


Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is a major public health threat globally. It is estimated that one-third of the world is infected with Mtb, and roughly 10% of these individuals progress to active TB disease. TB pathogenesis follows a two-stage process: first, uninfected individuals become exposed to infectious TB cases, and may progress to Mtb infection, though not all subjects exposed to TB become infected [Reference Ma1]. In the second stage, ~10% of Mtb-infected individuals progress to active TB disease. These two stages may have different genetic underpinnings. In 2015, 10.4 million developed active TB and another 1.8 million died from the disease (http://www.who.int/mediacentre/factsheets/fs104/en/). Ethiopia is one of the high TB burden countries. In 2014, the estimated prevalence of TB in Ethiopia was 200 per 100 000 and incidence of 207 per 100 000 [2].
Although TB is primarily a pulmonary disease (PTB) that is initiated by the deposition of Mtb contained in aerosol droplets onto lung alveolar surfaces, it is also characterized by a variable course of disease in terms of latency, reactivation and progression to active disease; variation in affected sites; and, variation in severity and mortality that can all be modulated by nutrition, age, sex and genetics [Reference Smith3]. Although Mtb is a necessary bacterial cause, it is not sufficient to cause the disease. In other words, active TB disease is not exactly an inevitable outcome of exposure to, or infection by, Mtb. Both innate and adaptive immunity mechanisms are involved in determining the outcome of Mtb infection. In this respect, different genetic polymorphisms have been found which are associated with increased susceptibility to and severity of TB. Some of these polymorphisms are functional, but for many of these no functional (immunologic) changes have been demonstrated yet, and these associations need further confirmation [Reference Van Crevel, Ottenhoff and Van der Meer4]. In addition, pathogenic diversity may play a role in disease progression disparity since some Mtb strains are reportedly more virulent than others, as defined by increased transmissibility, rapid pathogenesis, higher morbidity and mortality in infected individuals [Reference Ernst, Trevejo-Nunez and Banaiee5, Reference Filliol6]. However, results of genetic association studies have varied widely [Reference Stein7], likely due to complex multi-factorial nature of TB that impacts our ability to diagnose and characterize the disease phenotype in a manner that is amenable for genetic analysis. These difficulties translate into major gaps in our knowledge and, therefore, a comprehensive genetic model for TB has not yet emerged [Reference Stein8, Reference Glassroth9]. The genetic complexity of TB is understandable since immune responses to Mtb infection, molecular signaling patterns and the extent of tissue involvement change over the course of the disease, and it is unlikely that any single factor can adequately explain susceptibility to TB. Thus, numerous genetic variants have been variably associated with susceptibility to TB [Reference Azad, Sadee and Schlesinger10, Reference Sirugo11]. Despite these limitations after numerous twin, candidate-gene, as well as genome-wide linkage and association studies in different populations, the evidence for a human genetic component in predisposition to TB susceptibility is considered incontrovertible [Reference Galagan12, Reference Marlo and Eileen13].
In Africa, where both high human genetic variation and TB disease are still prevalent, genetic epidemiological investigations are crucial and the discovery of genetic associations with strong effect sizes that impact susceptibility/resistance to disease could guide public health policy [Reference Sirugo11, Reference Hill14]. Several genetic association studies of TB have been carried out in Africa which reported positive, suggestive or no association results [Reference Azad, Sadee and Schlesinger10, Reference Sirugo11]. For example, using the complementary approach of candidate gene analysis, case-control studies of West African samples have identified associations with variants in several genes, including SLC11A1 [Reference Awomoyi15, Reference Bellamy16]; IL1B [Reference Awomoyi17]; vitamin D receptor [Reference Bornman18, Reference Lombard19]; CD209 (DC-SIGN), PTX3 [Reference Olesen20]; and P2X7 genes [Reference Li21]. In East Africa, a combined linkage and association study of Ugandans has shown that IL10, interferon gamma receptor 1 (IFNGR1), and TNF alpha receptor 1 (TNFR1) variants are linked to active TB, but not with latent infection [Reference Stein8]. And, in the first genome-wide linkage scan for a major infectious disease in Africans, significant evidence of linkage was found on chromosomes Xq27 and 15q11 [Reference Bellamy22], further supporting the conclusion that TB susceptibility loci exist. A recent study in Ugandan populations based on tagSNPs that mostly fall in intronic regions of immunity genes identified TICAM2 and NOD1 as candidate genes among others [Reference Hall23]. There is only one previously published study that investigated genetic susceptibility to TB in Ethiopian populations [Reference Malik24]. The authors investigated polymorphisms in the SFTPA1 and SFTPA2 genes, both expressed in the lungs, and identified four polymorphisms that modify the risk of TB susceptibility.
In the present study, we focused on two innate immunity genes, NOD1 [Nucleotide-binding Oligmerization Domain containing 1] and TICAM2 [Toll/Interleukin-1 Receptor Domain-Containing Adaptor Molecule 2], based on a recent finding [Reference Hall23] in an East African population that indicated significant statistical association and another study demonstrating a biological plausibility of TICAM2-NOD1 synergistic action [Reference Hiroyuki25] and, thus, were deemed to warrant an effort to replicate those findings in an independent population. TICAM2 is a member of the pattern-recognition-receptors called Toll-like-receptors (TLRs) that sense pathogen-associated-molecular-patterns in the extracellular environment as well as in endosomes. TLRs are primarily expressed in immune cells, such as macrophages, monocytes, B-lymphocytes, and dendritic cells. TICAM2, also known as TRAM, mediates innate immune responses through extracellular recognition of microbes and by facilitating the expression of IFN-inducible genes [Reference Elson26–Reference Seya28]. Its physical structure and interaction properties have been elucidated recently [Reference Yoshiaki29]. In addition to the recent genetic association finding with TB [Reference Hall23], it was also shown to be associated with a candidate TB vaccine trial outcome [Reference Magali30]. NOD1 is a member of the intracellular pathogen-recognition-receptors, nod-like receptors (NLRs), which are important for the recognition of unique muropeptides of bacterial peptidoglycan fragments (derived predominantly from the cell walls of Mtb) or damage-associated-molecular-patterns that localize to the cytosol. Following microbial sensing, NOD1 directly recruits factors which initiate intracellular signaling cascade that leads to the activation of transcriptional responses culminating in the expression of a subset of innate immunity genes involved in inflammation (e.g., production of proinflammatory cytokines and chemokines), antimicrobial mechanisms and autophagy in cells responsible for eliminating Mtb including epithelial cells, alveolar macrophages and monocyte-derived macrophages [Reference Lilian and Dario31]. Mutations in several NLR members were found to be associated with the development of inflammatory disorders [Reference Luigi32–Reference Jun-young34] including TB [Reference Hall23].
There are several reasons for regarding Ethiopia as a ‘model human population’ for the study of the genetic profile of virtually any phenotype of interest. Several anthropologic, linguistic, and genetic studies have suggested that humans originated in the African continent [Reference Pagani35, Reference Pagani36] with the range and weight of evidence for the Ethiopian source predominating any other single alternative in Africa [Reference Semino37–Reference Poloni41]. In this regard, we have recently demonstrated using genome-wide data the existence of not only unparalleled overall human genetic diversity among present-day Ethiopian populations, but also that Ethiopians possess the greatest proportion of novel variation most of which are unshared (private) and rare [Reference Gurdasani42]. This finding can be explained away, in combination or alone, by the effects of neither genetic drift, population bottleneck, migration/admixture events, nor the extent and pattern of LD decay; but more logically by the longer mutational time-scale needed to generate the extent of observed diversity, and thus, the much older population history of present-day Ethiopians. These and other evidence posit Ethiopia within Africa as the most probable single source of modern humans. Therefore, it is reasonable to propose that the tremendous genetic diversity existing in Ethiopia can be harnessed towards the understanding of the genetic basis of various infectious and non-infectious diseases that afflict humanity. With respect to the current study of human genetic susceptibility to TB, Ethiopia is also ‘well situated’. There is increasing evidence indicating the common origin in Ethiopia of MTBC (mycobacterium-tuberculosis-complex) and humans and that MTBC has been co-evolving with anatomically modern humans. These conclusions are based on the observed congruence in their phylogeographies, the dating of major branching events, and the discovery of a unique lineage of MTBC localized only in Ethiopia [Reference Gagneux43]. Therefore, studies of the impact of Mtb–human interactions would best be investigated in an Ethiopian setting.
The primary hypothesis of the present study was that the reasons for non-replication of TB genetic association results may be explained by the lack of precise definition of TB phenotypes that frustrates the discovery of underlying gene-disease associations or lead to spurious conclusions. Clearly defined traits can help increase power to detect disease-predisposing loci and thus more informative than studying a heterogeneous lump of a complex phenotype. This study focused on finding a method of unraveling TB phenotype complexity by drawing a clear definition of the TB trait based on its known natural progression stages from exposure, to infection, and progression to active disease. This ensures that intermediate stages of the disease are included as phenotypes of interest, since they may represent distinct TB phenotypes with respective immuno-genetic profiles [Reference Vermund and Yamamoto44–Reference Rice46]. The recent identification of TICAM2 and NOD1 association with TB in Ugandan populations was made possible by a similar methodology [Reference Hall23]. It was also hypothesized that the Ethiopian setting could indirectly help to explore the possibility that Mtb-human co-evolution may result in differential TB genetic risk profiles across population categories. There is accumulated evidence indicating that (A) Mtb strain diversity is not only non-random across the globe but also associated with specific TB phenotypes, and (B) the existence of high Mtb strain diversity in Ethiopia including a unique strain [Reference Rebuma47–Reference Mulugeta49]. Accordingly, the current study will help to assess whether recent findings in an East African population, Uganda, based on similar methods of defining TB phenotypes replicate in an independent neighboring Ethiopian population and assess if there are common signatures of population-specific association with TB phenotypes.
Methods
Ethical considerations
The research proposal received Ethical Clearance from the relevant institutions in Ethiopia: the Ethical Review Committee of the Department of Biology at Addis Ababa University and the National Health Research Ethics Review Committee of the Federal Ministry of Science and Technology of Ethiopia (certificate#: RDHE/37-92/2010). Samples were collected after obtaining informed and signed consent.
Phenotyping (clinical characterization)
Cases of active PTB were diagnosed by hospital medical personnel as per the guidelines provided by the Federal Ministry of Health of Ethiopia [50] and all cases were undergoing treatment during recruitment. In Ethiopia the diagnosis of TB is based on the recommendations of the National TB and Leprosy Control Programme (NTLC) [51] as follows:
-
• Patients presenting with symptoms suggestive of PTB who had productive cough for 3 weeks or more with at least two positive sputum smears or one positive smear and X-ray findings consistent with active PTB are classified as smear-positive PTB cases.
-
• Patients presenting with cough of 3 weeks or more with initial three negative smears and no clinical response to a course of broad-spectrum antibiotics, three negative smear results after a course of broad-spectrum antibiotics, X-ray findings consistent with active PTB and decided by a clinician to be treated with anti-TB chemotherapy are classified as smear-negative PTB cases.
-
• Patients presenting with dry cough of 3 weeks or more are diagnosed based on strong clinical evidence and X-ray findings consistent with active TB.
Other diagnostic services such as Mycobacterial cultures and pathological services are not available for routine purposes and as a result, the NTLC advocates adherence to the above algorithm [Reference Mengistu, Tesfaye and Madeley52].
For each PTB case, all available and consenting unrelated household contacts, mostly spouses, were selected as controls. This ensured that exposure to TB could be known with certainty, thus reducing the potential for misclassification of controls [Reference Stein7]. The QuantiFERON®-TB Gold In-Tube (QFT®) kit was employed to test samples of asymptomatic household contacts of active TB patients for latent infection. This is an in vitro diagnostic method that specifically tests for latent Mtb infection using peptide antigens that are absent from all BCG strains and from most non-tuberculosis mycobacteria. The IFN-gamma level of the Nil tube is compared with the TB antigen and Mitogen tubes to determine infection status (Supplementary Table S1). All subjects were tested for HIV, and HIV-positive subjects were excluded from further analyses.
In this study, samples were taken from three regions representing some of the North, Central and Southern ethno-geographic categories (EGCs) of Ethiopia: Merhabet (Amhara ethnic group, Amharic speakers), Adigrat (Tigray ethnic group, Tigringna speakers), and Arbaminch (Gamo ethnic group, Gamingna speakers), respectively. The Amhara and Tigray populations have a Semetic background while the Gamo is Omotic (Fig. 1).

Fig. 1. The sample-populations were selected in a manner that traverses some of the major ethno-linguistic-geographic groups in Ethiopia: Tigray, Amhara, and Gamo.
Genotyping/exome sequencing
Genomic DNA was extracted from buffy coat samples using QIAGENE (FlexiGene) as per product instructions. DNA quality and quantity were determined using both Nanodrop and Qubit. Exonic regions of candidate genes were sequenced, alligned and variants called using Illumina MiSeq and the Homosapiens/UCSC/hg19 human genome reference panel at the Genome Sequencing Core Facility of Case Western Reserve University (Supplemental Table S2). Samples were randomly placed on sequencing plates so that there was no bias by case/control status or by ethnicity.
DNA sequence data quality control (QC)
QC thresholds were selected that maximized the number of individuals and markers/SNPs included while ensuring appropriate QC for both. For the per-individual QC, individuals with less than 90% genotyping rate (i.e., individuals missing genotypes for more than 10% of the total markers) were removed. In the per-marker QC, markers with genotyping failure rate of less than 95% (i.e., markers genotyped in less than 95% of all samples), markers which failed Hardy–Weinberg–Equilibrium (HWE) deviation test with p < 0.001, and those with minor allele frequency (MAF) less than 0.01 were all removed. Tests for significant bias in genotyping rate between cases and controls were all non-significant. Coverage for these specific exonic regions was 100%. The total number of exonic nucleotide sequences was 11 927 (TICAM2 = 3578, NOD1 = 8349) with an average genotyping rate of 0.86–0.92. Most of the filtered out markers (91–95%) were discarded because they were monomorphic and thus uninformative for downstream association analysis (Supplementary Table S3).
Genetic association analysis
Unrelated case-control phenotype categories were developed in an increasingly restrictive or exclusive manner. This set up helps to study the impact of phenotype definition and perform a sensitivity analysis by observing the trend of association test statistic across the phenotype classifications and is one of the major strengths of this study. First, all controls were ascertained by virtue of their close association with active pulmonary TB patients as household contacts/care-givers and, therefore, most likely to have been exposed to Mtb. Second, they have been living in generally TB-endemic communities under conditions conducive to TB exposure and transmission (e.g., housing and population density). Third, controls were deemed negative for symptoms of active TB after a physical examination by qualified clinicians thus forming the group ‘No Active TB’ meaning ‘no symptoms of active TB after Mtb exposure’. Fourth, IGRA was performed to ensure whether the infection has been established or not after exposure, i.e., discriminate between the phenotypes of ‘LTBI: susceptibility/progression to infection’ and ‘No LTBI: resistance to infection’, respectively. [LTBI: Latent-TB-Infection].
We constructed two primary case v. control test-models to examine whether these genetic variants predispose to TB disease or Mtb infection, respectively:
-
• Test-model 1: ‘Active TB’ v. ‘No Active TB’ (153 cases, 139 controls)
-
• Test-model 2: ‘LTBI’ v. ‘No LTBI’ (70 cases, 64 controls)
To avoid potential concerns with heterogeneity in the control group in test model 1, we conducted sensitivity analyses by constructing additional test models as follows:
-
• Test-model 3: ‘Active TB’ v. ‘No LTBI’ (153 cases, 64 controls)
-
• Test-model 4: ‘Active TB’ v. ‘LTBI’ (153 cases, 70 controls)
Several complementary analysis strategies were used to examine the association between the genetic variants in NOD1 and TICAM2 and TB phenotypes using the test models described above. All analyses were conducted using PLINK [Reference Purcell53]. First, we compared the distribution of the minor allele in cases and controls using Pearson's χ2 and Fisher's exact test. We corrected for multiple testing using a Bonferroni correction: a total of 94 variants were examined between this study and another one, resulting in a corrected significance level of α = 0.05/94 = 5.32E-04. Correction for multiple testing was also done based on the false discovery rate (FDR) estimation algorithm of Benjamini, Hochberg and Yekutieli as implemented in PLINK. We also coded the genotypes according to dominant, additive, and recessive models to examine mode of inheritance; we found that the additive model was always the most significant (data not shown) so we proceeded with this model for further analysis. Allelic association tests were also conducted stratified by ethnic group as well as in the overall study population. Then, we conducted a logistic regression analysis, using additive coding of the SNP genotypes with the Cochrane–Armitage test assuming that each extra copy of the ‘risk’ allele increases risk equally, and included sex, age, and ethnicity as covariates in this model. To test for potential population stratification, we compared allele frequencies among the ethnic groups using a χ2 test, and also used multidimensional scaling (MDS) to empirically visualize and assess population stratification based on both self-declared ethnicity (EGC) of subjects and identity-by-state (IBS) of SNPs. PLINK employs complete linkage agglomerative clustering, based on pair-wise SNP IBS distance.
Linkage-disequilibrium patterns within the twogenes were plotted using Haploview's ‘Gabriel et al. algorithm’ based on QC-passed SNPs in 292 individuals included in the largest primary test-model: Active TB v. No Active TB (Figs 2 and 3 for NOD1 and TICAM2, respectively). Haplotype association analysis for both an omnibus and haplotype-specific tests were performed that compared the distribution of haplotypes in cases v. controls.

Fig. 2. LD pattern for NOD1: Pair-wise r 2 are plotted for NOD1 based on 23 QC-passed SNPs in 292 individuals included in Active TB v. No Active TB test-model.

Fig. 3. LD pattern for TICAM2: Pair-wise r 2 are plotted for TICAM2 based on 11 QC-passed SNPs in 292 individuals included in Active TB v. No Active TB test-model.
Results
In this study, 153 TB cases and 134 contacts without TB were enrolled (Table 1). Among the non-TB cases, 70 were QFT positive and 64 were QFT negative. The QFT negative cases were more likely to be female and younger. Most of the TB cases came from the Arbaminich (Gamo) ethnicity.
Table 1. Demographic characteristics of participants

We identified one novel variant in TICAM2 and two novel variants in NOD1. Results of SNP association tests by each phenotype contrast are shown in Table 2. Detailed results for the primary test-models, as well as for the sensitivity analyses, are presented in Supplementary Tables S4 and S5. Only additive coded models attained statistical significance. Two results in NOD1 achieved statistical significance after multiple-test correction: rs751770147 (p = 7.28E-05) and chr7:30477156(T) (p = 1.0E-04), a novel variant. These results were attained in the comparison of active TB cases to non-TB controls (test model 1). Nominally significant association results for these same two variants were also seen in sensitivity analyses for the contrast of Active TB to no LTBI, and also Active TB to LTBI (Supplementary Tables S4 and S5), providing support that these variants are associated with increased risk for TB. One of these variants, rs151170709, is a missense variant, with p < 0.05. Two other variants in NOD1 showed similar trends with increased risk for TB, and four variants in NOD1 also showed protective effects, though these were not significant after correction for multiple testing. In addition, two variants in TICAM2 were nominally associated with increased risk for TB as seen by at least two of the contrasts, and another two variants were associated with protection against TB in two models, though these results did not attain statistical significance after multiple testing corrections. One of these variants was a novel variant. However, it is to be noted that p < 0.05 is sufficient for declaring replication. Only one variant, in NOD1, was nominally associated with risk of LTBI compared with no LTBI. Minor allele frequencies for phenotype-associated SNPs are provided in Table 3.
Table 2. Summary results of SNP-based association tests: primary analysis
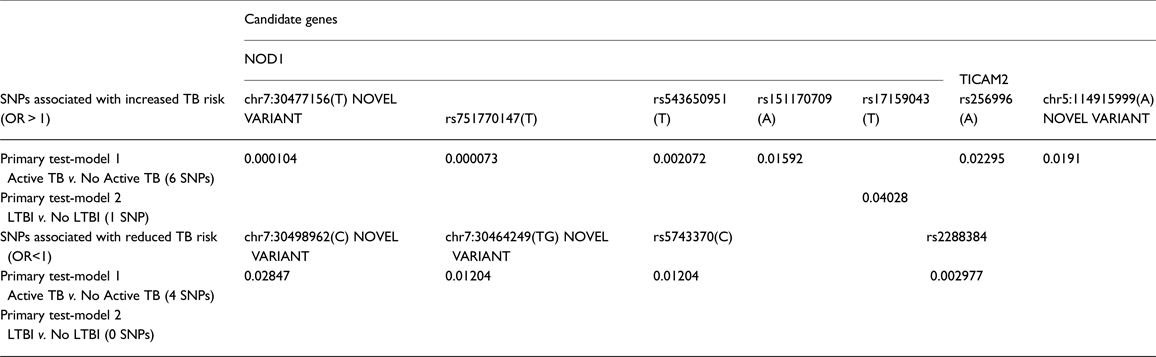
Table 3. Allele frequencies of TB phenotype-associated SNPs
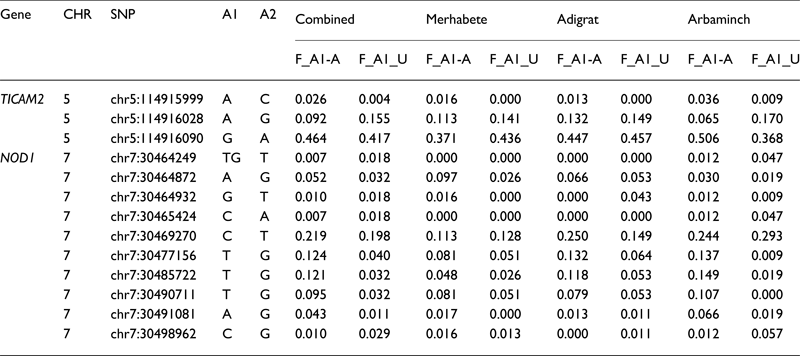
Note: A1/A2, minor/major allele; F_A1-A, frequency of minor allele in affecteds; F_A1-A, frequency of minor allele in unaffecteds.
Adjustment for the effect of sex, age and population stratification (EGC)
While most SNPs survived covariate analyses (Supplementary Tables S6–S9), those SNPs with already moderate association signals in the logistic regression model failed to retain significance. This is most probably because of sample size reduction and a concomitant loss of power to detect associations. For example, the Merhabete-Adigrat EGCs together constitute the least sample size and Adigrat-Arbaminch the largest; similarly, there was a successive reduction in sample size in the test-model construction. However, one cannot ignore population stratification as a possible confounder.
Accounting for possible population stratification
In as much as the actual ancestry of each individual can be inferred based on self-declared ethnicity or IBS, various tests were performed in order to directly assess and account for any possible stratification effect on the outcome of the association test statistic: population-specific tests of association (tests within each EGC); stratified analysis of association based on self-declared ethnicity (EGC) and based on assignment of individuals to homogeneous clusters based on IBS or empiric SNP data (Cochran–Mantel–Haenszel test); test for heterogeneous association (Breslow–Day test); and, test of allele frequency difference between EGCs (χ2 test) (Supplementary Tables S6–S10). Overall, while the top SNPs in this study associated with susceptibility phenotypes survived both the IBS and EGC-based tests of association, the IBS-based stratified analysis identified more SNP-phenotype association than the EGC-based test.
All phenotype-associated markers which survived the Bonferroni adjustment for multiple testing survived the FDR-adjustment as well (Supplementary Table S11).
These results, coupled with the observation that the association of the top SNP in this study, rs751770147, was replicated in both the Adigrat and Arbaminch populations with relatively higher sample sizes but not in the Merhabete population with the least sample size, may indicate that the effect of sample size could be a more probable explanation for the varied test results of the population-specific association tests.
LD structure
LD structure of the exonic regions was examined using Haploview which showed little LD in both genes suggesting a minor role for SNP co-segregation for the observed pattern of association (Figs 2 and 3 for NOD1 and TICAM2, respectively).
Haplotype analyses
The omnibus haplotype association test, which examines the difference in haplotype distribution between cases and controls, was significant for TB v. no TB in TICAM2 (p = 0.0178) and NOD1 (p = 0.0381) (Supplementary Tables S12 and S13). This test statistic was particularly significant in the contrast between TB and LTBI for NOD1 (p = 0.000359), providing additional evidence that this gene is associated with progression to active TB disease. In general, despite the apparently low LD between the TB phenotype-associated SNPs, the haplotype analysis further supports the single SNP association test results.
Discussion
Our results replicated the association of NOD1 and TICAM2 genes with TB in Ethiopian populations. We found two variants in NOD1 that were significantly associated with TB risk, and additional variants that were nominally associated. Furthermore, one of the most significantly associated SNPs in NOD1 was novel. Though we did not observe a statistically significant association between TB and TICAM2 variants after accounting for multiple testing, these nominally significant results still provide an independent replication of a previous report [Reference Hall23]. By contrasting TB disease with no disease and LTBI v. no LTBI, our study design helped to unravel a pattern of SNP-phenotype associations to determine which stage of TB pathogenesis was associated with these variants. In our analyses, we found consistent evidence of association of NOD1 variants with active TB in both our primary and sensitivity test models, and two of these results were significant after multiple-test correction. We did not observe notable associations with LTBI as the case phenotype, except for a single nominally associated NOD1 SNP, though some of the individuals in this category may progress to active TB later in time by virtue of possessing any of the susceptibility variants. The relatively smaller sample size in this test-model may also have reduced the power to detect association signals.
In addition to the careful phenotypic characterization of the control population, there are other aspects of this study that further enhance its validity. First, although other transcription-regulatory regions are also important, we focused on padded full exon resequencing of the two candidate genes of interest, enabling the discovery of novel genetic variants with a higher probability of translation. Second, we recruited individuals from multiple ethnic groups, and examined possible population stratification effects. It is hoped the design and conduct of this study, with particular emphasis on disease trait definitions that incorporate intermediate phenotypes, will serve as a genetic epidemiological model to future studies of complex disease traits. Although there were slight evidence suggestive of the presence of some population-specific genetic signatures, there was no strong indication of population stratification that would lead the authors of this study to put forward a recommendation of the prohibition of sampling from different ethno-geographic backgrounds and pooling data together particularly for studies based on exonic data of candidate genes.
Although it is typical in replication studies to perform a meta-analysis, we did not do so here for two reasons. First, to our best knowledge, this is only the first attempt at replication of these two genes. Second, we have taken an alternative approach that would make a meta-analysis with the data from the original work [Reference Hall23] nearly impossible. Rather than analyzing the same exact SNPs as in the original report, we conducted exon sequencing with some flanking buffers. Although this enabled us to identify novel variants in their association with TB, the gaps between the exons meant that we could not capture all the SNPs in the original study, i.e., the percentage of matching sequences between the two studies was low (6 SNPs in TICAM2 and 34 SNPs in NOD1) which were further reduced by QC filtering. This effectively precluded a comprehensive comparative assessment at finer levels beyond the overall gene level. Nonetheless, we were able to capture one SNP in NOD1, rs17159043, which showed signals of association with TB in both the original study (unadjusted p value of 0.009) and the current replication study (unadjusted p value of 0.040 in LTBI v. No LTBI).
The relatively small sample size of this study is a limitation as it affects statistical inference and declaration of significance. Particularly, the progressively stricter definition of case-control phenotypes across test-model constructs comes at the cost of reduced sample size. This difference in sample size could explain in part the difference in the observed pattern of SNP-phenotype associations between the test-models. In addition, mycobacterial growth in culture, the gold standard for TB diagnosis, is not routinely used in Ethiopia, though the diagnostic criteria implemented are still quite rigorous. Lastly, there were quite a few subjects that tested HIV positive or had indeterminate Quantiferon results, which eliminated them from some analyses, thus reducing power. Therefore, future research with larger sample size is warranted.
In conclusion, we have provided an independent replication of association between TB and variants in TICAM2 and NOD1. By conducting QFT to assess latent infection status, we were able to demonstrate that a higher proportion of these variants are associated with susceptibility to active TB disease, not a latent infection. Some variants in both genes were also associated with reduced risk to active TB. We examined multiple ethnic groups in Ethiopia, and found that our association results are robust to population stratification. We also identified novel variants in these genes. As Ethiopia is considered to be the origin of both humanity and Mtb, these findings are important for understanding Mtb-human co-evolution and the genetic underpinnings of TB in general. Our findings also support the hypothesis that the Ethiopian setting could indirectly help to explore the possibility that Mtb-human co-evolution may result in differential TB genetic risk profiles across population categories in that while our Ethiopian study replicated the Ugandan findings at the gene level, the associated SNPs in overlapping loci are not shared and some are novel. Therefore, while the Ugandan and Ethiopian findings indicate, on the one hand, a common signature of association at the gene level in the two populations, on the other hand, at the SNP level, the results suggest at the possible existence of population-specific signatures of association which may be driven by differential Mtb-human co-evolution. This phenomenon of population-specific adaptation is not uncommon and has been previously reported in Ethiopian populations with regard to genetic and physiological adaptations to high altitude (hypoxia) [Reference Beall54, Reference Scheinfeldt55]. The results of this study could encourage investigators who may be intimidated by the ‘burden’ of the high genetic diversity in African populations. The conduct of the study, interpretation of the results and any limitations should be informative to the efforts of the Human Heredity and Health in Africa initiative [56] at enriching region-specific health-related genomic knowledge.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/gheg.2017.17
Acknowledgement
The authors are grateful to all the participants of this study and applaud their high spirits of voluntarism and dedicate this work to them. They are grateful to the staff of Alem Ketema Hospital in Merhabete, Adigrat Hospital in Adigrat, and Arbaminch Hospital in Arbaminch for their unreserved cooperation and professional support in appropriate sample ascertainment, recruitment, interview, and collection of blood-samples. The authors also wish to acknowledge Keith Chervenak in the Tuberculosis Research Unit at Case Western Reserve University for his assistance running Quantiferon, and Dr. Alex Miron, Simone Edelheit, and Milena Rajak for sequencing the samples. For their various support and encouragement, the authors thank: the Department of Microbial, Cellular, and Molecular Biology and the Institute of Biotechnology of Addis Ababa University; Health and Nutrition Research Institute and Holetta Agricultural Research Institute of Ethiopia; and, all the staff at the Center for Research on Genomics and Global Health at NIH, USA, particularly Charles Rotimi, Adebowale Adeyemo, Amy Bently, and Fasil Ayele Tekola. This work was supported by C. M. S (grant # R01HL096811, National Institutes of Health).
Conflict of Interest
None.









 Open access
Open access