


Introduction
Dietary lipids have the capacity to regulate many aspects of metabolism in a manner dependent on chain length and the number, position and geometry of the double bonds within the fatty acids they contain. One of the most profound examples of such regulation is the impact of dietary fat on cholesterol and lipoprotein metabolism (Katan et al., Reference Katan, Zock and Mensink1994). It is well established that diets rich in saturated fatty acids are associated with elevated plasma concentrations of low-density lipoprotein (LDL) cholesterol. By contrast, enriching the diet with n-6 polyunsaturated fatty acids (PUFA) reduces LDL cholesterol. PUFA also have a major impact on the activity of enzymes associated with carbohydrate metabolism and lipid biosynthesis and oxidation (Sampath and Ntambi, Reference Sampath and Ntambi2005). The role of fatty acids as substrates in these pathways and their conversion to eicosanoids, with different levels of activity depending on the structure of the parent molecule, represents two mechanisms whereby they exert their effect. However, the last decade has seen a growing body of evidence to suggest that fatty acids and/or their derivatives also have direct effects on the expression of genes for proteins regulating carbohydrate and lipid metabolism (Jump and Clarke, Reference Jump and Clarke1999; Pégorier et al., Reference Pégorier, Le May and Girard2004; Jump et al., Reference Jump, Botolin, Wang, Xu, Christian and Demeure2005; Sampath and Ntambi, Reference Sampath and Ntambi2005; Davidson, Reference Davidson2006). This review will explore the emerging mechanisms whereby dietary fatty acids can directly influence gene expression.
Fatty acids and gene transcription
Dietary long-chain fatty acids enter the blood stream primarily as components of chylomicron triacylglycerol. The bulk of these will be released by the action of lipoprotein lipase in the capillaries of tissues such as adipose tissue and muscle, and will then be transported into cells either by passive diffusion or via transporter-mediated uptake (Kalant and Ciaflone, Reference Kalant and Cianflone2004). A significant portion of chylomicron triacylglycerol, together with phospholipid and cholesterol ester, will remain with the chylomicron remnant and be removed from the circulation by the liver. The fatty acids associated with these lipids will then be released during lysosomal degradation of the lipoprotein particles. Within the cell the bulk of these fatty acids will be converted into fatty acyl-CoA derivatives and channelled into various metabolic pathways. While it is clear that the bulk of such fatty acids are either esterified or oxidised (Jump and Clarke, Reference Jump and Clarke1999), there is growing evidence that fatty acids or their derivatives can accumulate in different intracellular compartments. For example, specific fatty acid binding proteins have been shown to transport fatty acids, or their derivatives, directly to the nucleus (Wolfrum et al., Reference Wolfrum, Borrmann, Borchers and Spener2001). Changes in the size and composition of these pools of fatty acids may regulate gene expression by modulating transcriptional and post-transcriptional events. In particular, much recent research has focused on the impact of fatty acids and their derivatives on the activity or abundance of specific transcription factors involved in regulating the expression of genes for key enzymes, receptors and transport proteins (Sampath and Ntambi, Reference Sampath and Ntambi2005; Deckelbaum et al., Reference Deckelbaum, Worgall and Seo2006).
Molecular mechanisms involved in fatty acid regulation of gene expression
There are at least four transcription factor families that play a major role in the fatty acid-induced regulation of gene transcription. These include peroxisome proliferator-activated receptors (PPARs), liver X receptor (LXR), hepatocyte nuclear factor 4 (HNF-4) and sterol regulatory binding proteins (SREBPs). All of these, except SREBP, are members of the nuclear hormone receptor superfamily of transcription factors.
Nuclear receptors
Nuclear receptors are a class of ligand-activated intracellular transcription factors which up- or downregulate the expression of genes. Approximately 50 such receptors have been identified in humans (Robinson-Rechavi et al., Reference Robinson-Rechavi, Carpentier, Duffraisse and Laudet2001). A subset of these, including PPAR and LXR, bind to specific motifs (response elements) within the promoters of genes as heterodimers with the retinoid X receptor (RXR). These response elements consist of direct repeats of nucleotide sequences separated by one or more nucleotides. It is now believed that these heterodimers can bind to response elements in the absence of ligand and may actually inhibit gene transcription through association with co-repressors. Ligand binding appears to initiate conformational changes which displace co-repressors and facilitate interaction with co-activators (Glass and Rosenfeld, Reference Glass and Rosenfeld2000). Through a reorganisation of the chromatin and modification or recruitment of a range of factors involved in gene expression this leads to activation of transcription. Figure 1 illustrates the potential regulation of gene expression by LXR.
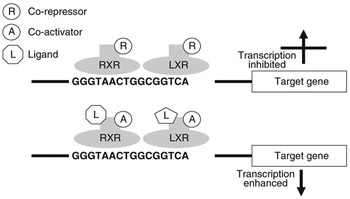
Figure 1 Regulation of gene transcription by LXR. LXR binds to the LXR response element of target genes as a heterodimer with RXR. When this dimer is bound in the absence of ligand, they may interact with co-repressors and gene transcription will be inhibited. When a ligand interacts with one or both of the receptors this will change their conformation, displacing co-repressors and attracting co-activators. As a result gene expression will be stimulated.
Peroxisome proliferator-activated receptors
PPARs are the most extensively characterised nuclear receptors that are regulated by fatty acids (Desvergne and Wahli, Reference Desvergne and Wahli1999). There are three isoforms, PPARα, PPARβ (also known as δ) and PPARγ. PPARα is expressed in liver, intestine and kidney. PPARγ (subforms γ1, γ2 and γ3 arising from alternative splicing of one gene) is mainly expressed in adipose tissue and macrophages, while PPARβ is ubiquitously expressed.
PPARs were initially identified for their capacity to be activated by a diverse range of compounds, including fibrates and xenobiotics, known as peroxisome proliferators (Issemann and Green, Reference Issemann and Green1990). In rodents the response to such activation is a proliferation and increase in size of peroxisomes in the liver. In turn this leads to a marked increase in the peroxisomal oxidation of fatty acids. Such peroxisome proliferation is ultimately associated with an increased incident of hepatocarcinoma (Lake, Reference Lake1995). However, it has been subsequently found that such PPAR agonists do not induce peroxisome proliferation (or cancer) in a number of species including guinea pigs, primates and humans (Cattley et al., Reference Cattley, DeLuca, Elcombe, Fenner-Crisp, Lake, Marsman, Pastoor, Popp, Robinson, Schwetz, Tugwood and Whali1998; Choudhury et al., Reference Choudhury, Chahal, Bell, Tomlinson, Roberts, Salter and Bell2000). In fact, fibrates, known PPARα ligands, have been commonly used in the treatment of elevated plasma lipids for many years (Watts and Dimmitt, Reference Watts and Dimmitt1999). The ligand activation of PPARα is now known to be associated with transcriptional upregulation of a wide range of genes for proteins associated with fatty acid oxidation and lipoprotein metabolism, including acyl-CoA oxidase, carnitine palmitoyl transferase 1, lipoprotein lipase, apolipoproteins AI and CIII (Brown and Plutzky, Reference Brown and Plutzky2007). A potential role for PPARα in inhibiting the expression of genes for pro-inflammatory proteins (including vascular cell adhesion molecule-1, CX-1 and interleukin-6) in the vascular endothelium has received considerable attention (Marx et al., Reference Marx, Duez, Fruchart and Staels2004).
PPARγ was originally identified as an important regulator of the aP2 gene, an adipocyte specific intracellular lipid binding protein (Tontonoz et al., Reference Tontonoz, Hu, Graves, Budavari and Spiegelman1994). It has now been established as an important regulator of adipocyte differentiation and regulator of adipose tissue metabolism. The insulin sensitising class of drugs commonly known as glitazones are agonists of PPARγ (Lehman et al., Reference Lehman, Moore, Smith-Oliver, Wilkinson, Willson and Kliewer1995).
The least well characterised of the PPARs is PPARβ. Its ubiquitous expression suggests a fundamental role in regulating gene expression. It is most abundantly found in the small intestine, colon, heart, adipose tissue and brain (Brown and Plutzky, Reference Brown and Plutzky2007).
A surprisingly diverse range of naturally occurring and synthetic compounds appear to act as ligands for the various PPAR isoforms. This includes both saturated and unsaturated fatty acids, though in general PUFA are more potent. All three PPAR subtypes have been shown to bind both n-3 and n-6 PUFA (Krey et al., Reference Krey, Braissant, L’Horset, Kalkhoven, Perroud, Parker and Whali1997; Kliewer et al., Reference Kliewer, Sundeth, Jones, Brown, Wisely, Koble, Devchand, Whali, Willson and Lenhard1997). In general, PUFA bind best to PPARα followed by PPARγ and PPARβ (Krey et al., Reference Krey, Braissant, L’Horset, Kalkhoven, Perroud, Parker and Whali1997). Furthermore, a range of eicosanoids, derived form n-3 and n-6 PUFA, have also been shown to be PPAR ligands, often with greater affinity than their parent molecules (Krey et al., Reference Krey, Braissant, L’Horset, Kalkhoven, Perroud, Parker and Whali1997).
Liver X receptors
LXRs function as sensors of cholesterol in the nucleus and they are activated by elevated levels of intracellular cholesterol in many different cell types. LXRα and LXRβ (also known as NR1H3 and NR1H2, respectively) were originally thought of as ‘orphan’ nuclear receptors as their natural ligands were not known. The naturally occurring oxysterols 22-R-hydroxycholesterol and 24,25-epoxycholesterol and long-chain fatty acids have since been identified as ligands for LXR (Janowski et al., Reference Janowski, Grogan, Jones, Wisely, Kliewer, Corey and Mangelsdorf1999). LXRα is highly expressed in the liver but also at lower levels in adrenal glands, intestine, adipose, macrophages, kidney and lung whereas LXRβ is more ubiquitously expressed (Repa and Mangelsdorf, Reference Repa and Mangelsdorf2000). LXRs bind to response elements also as heterodimers with RXR. LXRs regulate the expression of genes involved in sterol and fatty acid metabolism, particularly hepatic bile acid synthesis. The rate-limiting enzyme in the synthesis of bile acids, 7α cholesterol hydroxylase (CYP7A) was one of the first genes to be identified as being directly regulated by LXRs (Peet et al., Reference Peet, Turley, Ma, Janowski, Lobaccaro, Hammer and Mangelsdorf1998). A whole range of genes involved in lipoprotein metabolism, including adenosine triphosphate-binding cassette protein A1 (ABCA1, involved in reverse cholesterol transport), apolipoprotein E, lipoprotein lipase, cholesterol ester transfer protein and phospholipid transfer protein, have since been found to be regulated by LXRs (Geyeregger et al., Reference Geyeregger, Zeyda and Stulnig2006). LXRs also play an important role in the regulation of genes for proteins involved in other aspects of metabolism including lipogenesis (Peet et al., Reference Peet, Turley, Ma, Janowski, Lobaccaro, Hammer and Mangelsdorf1998) and carbohydrate metabolism (Cao et al., Reference Cao, Liang, Broderick, Oldham, Beyer, Cao, Liang, Broderick, Oldham, Beyer, Schmidt, Zhang, Stayrook, Suen, Otto, Miller, Dai, Foxworthy, Gao, Ryan, Jiang, Burris, Eacho and Etgen2003; Laffitte et al., Reference Laffitte, Chao, Li, Walczak, Hummasti, Joseph, Castrillo, Wilpitz, Mangelsdorf, Collins, Saez and Tontonoz2003). The role of LXR in regulating lipogenesis and its interaction with SREBP1c are discussed below.
Hepatocyte nuclear factor 4α
HNF-4α is another member of the nuclear receptor superfamily and, while as its name suggests it is highly expressed in liver, it is also found in kidney, intestine and pancreas (Sladek et al., Reference Sladek, Zhong, Lai and Darnell1990). As conventional gene knock-out is lethal, Hayhurst et al. (Reference Hayhurst, Lee, Lambert, Ward and Gonzalez2001) addressed the role of HNF4α in mature hepatocytes using a conditional gene knockout. Mice lacking hepatic HNF-4α exhibit major perturbations of lipid metabolism with increased accumulation of lipid in the liver and reduced plasma cholesterol and triacylglycerol concentrations. This was associated with changes in expression of a range of genes involved in lipid and lipoprotein metabolism including apolipoproteins AII, AIV, CII and CIII, the microsomal triglyceride transfer protein and CYP7A1. HNF-4α has been shown to bind long-chain fatty acyl CoA with high affinity (Hertz et al., Reference Hertz, Magenheim, Berman and Bar-Tana1998). Binding of saturated fatty acyl-CoAs activates HNF-4α while unsaturated fatty acyl-CoAs inhibit HNF-4α transcriptional activity. HNF-4α binds to DR1 response elements as a homodimer and may potentially compete with PPAR/RXR heterodimers for DNA binding (Pégorier et al., Reference Pégorier, Le May and Girard2004). It has also been shown that fibrates can be converted to CoA-thioesters that bind HNF-4α to inhibit its transcriptional activity, thus providing another level of competition between HNF-4α and PPARs (Hertz et al., Reference Hertz, Sheena, Kalderon, Berman and Bar-Tana2001).
Sterol regulatory element binding proteins
The SREBPs play a major role in regulating the expression of genes associated with lipid and lipoprotein metabolism (Brown and Goldstein, Reference Brown and Goldstein1999; Horton et al., Reference Horton, Goldstein and Brown2002; Eberlé et al., Reference Eberlé, Hegarty, Bossard, Ferré and Foufelle2004). They are membrane-bound members of the basic helix–loop–helix leucine zipper family of transcription factors. Two SREBP genes produce three separate proteins (SREBP1a and 1c from one and SREBP2 from the other). The SREBP1 and SREBP2 proteins share 47% homology and SREBP1a and 1c differ in their first exon. SREBP1a and SREBP2 exhibit higher levels of transcriptional activation due to the presence of a longer transcriptional activator domain (Shimano et al., Reference Shimano, Horton, Shimomura, Hammer, Brown and Goldstein1997). While in most cell lines SREBP2 and SREBP1a predominate, in most tissues, SREBP1c mRNA concentrations exceed those for SREBP1a (Shimomura et al., Reference Shimomura, Shimano, Horton, Goldstein and Brown1997a and 1997b). Each of the SREBP proteins is synthesised in an immature form which resides on the endoplasmic reticulum (ER). Activation is by a two-step proteolytic process that occurs in the Golgi apparatus (Brown and Goldstein, Reference Brown and Goldstein1999; Figure 2a). SREBPs are transported to the Golgi through the action of an escort protein called SCAP (Goldstein et al., Reference Goldstein, DeBose-Boyd and Brown2006). SCAP contains binding sites for COPII proteins which cluster the SCAP-SREBP complex into COPII-coated vesicles (Sun et al., Reference Sun, Li, Goldstein and Brown2005). These vesicles then bud from the ER and fuse with the Golgi. Increases in cellular cholesterol cause conformational changes in SCAP resulting in retention of SREBP in the ER (Nohturfft et al., Reference Nohturfft, Yabe, Goldstein, Brown and Espenshade2000; Figure 2b). This has now been shown to be as a result of binding of SCAP to two ER anchor proteins called Insig 1 and 2 (Yang et al., Reference Yang, Espenshade, Wright, Yabe, Gong, Aebersold, Goldstein and Brown2002). The binding of Insig to SCAP inhibits its interaction with CopII proteins, and thus retards its movement to the Golgi (Goldstein et al., Reference Goldstein, DeBose-Boyd and Brown2006). This leads to reduced proteolysis and a decrease in the nuclear active form of SREBP. This sensitivity to changes in intracellular sterol concentrations may not apply to all three SREBP isoforms. While evidence from studies using cell lines indicate that both SREBP1a and SREBP2 display such sensitivity to sterols, in vivo studies indicate SREBP1 may not respond to sterol depletion (Sheng et al., Reference Sheng, Otani, Brown and Goldstein1995).
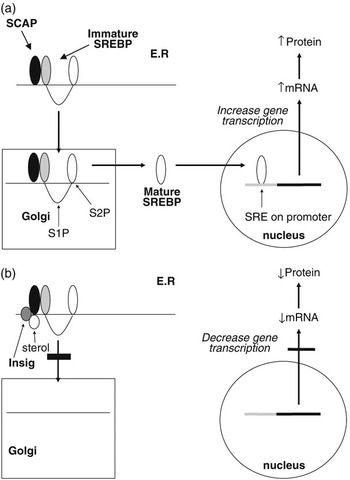
Figure 2 Regulation of gene transcription by SREBPs. Immature SREBP resides on the endoplasmic reticulum in association with SCAP. When cellular sterol concentrations are low (a) then SCAP escorts the SREBP to the Golgi apparatus where it is acted upon by two proteases (S1P and S2P) to release the active mature form of SREBP. This migrates to the nucleus where it interacts with the sterol regulatory element of target genes to upregulate gene transcription. In the presence of high cellular sterol concentrations, (b) the sterol interacts with the protein Insig which acts to anchor the SCAP-SREBP complex in the endoplasmic reticulum and thereby inhibit SREBP maturation. As a result the transcription of target genes is downregulated.
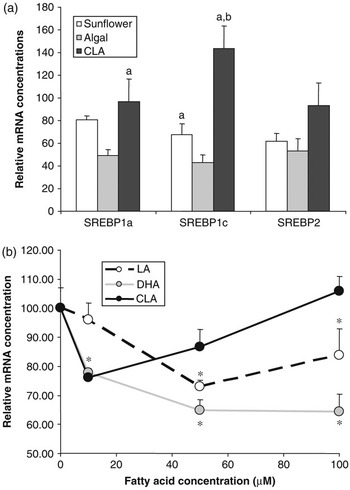
Figure 3 Effect of polyunsaturated fatty acids on SREBP expression. (a) Golden Syrian hamsters were fed chow supplemented with 0.12% cholesterol with or without 5% added fat for 4 weeks. Added fat included sunflower oil (rich in linoleic acid), algal oil (rich in DHA) or CLA (a mixture of approximately 50% c-9,t-11and 50% t-10,c-12 isomers). At the end of the trial, RNA was extracted from liver and SREBP1a, 1c and 2 mRNA concentrations were determined by quantitative nuclease protection assay. Data are presented as a percentage of the mean mRNA concentration in animals fed chow+cholesterol without added fat (control). Compared with control animals, those fed sunflower oil showed no significant difference in mRNA concentration for any of the SREBPs, those fed algal oil had lower levels of mRNA for all three SREBPs and those fed CLA had higher levels SREBP1c mRNA (P < 0.05). Bars sharing common letters were significantly different from each other (P < 0.05) (S. Cooper et al., unpublished data). (b) The human SREBP1c promoter was cloned and inserted into a vector containing the firefly luciferase gene. This was then transfected into H4IIE (Rat hepatoma) cells which were then treated with the indicated quantities of fatty acids bound to albumin. Fatty acids included linoleic acid, DHA and c-9,t-11CLA isomer. SREBP1c promoter activity was assessed by determining the luciferase activity of the transfected cells. *Significantly different from cells incubated in the absence of added fatty acid (E. J. Tarling et al., unpublished data).
The transcriptionally active SREBP translocates to the nucleus where it binds to sterol regulatory elements (SREs) on target genes. SRE nucleotide sequences vary considerably between genes but are commonly represented by the sequence 5′-TCACNCCAC-3′ (Kim et al., Reference Kim, Spotts, Halvorsen, Shih, Ellenberger, Towle and Spiegelman1995). While there is a sizeable overlap in the specificities of the individual SREBP isoforms, studies in transgenic mice over-expressing truncated nuclear active forms of SREBP in liver have shown specific roles for each isoform (Horton et al., Reference Horton, Goldstein and Brown2002). Over-expression of SREBP2 results in a vast increase in cholesterol synthesis and SREBP2 has been shown to regulate at least 11 of the enzymes involved in the cholesterol synthetic pathway (Horton et al., Reference Horton, Shimomura, Brown, Hammer, Goldstein and Shimano2000). Over-expressing the nuclear form of SREBP1a results in the accumulation of cholesterol and triglycerides in the liver (Shimano et al., Reference Shimano, Horton, Hammer, Shimomura, Brown and Goldstein1996) while over-expressing SREBP1c causes an increase in liver triglycerides without a concomitant increase in cholesterol (Shimano et al., Reference Shimano, Horton, Shimomura, Hammer, Brown and Goldstein1997). This suggests SREBP1c primarily regulates the expression of genes involved in fatty acid synthesis while SREBP1a can regulate enzymes of both the cholesterol and fatty acid synthetic pathways. SREBP1c mRNA and protein levels, and not those of SREBP2, are increased on a high carbohydrate diet or on re-feeding fasted mice, correlating with changes in lipogenic enzyme mRNA levels and dietary PUFA decrease nuclear SREBP-1 protein levels without affecting SREBP2 (Horton et al., Reference Horton, Bashmakov, Shimomura and Shimano1998). SREBP2 expression is primarily regulated at a post-translational level, through modulation of cleavage of its membrane-bound precursor to release the active nuclear form, whereas SREBP1 expression is primarily regulated at the level of gene transcription (Horton et al., Reference Horton, Goldstein and Brown2002; Eberlé et al., Reference Eberlé, Hegarty, Bossard, Ferré and Foufelle2004).
Regulation of gene expression by dietary lipids
Regulation of lipogenic gene expression by PUFA
The ability for polyunsaturated fatty acids to downregulate the expression of genes for enzymes involved in fatty acid synthesis, including ACC, FAS and SCD, has been recognised for some time (Jump and Clarke, Reference Jump and Clarke1999; Sampath and Ntambi, Reference Sampath and Ntambi2005). These genes appear to be repressed by both n-3 and n-6 PUFA though response to the former may be more profound. More recently, there has been considerable interest in the ability of conjugated linoleic acids (CLA) to specifically reduce lipid deposition in adipose tissue (Wang and Jones, Reference Wang and Jones2004). While at least part of these responses may be mediated by PUFA activation of PPARα (see below), experiments in PPARα knockout mice clearly indicate that PUFA still repress the expression of ACC and FAS (Ren et al., Reference Ren, Thelen, Peters, Gonzalez and Jump1997). This has led to the search for alternative mechanisms.
The role of SREBP1c in regulating lipogenic gene expression makes it an obvious candidate for mediating the effects of PUFA on lipogenesis. Cell culture studies have clearly shown that PUFA have the ability to reduce SREBP1a and 1c mRNA and SREBP1 protein concentrations (Worgall et al., Reference Worgall, Sturley, Soe, Osborne and Deckelbaum1998). In our own experiments in the hamster feeding PUFA reduces mRNA for all three SREBPs in a manner that is dependent on the nature of the PUFA (Figure 3a). Docosahexaenoic acid (DHA, C22:6n-3) exerts a more potent effect than linoleic acid (C18:2n-6). Intriguingly, a mixture of c-9,t-11and t-10,c-12 isomers of CLA appeared to actually increase SREBP1c mRNA concentrations. This effect of PUFA on SREBP mRNA concentrations has been attributed to both repression of gene transcription and increased turnover of the mRNA. To investigate further the impact of PUFA on SREBP1c gene transcription, we have recently cloned the human SREBP1c promoter (Tarling et al., Reference Tarling, Salter and Bennett2004) and conducted a series of reporter gene experiments. The SREBP1c promoter was attached to a luciferase reporter gene and transfected into H4IIE rat hepatoma cells. Cells were then treated with various concentrations of PUFA. It can be seen that, again DHA showed a more potent repression of SREBP1c promoter activity compared linoleic acid. Interestingly, c-9,t-11(but not t-10,c-12) CLA actually stimulated promoter activity at higher concentrations. Taken together these data provide further evidence that PUFA may directly regulate SREBP gene transcription and that the specific structure of the fatty acid impacts on its effects.
How does dietary PUFA regulate SREBP expression? Recent evidence suggests that, at least part of this effect may be mediated through LXR. PUFA have been reported to inhibit the interaction of oxysterols with LXR and thereby inhibit its activation of target genes, including SREBP1c itself (Hannah et al., Reference Hannah, Ou, Luong, Goldstein and Brown2001; Yoshikawa et al., Reference Yoshikawa, Shimano, Amemiya-Kudo, Yahagi, Hasty, Matsuzaka, Okazaki, Tamura, Iizuka, Ohashi, Osuga, Harada, Gotoda, Kimura, Ishibashi and Yamada2001 and Reference Yoshikawa, Shimano, Yahagi, Ide, Amemiya-Kudo, Matsuzaka, Nakakuki, Tomita, Okazaki, Tamura, Iizuka, Ohashi, Takahashi, Sone, Osuga, Gotoda, Ishibashi and Yamada2002). Both the mouse and the human SREBP1c promoters have been shown to contain potential LXREs (Tarling et al., Reference Tarling, Salter and Bennett2004). Indeed, we have shown the increased activity of human SREBP1c promoter in the presence of a specific LXR agonist (E. J. Tarling et al., unpublished data). Thus, inhibition of the association of LXR with its oxysterol ligands by PUFA may repress activation of transcription of the SREBP1c gene by LXR. Another potential mechanism is the inhibition of LXR transcriptional activity by PUFA activation of PPARs. Over-expression of PPARα and PPARγ in HEK-293 cells has been shown to inhibit SREBP1c promoter activity, and this effect was abolished by mutating the putative LXREs in the SREBP1c promoter (Yoshikawa et al., Reference Yoshikawa, Ide, Shimano, Yahagi, Amemiya-Kudo, Matsuzaka, Yatoh, Kitamine, Okazaki, Tamura, Sekiya, Takahashi, Hasty, Sato, Sone, Osuga, Ishibashi and Yamada2003). It has been suggested that this was a result of increased competition between PPAR and LXR for association with its transcriptional partner RXR. The downregulation of expression of SREBP1a and SREBP2 we have observed in the hamster cannot as yet be explained by reduced LXR activity, as neither promoter have been reported to contain LXREs. Earlier work has suggested that PUFA may reduce SREBP mRNA stability as well as reducing gene transcription (Xu et al., Reference Xu, Teran-Garcia, Park, Nakamura and Clarke2001). There is also more recent data to suggest PUFA may affect the nuclear abundance of SREBP isoforms by regulating proteasomal degradation of these proteins (Botolin et al., Reference Botolin, Wang, Christian and Jump2006). Thus, it appears that the regulation of lipogenic gene expression by dietary PUFA is a result of a complex interaction between a number of transcription factors including SREBPs, PPARs and LXR.
The hypotriglyceridemic effects of n-3 PUFA
Long-chain n-3 PUFAs of marine origin have long been known to reduce plasma triacylglycerol concentrations (Harris, Reference Harris1997). Despite extensive investigation the mechanisms underlying these effects remain to be fully elucidated (Davidson, Reference Davidson2006). Suppression of hepatic lipogenesis through reduced LXR-mediated transcription of SREBP1c, as described above, may certainly be one potential mechanism. Indeed, as can be seen in Figure 3a, DHA-rich algal oil appears to be considerably more effective at reducing hepatic SREBP1c mRNA concentrations than linoleic acid-rich sunflower oil. However, n-3 PUFA appear to exert effects on plasma triacylgycerol concentration even when subjects are consuming diets rich in fat and, presumably, when rates of hepatic lipogenesis are relatively low. The potential for these fatty acids, or their derivatives, to stimulate fatty acid oxidation through activation of hepatic PPARα has long been suggested to represent another potential mechanism. As already discussed, fatty acids are ligands for the PPARs and in general n-3 PUFA are more potent activators of PPARα than n-6 PUFA. Feeding fish oil to rodents has been shown to increase the transcription of enzymes involved in fatty acid oxidation (Nakatani et al., Reference Nakatani, Kim, Kaburagi, Yasuda and Ezaki2003). However, PPARα-knockout mice still display a dose-dependent reduction in plasma triacylglycerol in response to fish oil, suggesting that PPARα has only a minor role in this response (Dallongeville et al., Reference Dallongeville, Bauge, Tailleux, Peters, Gonzalez, Fruchart and Staels2001). It has also been suggested that n-3 PUFA may have effects on the activity of HNF-4α and this may be responsible for some of the hypotriglyceridemic effects of these fatty acids (Davidson, Reference Davidson2006).
Regulation of gene expression by saturated fatty acids
Evidence of direct regulation of gene expression by saturated fatty acids is considerably less compelling. Saturated fatty acids profoundly influence the activity of a range of proteins involved in lipid and lipoprotein metabolism. However, in many cases this may be an indirect effect. For example, diets rich in saturated fatty acids may alter the concentrations of cholesterol and its derivatives in the liver (Spady et al., Reference Spady, Woollett and Dietschy1993). Saturated fatty acids have been reported to suppress the esterification of cholesterol in the liver, thereby leading to an accumulation of metabolically active free cholesterol or perhaps oxygenated derivatives (Spady et al., Reference Spady, Woollett and Dietschy1993). This effect is specific to saturated fatty acids with 12–16 carbon atoms, with shorter chain fatty acids and stearic acid (C18:0) having no effect (Woollett et al., Reference Woollett, Spady and Dietschy1992). These may then regulate gene expression through modulating the activity of SREBPs or LXR. This has been suggested to be a potential mechanism through which saturated fatty acids reduce LDL receptor expression and thereby increase plasma LDL cholesterol concentrations (Spady et al., Reference Spady, Woollett and Dietschy1993).
One mechanism whereby saturated fatty acids may have a direct effect on gene expression is through modulating the activity of HNF-4α. As described above, there is evidence to suggest that saturated fatty acid, as coenzyme A derivatives, may modulate HNF-4α- regulation of gene expression (Hertz et al., Reference Hertz, Magenheim, Berman and Bar-Tana1998). However, direct evidence of dietary fatty acids acting through such a mechanism is still lacking.
It has recently been reported that a diet rich in saturated fat increased expression of the transcriptional co-activator PGC1β (Lin et al., Reference Lin, Yang, Tarr, Wu, Handschin, Li, Yang, Pei, Uldry, Tontonoz, Newgard and Spiegelman2005). This in turn was shown to enhance the transcriptional activity of SREBP1c. Modulation of the activity of co-activators, or indeed co-repressors by dietary lipids, opens up another intriguing level of nutrient:gene interaction.
Conclusions
It is clear that dietary lipids have the potential to modulate metabolism through altering the expression of genes for key proteins. The discussion above outlines growing evidence for potential mechanisms whereby they may exert such effects. However, much of this evidence emerges from cell culture studies where single fatty acid entities can be added and effects measured or animal experiments where diets of clearly defined fatty acid composition have been used. Translating these results into a situation when complex mixtures of fatty acids are normally consumed and join the already multifarious mixture of fatty acids and their derivatives already present in the body, represents a major challenge for future research.



