inconsistency in regression coefficients between the two validation sets could reflect difference in correlation coefficients between random sample (cows) and selected sample (bulls). Mäntysaari et al. (Reference Muir, Van Doormaal and Kistemaker2010) pointed out that selection of test bulls will reduce the regression coefficient and reliability of GEBV.
Table 4 Regression coefficients of GEBV on DRP for genomic prediction using Danish bull reference population (DK), joint DK-US bull reference population (DKUS), Danish bull and cow reference population (DKCOW), DK-US bull and cow reference population (DKUSCOW), based on validation on cows
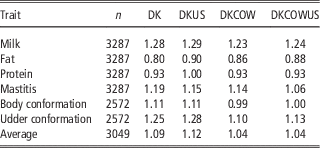
GEBV=genomic estimated breeding value; DRP=de-regressed proofs.
Gain in prediction reliability from including US bulls in reference population
Including US Jersey bulls in the reference population resulted in a large increase in accuracy of genomic prediction. In validation on bulls (Table 2), six of the eight traits benefitted from the joint reference population. The gains by including US Jersey bulls in the reference population ranged from 1.0 percentage points for fat to 10.5 percentage points for udder conformation. However, there was a loss in reliability by 1.5 percentage points for fertility and 1.1 percentage points for longevity. A possible reason for the loss of reliability could be that the definitions of the two traits were not the same in Danish and US Jersey populations. Averaged over all eight traits, reliability of GEBV using the joint reference population was 3.0 percentage points higher than the reliability of GEBV using the Danish bull reference population alone.
The gain by including about 1100 US Jersey bulls in the reference population was more pronounced in validation on cows. As shown in Table 3, reliability of GEBV using the joint DK-US bull reference population was higher than using the Danish bull reference population alone for all six traits. The gain was >6.0 percentage points, except for mastitis which gained 1.5 percentage points, leading to an average gain of 6.6 percentage points. For these six traits the average gain in validation reliability on bulls was 4.4 percentage points. On the other hand, the joint reference population did not reduce bias of GEBV. The regression coefficients of DRP on GEBV were in general similar to those when using the Danish bull reference population, that is, generally <1 in validation on bulls (Table 2) and slightly >1 in validation on cows.
Gain in prediction reliability from including cows in the reference population.
Including about 4800 cows in the reference population led to a large increase in reliability of GEBV (Table 3). Compared with genomic predictions using the Danish bull reference population, using the reference population comprising Danish bulls and cows increased the reliabilities of GEBV by >11 percentage points for the three production traits and by 5.7 percentage points for udder conformation. However, there was only a slight increase of reliability for mastitis (0.8 percentage points) and a slight decrease for body conformation (−1.9 percentage points). Averaged over the six traits in validation on cows, the increase of reliabilities was 8.2 percentage points.
When the reference population already included US bulls, the further gain from including cows became relatively smaller. There was still large gain for the three production traits but no gain for mastitis and body conformation. The gain averaged over the six traits was 3.9 percentage points. Similarly, when the reference population already included cows and Danish bulls, the further gain from including the US bulls was reduced also, leading to an average gain of 2.3 percentage points. Consequently the gain from adding both cows and US bulls, averaged over the six traits, was 10.5 percentage points.
Including cows in the reference population had a small influence on the unbiasedness of GEBV. The regression coefficients of DRP on GEBV from the reference population including cows ranged from 0.80 for fat to 1.28 for milk, while the regression coefficients from the Danish bull reference population were between 0.86 for fat and 1.23 for milk. For all six traits, the regression coefficients for the former scenario deviated slightly less from 1, compared to those for the latter scenario. On the average, the absolute deviation from 1 was 0.115 when using the reference population including cows, while 0.183 when using the Danish bull reference population. These results indicated that the reference population including cows led to a slight improvement in unbiasedness of genomic predictions.
Consistency in genome and genetic relationship between Danish and US Jersey populations
As shown in Table 5, there was a high consistency between Danish and US Jersey populations. The two populations had a similar degree of linkage disequilibrium, a high correlation in linkage disequilibrium up to 0.94, and a high correlation of allele frequency up to 0.91. The accumulative frequency of maximum genomic relationship coefficient and the average of top 10 relationship coefficients between a bull in validation data and US bulls are shown in Figures 1 and 2. The maximum relationship between a Danish validation bull and US bulls ranged from 0.1 to 0.54 with a median of 0.22, that is, 50% of Danish validation bulls had a maximum relationship equal to or over 0.22 with one or more US bulls. The median is near the relationship between half-sibs. Correspondingly, the averages of top 10 relationships range from 0.06 to 0.34 with a median 0.16, that is, 50% of Danish test bulls had an average relationship with the closest 10 US bulls equal to or over 0.16. This is equivalent to half of the Danish validation bulls having >10 cousins in the US reference data.

Figure 1 Cumulative frequency against maximum relationship coefficient between a Danish validation bull and the US reference bulls.

Figure 2 Cumulative frequency against average of top 10 relationship coefficients between a Danish validation bull and the US reference bulls.
Table 5 Degree of linkage disequilibrium (LD) between adjacent SNPs for Danish and US Jersey populations, correlation of LD and correlation of allele frequency between populations
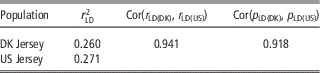
SNP=single nucleotide polymorphism.
Discussion
This study investigated the gain in reliability of genomic prediction by sharing reference data and adding cows to the reference population. In validation of eight traits on Danish bulls, a joint reference population combining Danish and US reference bulls led to an average increase in reliability of 3 percentage points, compared with genomic prediction using the Danish bull reference population alone. In validation of six traits on Danish cows, the average gain from US reference bulls was 6.6 percentage points, and the average gain from inclusion of cows in reference population was 8.2 percentage points. However, the gains from US bulls and from cows were not accumulative, and the total gain was 10.5 percentage points in the validation on cows. There are at least two reasons that can cause non-accumulative gains in reliability. One is that the information sources (cow information and US bull information here) are not independent (Harris and Johnson, Reference Harris and Johnson1998), and the other is that the increase of reliability with increase of reference population size is not linear (Daetwyler et al., Reference Daetwyler, Villanueva and Woolliams2008; Goddard, Reference Goddard2009; Goddard and Hayes, Reference Goddard and Hayes2009).
Improving genetic prediction by sharing reference data
Sharing reference data is an efficient approach to increase the size of a reference population and consequently improve the accuracy of genomic predictions. Previous studies have reported that the reliability of genomic prediction can be increased by using a joint reference population combining reference animals from other populations. The reliabilities of GEBV increased by 10 percentage points when using a joint reference data combining reference bulls of four European Holstein populations, compared with those obtained from national reference population alone (Lund et al., Reference Lund, Su, Nielsen and Aamand2011). A large improvement was also realized when combining Holstein populations in North America (Schenkel et al., Reference Schenkel, Sargolzaei, Kistemaker, Jansen, Sullivan, Van Doormaal, VanRaden and Wiggans2009; Muir et al., Reference Mäntysaari, Liu and VanRaden2010). Reliability was 3.2 percentage points higher for Brown Swiss cattle using a joint reference population including foreign bulls in the US domestic prediction (VanRaden et al., Reference VanRaden, Olson, Null, Sargolzaei, Winters and van Kaam2012).
Inclusion of about 1150 US Jersey bulls resulted in a large increase in accuracy of genomic prediction. The results were consistent with the validation of Danish bulls in US scale (i.e., performance in US) using joint US-Danish reference population (Wiggans et al., Reference Wiggans, Su, Cooper, Nielsen, Aamand, Guldbrandtsen, Lund and VanRaden2015). At least two reasons can explain this large gain. First, the Danish reference population was small; the inclusion of US Jersey bulls doubled the size of reference population. Large benefit from including reference animals of another population to a small national reference population has been reported by Zhou et al. (Reference Zhou, Heringstad, Su, Guldbrandtsen, Meuwissen, Svendsen, Grove, Nielsen and Lund2013), who added 4400 Danish progeny-tested Holstein bulls to the Chinese reference population, which comprised 1500 Chinese Holstein cows. The gain from the inclusion of Danish bulls was 29 percentage points for Chinese Holstein bulls and 7 percentage points for Chinese Holstein cows.
The second reason for the large gain in prediction accuracy from US Jersey was that there was a high consistency in genome and a strong genetic link between Danish Jersey and US Jersey. Semen of US Jerseys has been used in the Danish Jersey population for a long time, especially during the period from 1985 to 1995. Today the US Jersey breed proportion is about 38% in the Danish Jersey population (http://www.vikinggenetics.com.au/breeds/viking-jersey/about-viking-jersey). Consequently, the correlation of linkage disequilibrium between the two populations was high up to 0.94, and the correlation of allele frequency was 0.91. Furthermore, half of the Danish validation bulls have a relationship coefficient of at least 0.22 with one or more US reference bulls. The importance of relationship between populations for genomic prediction across populations has been reported in many previous studies (Brondum et al., Reference Brondum, Rius-Vilarrasa, Stranden, Su, Guldbrandtsen, Fikse and Lund2011; Zhou et al., Reference Zhou, Heringstad, Su, Guldbrandtsen, Meuwissen, Svendsen, Grove, Nielsen and Lund2013 and Reference Zhou, Ding, Zhang, Wang, Lund and Su2014b).
Improving genetic prediction by including cows in reference population
In dairy cattle the reference population for genomic prediction usually consisted of progeny-tested bulls since they have a large group of daughters with records, thus reliable phenotypic information. However, the phenotypic information of cows is also helpful. Though cow information can be summarized as daughter group means or DRP of sires which are usually in the reference population, this could result in a loss of information from the variation between daughters. Previous studies have reported that a cow reference population leads to moderate reliability of genomic predictions (Ding et al., Reference Ding, Zhang, Li, Wang, Wu, Sun, Yu, Liu, Wang, Zhang, Zhang, Zhang and Zhang2013; Li et al., Reference Li, Wang, Huang, Li, Zhang and Ding2014). A more common approach is to add genotyped cows to the bull reference population. Calus et al. (Reference Calus, de Haas and Veerkamp2013) investigated accuracy of genomic prediction using 1609 cows and 296 bulls as reference animals, and reported that the combined cow and bull reference population resulted in a prediction accuracy higher than using cow reference population alone and much higher than using bull reference population alone. Cooper et al. (Reference Cooper, Wiggans and VanRaden2015) reported that adding 30 852 cows to the bull reference population (21 833 bulls) increased reliability by 0.4 percentage points for validation bulls and 4.4 points for validation cows. In a simulation study with 60 progeny-tested bulls and 2000 or 4000 cows as reference population, the reliability of genomic prediction using the combined bull and cow reference population was nearly twice as high as using the bull reference population alone (Thomasen et al., Reference Thomasen, Sorensen, Lund and Guldbrandtsen2014). The large increase of prediction accuracy by including cows in reference population was also reported in another simulation study (Buch et al., Reference Buch, Kargo, Berg, Lassen and Sorensen2012).
In the current study, inclusion of about 4800 cows in the reference population increased reliability of GEBV by 8.2 percentage points. It can be argued that the gain may be overestimated, because most cows in the reference population were contemporaries of the validation cows, which may be favorable for the prediction of the validation cows. However, in this study all half and full sibs of the validation cows as well as their offspring were excluded from the reference population. Therefore, the overestimation of the gain from cows is not expected to be an issue. The large gain from inclusion of cows in the reference population could be due to the fact that the bull reference population was small (about 1250 bulls). The inclusion of cows actually greatly increased the size of the reference population. When the reference population already included US bulls, the further gain from cows decreased to 3.9 percentage points. This indicates that inclusion of cows in the reference population greatly benefits populations with a small reference data set, but may not necessary largely benefit populations with large reference data sets. It should be pointed that only half of the cows available were used as reference animals in this study. The remaining cows were used as either in the validation set or deleted because they were the close relatives of the validation cows. In practical genomic evaluation a larger gain from including cow information would be obtained since all these cows can be used as reference animals.
Some previous studies have detected bias of genomic prediction when including cows in reference population (Wiggans et al., Reference Wiggans, Cooper and VanRaden2010 and Reference Wiggans, Cooper, VanRaden and Cole2011; Dassonneville et al., Reference Dassonneville, Fritz, Ducrocq and Boichard2012a). However, in the current study, inclusion of cows in reference population actually slightly reduced bias of GEBV. This is due to the fact that most cows in the analysis were from herds with good data registration where all cows available were genotyped. Therefore, bias due to preferential treatment of bull’s dam is not an issue in current study.
Many countries have genotyped cows either to increase the size of reference population or to select females or bull dams. To reduce the cost of genotyping, cows are usually genotyped with a low density chip. Previous studies have reported that the accuracy of imputation from low density panel (7k) to Bovine SNP50 panel (54k) is over 97% (Boichard et al., Reference Boichard, Chung, Dassonneville, David, Eggen, Fritz, Gietzen, Hayes, Lawley, Sonstegard, Van Tassell, VanRaden, Viaud-Martinez, Wiggans and Bovine2012; Dassonneville et al., Reference Dassonneville, Baur, Fritz, Boichard and Ducrocq2012b; Su et al., Reference Su, Madsen, Nielsen, Mäntysaari, Aamand, Christensen and Lund2014). This indicates that genotyping cows for genomic prediction is feasible.
Alternative approaches to improve genomic prediction for small breeds
In addition to sharing reference data and including cows in the reference population, there are many alternative approaches that may improve accuracy of genomic prediction for numerically small breeds. One approach is to use a single-step model for genomic prediction (Legarra et al., Reference Legarra, Aguilar and Misztal2009; Aguilar et al., Reference Aguilar, Misztal, Johnson, Legarra, Tsuruta and Lawlor2010; Christensen and Lund, Reference Christensen and Lund2010). Single-step models have the advantage that they directly use information of both genotyped and non-genotyped animals by integrating genomic, pedigree and phenotype information in a single-step procedure. Makgahlela et al. (Reference Makgahlela, Stranden, Nielsen, Sillanpaa and Mantysaari2014) predicted GEBV using a single-step model in which DRP of all cows in the Nordic Red population were used as response variables. It allowed using information of all animals, especially directly using dam information to predict breeding value of an individual. The single-step approach increased reliability by 5 to 8 percentage points for yield traits, compared with a GBLUP model using only DRP of genotyped bulls as the response variable (Makgahlela et al., Reference Makgahlela, Stranden, Nielsen, Sillanpaa and Mantysaari2013).
Another alternative is to use a multi-breed reference population that combines information from numerically large breeds. However, previous studies have reported that multi-breed reference population can improve reliability of genomic prediction if the breeds involved have a genetic link (Brondum et al., Reference Brondum, Rius-Vilarrasa, Stranden, Su, Guldbrandtsen, Fikse and Lund2011; Zhou et al., Reference Zhou, Lund, Wang and Su2014a), and very little effect on accuracy of genomic prediction for the genetically distant breeds (Karoui et al., Reference Karoui, Jesus Carabano, Diaz and Legarra2012; Zhou et al., Reference Zhou, Ding, Zhang, Wang, Lund and Su2014b). One of the reasons that no or very little gain is observed from using multi-breed genomic prediction for genetically distant breeds could be due to differences in linkage disequilibrium between breeds. A possible solution could be to detect causal variants based on sequence data. This would eliminate the reliance on linkage disequilibrium, and thus the information of other breeds can be efficiently used for genomic prediction through the covariance structure of the detected causal variants.
Conclusions
Both sharing reference data and including cows in the reference population greatly increased reliability of genomic prediction in Danish Jersey. The gain in reliability of GEBV from the two approaches was >10 percentage points. The results indicate that sharing reference data and including cows in the reference population are efficient approaches to increase reliabilities of genomic predictions and thus increase genetic gain, especially for populations where the number of progeny-tested bulls is small. Therefore, by efficiently using information recourses, genomic prediction for numerical small breeds is promising.
Acknowledgment
This work was performed within the project “Genomic in herds”, funded by VikingGenetics and Nordic Cattle Genetic Evaluation.








 Open access
Open access