



Sarcopenic obesity is an important health challenge, characterised by the double burden of diminished skeletal muscle mass concurrent with excess adiposity(Reference Zamboni, Mazzali and Fantin1,Reference Baumgartner2) . Population ageing, coupled with the ever-rising obesity incidence has led to significant proportion of adults who display excess adiposity combined with reduced skeletal muscle mass(Reference Kim, Yang and Yoo3,Reference Batsis, Mackenzie and Emeny4) . Currently, overweight and obesity affects 50 % of the European population, accounting for about 80 % of type 2 diabetes (T2D) cases(Reference Tsigos, Hainer and Basdevant5). Obesity is typified by increased lipid accumulation in adipose tissue, with peripheral ectopic fat deposition in the liver and skeletal muscle(Reference Reilly and Saltiel6,Reference Longo, Zatterale and Naderi7) . Sarcopenia is associated with the progressive loss of muscle mass and strength, typically coupled with ageing(Reference Cruz-Jentoft, Bahat and Bauer8). It is estimated that the prevalence of sarcopenia ranges from 1 to 29 % in community-dwelling individuals over the age of 50 years(Reference Cruz-Jentoft, Landi and Schneider9). The risk of adverse outcomes associated with sarcopenia, including falls, physical disability and frailty, can be greatly perpetuated by coincident obesity(Reference Follis, Cook and Bea10–Reference Hirani, Naganathan and Blyth12). Indeed, sarcopenic obesity has additional comorbidities also associated with obesity, namely CVD, T2D(Reference Baumgartner2,Reference Villareal, Banks and Siener13) , as well as all-cause mortality(Reference Baumgartner, Wayne and Waters11,Reference Zhang, Xie and Dou14) .
Sarcopenic obesity is characterised by the double burden of diminished skeletal muscle mass and excess adiposity(Reference Baumgartner2). This review is specifically intended to focus on the interaction between inflammation and metabolism, specifically focusing on the impact of adiposity in sarcopenic obesity. Despite a wealth of literature on both obesity and sarcopenia, there is substantial heterogeneity in the diagnostic criteria used to define sarcopenic obesity(Reference Prado, Wells and Smith15–Reference Cruz-Jentoft, Baeyens and Bauer19). Nevertheless, more recent efforts have sought to establish potential synergism between the causal factors and mechanisms underlying the two coincident conditions(Reference Batsis and Villareal16,Reference Kalinkovich and Livshits20–Reference Cleasby, Jamieson and Atherton24) .
Fig. 1 illustrates our conceptual model of the potential impact of different lifestyle determinants on risk of sarcopenic obesity, wherein there has been a surge in research investigating whether co-regulatory link(s) exist between reduced muscle mass and obesity, v. a coincidental association(Reference Batsis and Villareal16,Reference Kalinkovich and Livshits20,Reference Cleasby, Jamieson and Atherton24,Reference Jo, Lee and Park25) . Ageing is typically characterised by increased adiposity and reduced physical activity(Reference Baumgartner2,Reference Kruger, Carlson and Buchner26,Reference Taylor, Cable and Faulkner27) . In addition to ageing, suboptimal dietary patterns and physical inactivity play a role(Reference Deutz, Bauer and Barazzoni28). From the nutritional perspective, inadequate dietary protein intake coupled with excess dietary energy intake, rich in SFA, are two potential factors in sarcopenic obesity(Reference Deutz, Bauer and Barazzoni28–Reference Rasaei, Kashavarz and Yekaninejad33).
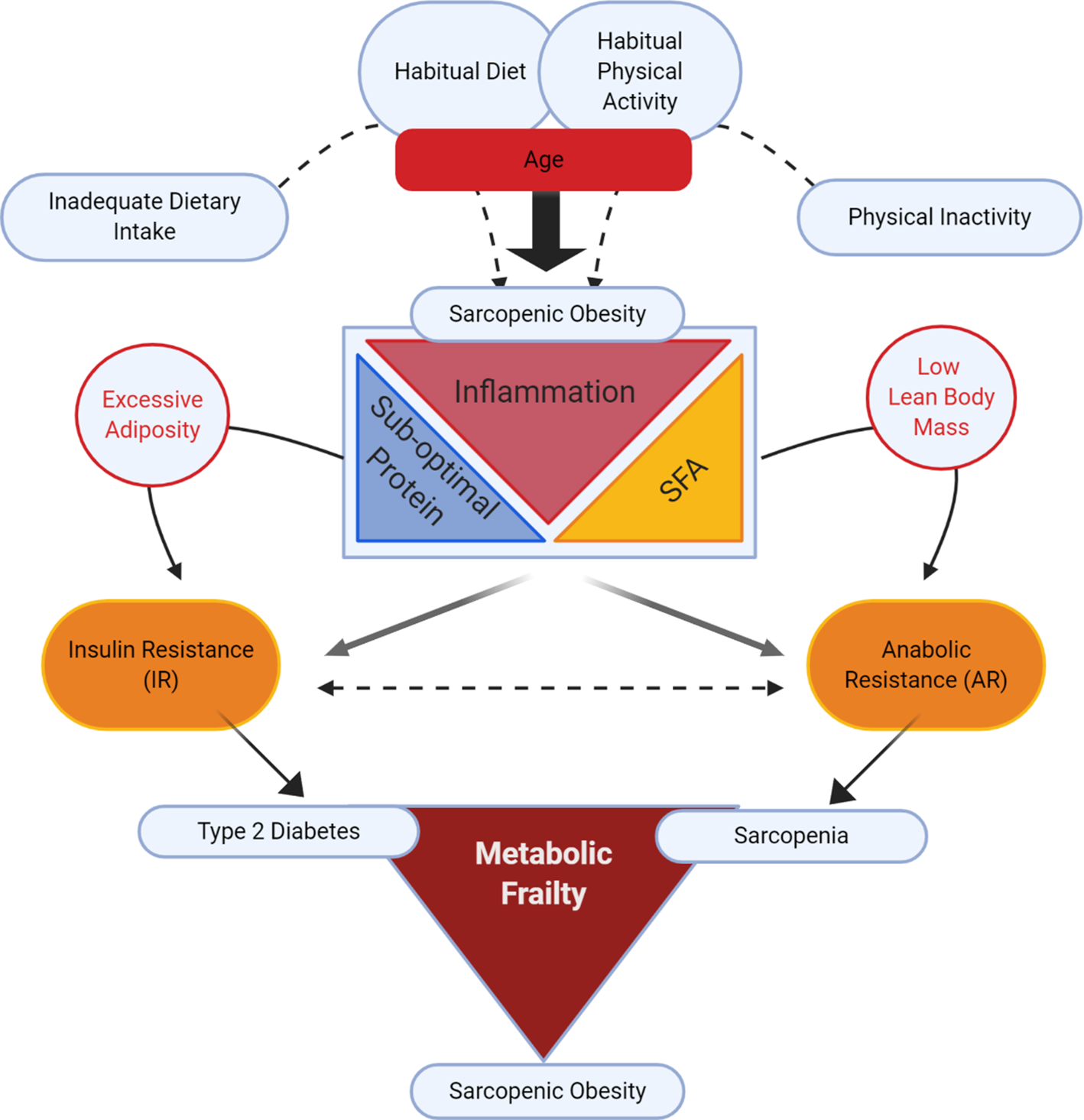
Fig. 1. (Colour online) Lifestyle determinants of sarcopenic obesity: impacting insulin resistance v. anabolic resistance.
From the mechanistic perspective, both obesity and sarcopenia are associated with a sub-acute, chronic pro-inflammatory state that probably impedes metabolic processes, disrupting both adipose and skeletal functionality(Reference Cleasby, Jamieson and Atherton24,Reference Dalle, Rossmeislova and Koppo34–Reference Beyer, Mets and Bautmans36) . The conceptual model in Fig. 1 purports that sub-acute chronic inflammation may interact with excess dietary and/or endogenous SFA, and that inadequate protein intake may augment the risk of sarcopenic obesity. Indeed, we speculate that two important and concurrent molecular processes may be at play, namely insulin resistance (IR) and anabolic resistance (AR), both of which are possibly largely impacted by inflammation(Reference Dalle, Rossmeislova and Koppo34–Reference Ferrucci and Fabbri38). IR is defined as sub-optimal insulin-stimulated glucose uptake into metabolic tissues, including skeletal muscle and adipose tissue(Reference Shulman39,Reference Samuel and Shulman40) . Obesity-related inflammation is a key factor contributing to IR (for review, see(Reference Ralston, Lyons and Kennedy41)). AR is defined as the blunted response of skeletal muscle protein synthesis (MPS) to anabolic stimuli such as protein intake(Reference Moore, Churchward-Venne and Witard42) and muscle contraction(Reference Kumar, Atherton and Selby43). A number of factors may contribute to AR including impaired protein digestion and absorption, sub-optimal muscle amino acid sensing and dysregulated cell signalling in myotubes in response to anabolic hormones (insulin and insulin-like growth factor-1)(Reference Haran, Rivas and Fielding44–Reference Moro, Ebert and Adams48). In sarcopenic obesity, increased adiposity with IR likely coincides with age-related AR(Reference Cleasby, Jamieson and Atherton24,Reference Meex, Blaak and van Loon49) . Thus together IR and AR may act synergistically to develop a state of metabolic frailty, that can culminate in a triad of T2D, sarcopenia and sarcopenic obesity(Reference Meex, Blaak and van Loon49).
Insulin resistance v. anabolic resistance: two sides of the same coin?
Considering the metabolic and molecular processes associated with sarcopenic obesity, it is plausible that IR and AR are two inter-related co-existing processes in sarcopenic obesity(Reference Khadra, Itani and Tannir50). As illustrated in Fig. 1, AR and IR co-exist but have separate effects on skeletal muscle, resulting in sarcopenia(Reference Breen and Phillips51) and T2D, respectively. When AR and IR exist simultaneously, they may evoke the development of sarcopenic obesity. Obesity not only causes IR, but also within the context of sarcopenic obesity, it likely also promotes musculoskeletal AR(Reference Beals, Burd and Moore52–Reference Tardif, Salles and Guillet54), ultimately contributing to muscle atrophy with increased adiposity(Reference Zhu, Tian and Torigoe55). Thus, in sarcopenic obesity, IR and AR may represent two sides of the same coin wherein the occurrence of both muscle atrophy with AR, and IR in adipose and skeletal muscle, leads to sarcopenic obesity.
Muscle homoeostasis is maintained primarily through a balance between the processes of MPS and muscle protein breakdown (MPB). Anabolic stimulus such as exercise or nutrition triggers skeletal muscle by activating the complex mTORC1(Reference Bond56). Active mTORC1 results in the phosphorylation of substrates 4EBP1 and P70S6K1, enhancing translation of mRNA to protein(Reference Sandri57), a critical step in MPS that is interrupted in AR and sarcopenia. Simultaneously, MPB-related pathways are supressed(Reference Zhao, Zhai and Gygi58) by mTORC1 and Akt. In addition to Akt activation of mTORC1, Akt controls the forkhead box O (FOXO) family of transcription factors including FOXO1 and FOXO3a(Reference Leger, Cartoni and Praz59). Stimulation of Akt by anabolic stimuli inhibits FOXO, resulting in the deactivation of transcriptional E3 ubiquitin ligases, MuRF-1 and MAFbx. Without Akt activation, these ligases are involved in ubiquitination and proteasomal degradation of different substrates(Reference Bodine and Baehr60).
This imbalance between MPS and MPB, in favour of MPB, is a key contributor to loss of skeletal muscle mass(Reference Koopman and van Loon61). When comparing the elderly population to their younger counterpart, postprandial MPS rates are reduced, specially to small amino acid doses(Reference Moore, Churchward-Venne and Witard42), as well as to a bout of resistance exercise(Reference Kumar, Selby and Rankin62). This decreased MPS rate is linked to reduced mTORC1 downstream phosphorylation of P70S6K1 and 4EBP1 targets(Reference Fry, Drummond and Glynn63). The molecular signatures of ageing and physical inactivity in skeletal muscle level are similar(Reference McGlory and Phillips64) and are also often concomitant (i.e. overall older people are more sedentary than younger people)(Reference Wall, Dirks and Van Loon65). While bouts of exercise induce hypertrophy, muscle disuse (with the sum effect of ageing via AR) induce atrophy(Reference Wall, Dirks and Van Loon65–Reference Dirks, Wall and Nilwik68). Furthermore, the muscle of obese individuals also experiences an attenuated anabolic response to protein ingestion and muscle contraction, in comparison with their healthy counterpart (for review, see(Reference Beals, Burd and Moore69)). This state of AR may reflect inertia towards insulin, insulin-like growth factor-1, sub-acute inflammation and/or musculoskeletal lipid infiltration(Reference Beals, Burd and Moore69), which we will explore in more detail later. Interestingly from the muscle atrophy or MPB perspective, MuRF-1 and atrogin-1 are actively transcribed in models of atrophy, metabolic stress and inflammatory conditions(Reference Bodine and Baehr60) including (but not restricted to) fasting(Reference Lecker, Jagoe and Gilbert70,Reference Allen, Cleary and Lindsay71) , bed rest(Reference Ogawa, Furochi and Mameoka72), T2D(Reference Lecker, Jagoe and Gilbert70), cachexia(Reference Lecker, Jagoe and Gilbert70,Reference Siddiqui, Hassan and Harvey73,Reference Costelli, Muscaritoli and Bossola74) and ageing(Reference Altun, Besche and Overkleeft75–Reference Raue, Slivka and Jemiolo77). Essentially, we do not have full clarity in relation to the molecular processes that control AR. Indeed, AR does not explain sarcopenia in T2D, despite reduced mTOR pathway activity compared with healthy controls(Reference Cuthbertson, Babraj and Leese78). Thus, greater understanding in relation to if or how we can regulate the molecular processes and inter-relationship between AR and IR may be fundamental to understanding the extent to which we can attenuate age- and obesity-related sarcopenia.
Obesity and adipose tissue inflammation
White adipose tissue is a multifaceted organ which not only plays an important role in energy homoeostasis, but also acts as an endocrine organ, secreting paracrine factors to influence the activities of surrounding cells and subsequently regulate other metabolic organs. The contribution of obese adipose tissue in the generation of a chronic, sub-acute pro-inflammatory state is well-documented(Reference Reilly and Saltiel6,Reference Ralston, Lyons and Kennedy41,Reference Harford, Reynolds and McGillicuddy79) , therefore we provide a brief synopsis of key cellular and molecular alterations. In response to excess energy intake, pre-adipocytes and mature adipocytes increase in size (hypertrophy) and number (hyperplasia)(Reference Drolet, Richard and Sniderman80,Reference Spalding, Arner and Westermark81) to augment the storage capacity of excess dietary energy in the form of TAG(Reference Longo, Zatterale and Naderi7,Reference Gregoire, Smas and Sul82) . As obesity develops, white adipose tissue can become progressively dysfunctional(Reference Sun, Kusminski and Scherer83). First, adipose can lose the ability to effectively store excess dietary and endogenous lipids(Reference Spalding, Bernard and Näslund84). When TAG is not effectively stored in adipose, NEFA can spill over, increasing circulating lipid concentrations and causing inappropriate lipid deposition in peripheral metabolic tissues(Reference Yki-Järvinen85,Reference Karpe, Dickmann and Frayn86) , which has detrimental consequences on metabolic and skeletal health. Secondly, the adipose tissue-derived pro-inflammatory milieu can have detrimental effects on pre-adipocyte and adipocyte biology, which also contribute to adipose dysfunction(Reference Harford, Reynolds and McGillicuddy79). Pro-inflammatory cytokines such as IL-1β inhibit adipogenesis, or the ability of pre-adipocytes to mature into functional TAG-enriched adipocytes(Reference McGillicuddy, Reynolds and Finucane87). Thus, less functional adipocytes are present to carry the TAG load, leading to the typical hypertrophic adipose phenotype, greater potential for NEFA and TAG spill over and peripheral lipotoxicity(Reference Dulloo, Antic and Montani88).
With increasing obesity, the stroma vascular fraction between adipocytes becomes infiltrated with a range of immune cells, including adipose tissue macrophages (ATM), T-cells, mast and dendritic cells(Reference Harford, Reynolds and McGillicuddy79). Enhanced immune cell infiltration, with the associated pro-inflammatory milieu, has been implicated in the progression of whole-body glucose intolerance, IR and T2D(Reference Olefsky and Glass89). Immune cells, particularly macrophages, display great ‘plasticity’ wherein the nature of the cellular phenotype can switch between pro- and anti-inflammatory states, which largely reflect their biological environment(Reference Lumeng, Bodzin and Saltiel90). In obese adipose tissue, the majority of ATM display an M1 or ‘classically activated’ pro-inflammatory phenotype(Reference Lumeng, Bodzin and Saltiel90–Reference Kratz, Coats and Hisert92). These M1 ATM largely replace the M2 ATM or ‘alternatively activated’ macrophages which are present in lean adipose tissue and display an anti-inflammatory or pro-resolving secretome(Reference Ouchi, Parker and Lugus93). M1 ATM secrete a range of pro-inflammatory mediators including TNFα, IL-1β, IL-6, IL-8 CCR2, CCR5, MCP-1, MIP-1α, resistin and nitric oxide(Reference Olefsky and Glass89–Reference Castoldi, Naffah de Souza and Câmara91,Reference Trim, Turner and Thompson94) . Obese and dysfunctional adipose tissue also display ‘crown-like structures’ wherein necrotic adipocytes become surrounded by ATM and other immune cells(Reference Osborn and Olefsky95) and release a range of damage-associated molecular proteins(Reference Sun, Ji and Kersten96) which are recognised by nod-like receptor proteins(Reference Yang, Wang and Kouadir97). Nod-like receptor protein-3 activation then results in the maturation and activation of IL-1β, further contributing to adipose inflammation(Reference Vandanmagsar, Youm and Ravussin98). It is probable that a range of adipose-derived signals, including lipids, lipid derivatives, cytokines and/or adipokines, adversely impact skeletal muscle metabolism and inflammation(Reference Pillon, Bilan and Fink99), all of which are potentially relevant within the context of sarcopenic obesity.
Adipose tissue, adipokines and lipids: putative crosstalk with muscle metabolism
Fig. 2 illustrates a range of potential adipose-derived agents that may interrupt skeletal health with age and increasing adiposity. In sarcopenic obesity, it is well established that low protein intake and physical inactivity adversely affects skeletal health(Reference Deutz, Bauer and Barazzoni28,Reference Granic, Mendonça and Sayer30–Reference Xu, Houston and Locher32,Reference Katsanos, Kobayashi and Sheffield-Moore45,Reference McGlory and Phillips64) . In terms of adipose-derived signals, key processes include inflammation, lipid infiltration, lipotoxicity and impaired reactive oxygen stress responses(Reference Lumeng, Bodzin and Saltiel90,Reference Arner, Bernard and Salehpour100,Reference Mau and Yung101) . This review will mainly focus on the first two in order to align with the remit of our review, pertaining to the role of adiposity, inflammation and metabolism in sarcopenic obesity. Nevertheless, we fully acknowledge that cellular stress responses and inefficient nutrient sensing are equally important molecular processes. The pro-inflammatory cytokines, IL-1β and TNFα, secreted from obese ATM impact both adipose metabolism and inflammation. Chronic stimulation of pro-inflammatory signalling pathways, particularly MAPK, JNK and NF-κB(Reference Schutze, Wiegmann and Machleidt102), impedes insulin signalling, adipogenesis and adipocyte lipid storage, and augments adipocyte lipolysis, all of which reduce adipocyte capacity to store and manage excessive energy intake as an inert lipid depot(Reference Hotamisligil, Shargill and Spiegelman103–Reference McGillicuddy, Chiquoine and Hinkle107). This leads to lipid spill over from adipose to peripheral organs.
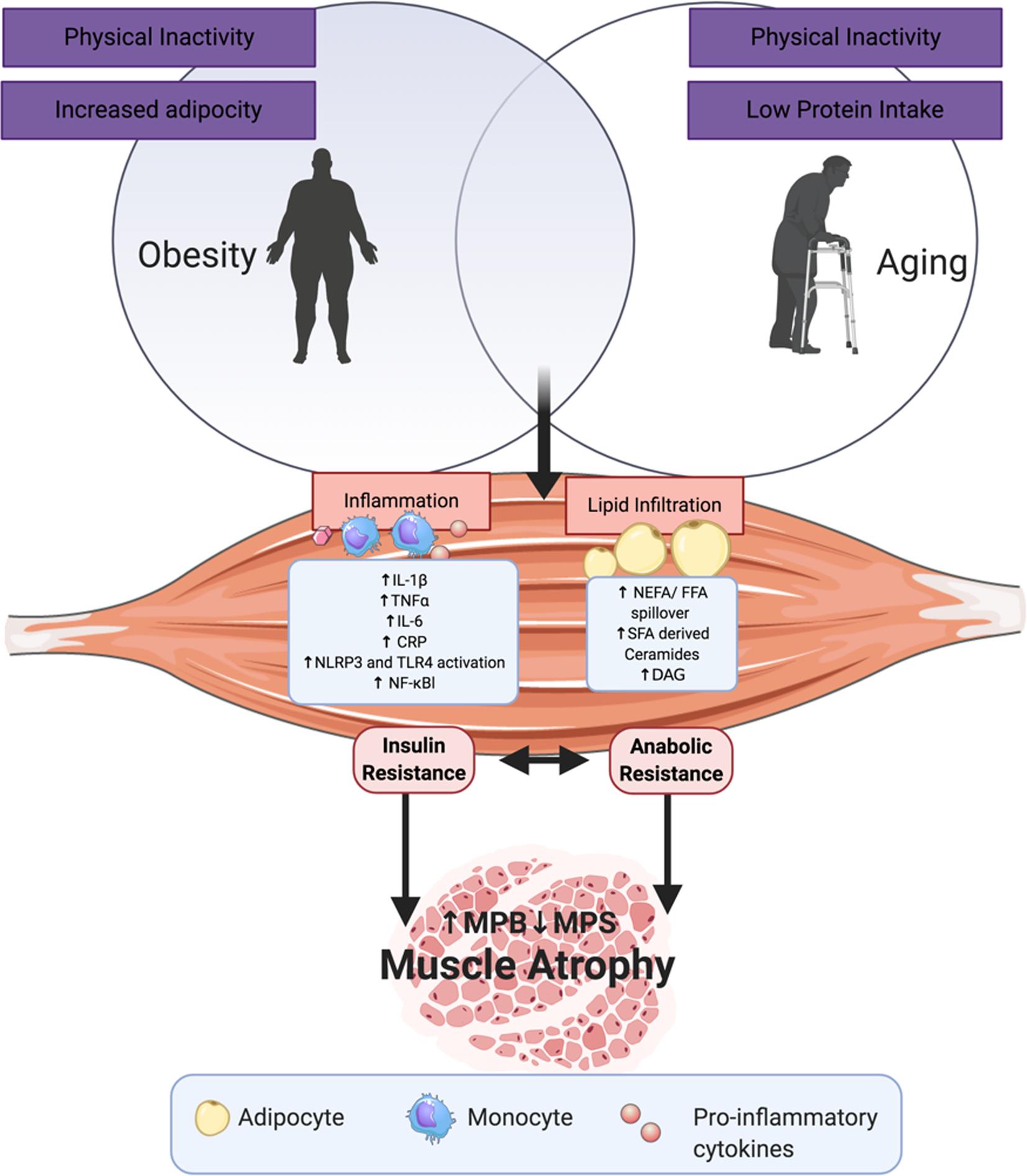
Fig. 2. (Colour online) Adipose- and ageing-derived detriments to skeletal muscle: some of the adipose-derived insults that interact with skeletal biology, lipid derivatives and adipose inflammatory mediators as those mentioned, as well as reactive oxygen species (ROS), reactive nitrogen oxide species (RNOS) and impaired nutrient sensing. CRP, C-reactive protein; MPS, muscle protein synthesis; MPB, muscle protein breakdown; DAG, diacylglycerol; NLPR, nucleotide-binding oligomerisation domain, leucine rich repeat and pyrin domain containing; TLR, Toll-like receptor.
Research demonstrates that lipid mediators, particularly those derived from SFA, are potent inflammatory agonists, via several potential mechanisms. SFA stimulate the pro-inflammatory responses via Toll-like receptor- and nod-like receptor protein-3-mediated pathways, which promote downstream NF-κB signalling to induce cytokine production (e.g. IL-1β, TNFα, IL-6 and MCP-1). These cytokines act in a paracrine manner to further recruit ATM into the stroma vascular fraction of adipose tissue and contribute to systemic low-grade inflammation wherein they may interrupt skeletal metabolism. Indeed, adults with sarcopenic obesity display elevated circulating concentrations of IL-6, TNFα and the acute phase protein C-reactive protein (CRP)(Reference Cesari, Kritchevsky and Baumgartner37).
The extent to which ATM-derived cytokines and/or adipocytokines have direct adverse metabolic effects on skeletal muscle in vivo is not fully deciphered. Age-related low-level chronic inflammation (sometimes termed inflammaging), characterised by elevated plasma/serum pro-inflammatory cytokine concentrations is often attributed to adipose tissue, which in turn may interrupt MPS (i.e. blunted via enhanced IR and AR) and MPB (i.e. via protein degradation promotion) ultimately leading to muscle protein catabolism(Reference Franceschi and Campisi108–Reference Roubenoff110). Inflammaging is typified by persistently high levels of circulating pro-inflammatory cytokines (e.g. TNFα, IL-6 and IL-1) and acute phase proteins (e.g. CRP)(Reference Schaap, Pluijm and Deeg111–Reference Jozsi, Dupont-Versteegden and Taylor-Jones114); while in the absence of obesity, sarcopenia in human subjects is characterised by higher CRP levels only(Reference Bano, Trevisan and Carraro35). Overall, higher levels of these pro-inflammatory markers are associated with lower muscle mass and strength in older participants(Reference Barbieri, Ferrucci and Ragno115–Reference Wåhlin-Larsson, Wilkinson and Strandberg119). However, there is not full clarity in relation to whether the pro-inflammatory phenotype is coincidental or causal with respect to the impact of adipose-derived inflammation on skeletal health.
The putative impact of such inflammatory insults may contribute to AR by blunting MPS. For instance, chronic CRP elevation is inversely related to lean muscle mass in older women(Reference Wåhlin-Larsson, Wilkinson and Strandberg119). However, it needs to be noted the CRP is an acute phase protein, secreted predominantly by the liver, although indicative of systemic inflammation. Mechanistically, human myotubes exposed to CRP resulted in reduced size and decreased MPS rates, as well as increased activation of AMPK and inhibition of Akt signalling, thus reducing mTORC1 activity(Reference Wåhlin-Larsson, Wilkinson and Strandberg119). Additionally, evidence from human- and murine-derived cells as well as in vitro studies show that the local expression of insulin-like growth factor-1 is blunted by pro-inflammatory cytokines TNFα and IL-1β, leading to impairment of glucose disposal into the muscle and the resistance of mTORC1 activation from IRS1 phosphorylation and its downstream cascade, an example of the IR and AR synergistic effect(Reference Jo, Lee and Park25,Reference Späte and Schulze120–Reference Steensberg, Keller and Starkie123) . NF-κB is activated by TNFα in rat and mouse skeletal muscle cells to ultimately induce protein degradation(Reference Guttridge, Mayo and Madrid124–Reference García-Martínez, Agell and Llovera126), but downstream activation of NF-κB has also been reported from excess muscle fatty acid infiltration (for review, see(Reference Jo, Lee and Park25)). As such, the impact of these cytokines may be both direct and indirect via lipid derivatives including ceramides. Indeed, Strle et al.(Reference Strle, Broussard and McCusker127) showed that ceramide, a SFA-derived second messenger in TNFα and IL-1β signalling pathways, was a key element in insulin-like growth factor-1 resistance and thus AR in vitro. High TNFα concentrations also result in the activation of the caspase cascade via reactive oxygen species production and TNFR1 activation. TNFR1 activation increases cell apoptosis(Reference Jo, Lee and Park25,Reference Marzetti, Wohlgemuth and Lees128) leading to the release of circulatory inflammatory peptides such as TNFα to continue the blunted MPS loop as well as the increase in plasma NEFA and thus intracellular fatty acids(Reference Jo, Lee and Park25).
Whilst our review is focused on the potential impact of adipose inflammation on IR and AR, it would be remiss not to highlight that inflammatory insults may play a role in skeletal degradation. Switching the focus to MPB upon local inflammatory stimuli, by binding to TNFR1, the role of TNFα-induced skeletal muscle loss in ageing mice was pinpointed to the NF-κB-mediated activation of E3 ligases (MAFbx and MuRF-1) and p38MAPK. By virtue of ubiquitination of MyHC proteins and induced malfunction of skeletal muscle satellite cells, NF-κB promoted protein degradation and impaired regeneration(Reference Jo, Lee and Park25,Reference Li, Yi and Yao129) (previous murine data supports these findings(Reference Guttridge, Mayo and Madrid124–Reference García-Martínez, Agell and Llovera126,Reference Wu, Cornwell and Jackman130) ). TNFα acts in a positive feedback loop to increase the transcription of additional TNFα, as well as IL-1β and IL-6, which feed into chronic inflammation and reactive oxygen species production, leading to NF-κB-mediated ubiquitination and protein degradation(Reference Jo, Lee and Park25). Importantly however, existing human evidence indicates MPB is not elevated in non-pathological ageing(Reference Raue, Slivka and Jemiolo77), although there is some evidence that the suppressive effect of insulin on MPB may be diminished at moderate insulin concentrations (about 15 μIU/ml) equivalent to those following a small, low glycaemic index meal(Reference Wilkes, Selby and Atherton131).
‘Lipid saturation’: lipid-derived adipose signals and skeletal health
Hypertrophic obese adipose tissue tends to become increasingly dysfunctional with increasing obesity, wherein excessive ‘lipid saturation’ of adipocytes leads to significant spill-over into peripheral tissues(Reference Yki-Järvinen85,Reference Karpe, Dickmann and Frayn86) , including the skeletal muscle compartment(Reference Zoico, Rossi and Di Francesco132). Lipotoxicity refers not only to the aforementioned ‘spill-over’ but also generation of lipid mediators, sphingolipids, TAG and diacylglycerols, all of which may adversely impact skeletal muscle biology(Reference Bandet, Tan-Chen and Bourron133,Reference Bergman and Goodpaster134) . Intramyocellular lipid accumulation, present as deposits of TAG, diacylglycerols or sphingolipids, are a characteristic of lipotoxicity. Elevated intramyocellular lipids are observed in obese, IR and T2D subjects(Reference Kelley, Slasky and Janosky135). Greater intramyocellular lipid content is associated with greater content of ceramides. Ceramide's potent lipotoxicity has been shown to impede intracellular anabolic signalling and is implicated in the progression of IR in skeletal muscle(Reference Lipina and Hundal136). For example, an in vitro study showed that ceramide treatment decreased small neutral amino acid transporter mediated sarcolemmal translocation and amino acid uptake in rat myotubes, leading to the suppression of p70S6KT389 phosphorylation and MPS(Reference Hyde, Hajduch and Powell137). It has been widely shown in vitro that palmitate treatment of C2C12 myoblasts results in ceramide accumulation(Reference Tardif, Salles and Guillet54), IR, upregulation of pro-apoptotic genes(Reference Bryner, Woodworth-Hobbs and Williamson138,Reference Turpin, Lancaster and Darby139) , diminished protein synthesis, augmented FOXO3a and upregulated eIF2a activation(Reference Tardif, Salles and Guillet54). Impeding ceramide synthesis results in improvements in mTOR signalling in vitro (Reference De Larichaudy, Zufferli and Serra140). Even though ceramide levels have been identified to be significantly higher in obese rats compared to their lean counterparts(Reference Rivas, Morris and Fielding141), in human subjects there were no identifiable differences between total ceramide levels in older v. younger individuals(Reference Rivas, Morris and Haran142). Although difference existed for specific ceramides (C16:0, C18:0 and C20:0), and C16:0 levels were negatively correlate with lower leg lean mass in both healthy young and elderly cohorts(Reference Rivas, Morris and Haran142).
Both high-fat diets(Reference Bachmann, Dahl and Brechtel66) and increasing plasma NEFA concentrations through lipid infusion(Reference Boden, Lebed and Schatz143) impair insulin sensitivity, and emerging evidence indicates that lipid overload may also induce AR(Reference Stephens, Chee and Wall53,Reference Beals, Sukiennik and Nallabelli144,Reference Smeuninx, McKendry and Wilson145) . Obese rats display reduced activation of MPS, due to chronic lipotoxicity in skeletal muscle(Reference Masgrau, Mishellany-Dutour and Murakami146). Stephens et al.(Reference Stephens, Chee and Wall53) demonstrated that an acute lipid infusion in the presence of physiological hyperinsulinaemia impaired the MPS response to the ingestion of 20 g amino acids in young men, in part via the suppression of p-4EBP1 signalling, an effector downstream of mTORC1. Interestingly, reversal of these attenuated anabolic effects was observed with a bout of strenuous exercise(Reference Smiles, Churchward-Venne and van Loon147), suggesting that physical activity may offset lipid-induced AR in skeletal muscle. There is accumulating evidence that obesity, which represents another model of lipid overload, suppresses the stimulation of MPS rates following dietary protein ingestion(Reference Beals, Sukiennik and Nallabelli144,Reference Smeuninx, McKendry and Wilson145) , combined feeding and resistance exercise(Reference Beals, Skinner and McKenna148) in human subjects. A recent study was the first to investigate the impact of high fat feeding on MPS in human subjects(Reference Tsintzas, Jones and Pabla149). This study reported that, in overweight and obese middle-aged men, neither the acute consumption of a high fat meal nor 2 weeks of overfeeding via a high-SFA diet had a detrimental effect on the MPS response to dietary protein ingestion. Surprisingly, when a dose of protein was consumed 4 h after the acute ingestion of a high fat meal, more of the amino acids from the consumed protein were incorporated into skeletal muscle. Notably, in this study the high fat feeding interventions did not induce IR. This suggests that IR may be necessary to produce lipid overload-induced AR in human subjects. It will be interesting for future studies to investigate whether longer-term high fat feeding interventions that result in IR also lead to the development of AR(Reference Tsintzas, Jones and Pabla149).
Dietary interventions relevant to sarcopenic obesity
The reduced responsiveness of MPS to protein feeding in older adults specifically occurs with the ingestion of low-to-moderate protein doses (i.e. 20 g protein/meal or less)(Reference Moore, Churchward-Venne and Witard42,Reference Wall, Gorissen and Pennings150) . This can be overcome by the consumption of larger quantities of high quality protein (about 30–40 g/meal)(Reference Moore, Churchward-Venne and Witard42). This indicates that older adults likely require higher protein intakes than younger adults to preserve skeletal muscle mass and potentially other aspects of muscle health. As obesity appears to exacerbate age-related AR to protein ingestion(Reference Smeuninx, McKendry and Wilson145) and amino acid infusion(Reference Murton, Marimuthu and Mallinson151), a higher protein intake may be especially important in obese older adults to prevent the development and/or progression of sarcopenic obesity. In particular, during diet-induced weight loss, which typically results in the loss of both fat-free mass (composed of about 45 % skeletal muscle) and fat mass(Reference Weinheimer, Sands and Campbell152), higher protein intakes have been hypothesised to attenuate losses in muscle mass(Reference Phillips, Chevalier and Leidy153).
A number of studies have explored the role of higher protein intakes (≥1 g/kg/d) in preserving muscle mass and function during weight loss in obese older adults(Reference Backx, Tieland and Borgonjen-van den Berg154–Reference Layman, Evans and Baum157), although studies in individuals with sarcopenic obesity are lacking. The current literature indicates that the preservation of muscle mass is not possible with a dietary intervention alone(Reference Backx, Tieland and Borgonjen-van den Berg154,Reference Smith, Commean and Reeds158) . For example, Backx et al. compared the effect of normal protein (0⋅9 g/kg/d) and higher protein (1⋅7 g/kg/d) intakes during 12 weeks of diet-induced weight loss in overweight/obese older men and women. The normal protein and higher protein diet groups exhibited similar reductions in lean mass and leg strength, and equivalent improvements in 400 m walking speed, following the weight loss intervention(Reference Backx, Tieland and Borgonjen-van den Berg154). In another study, obese postmenopausal women consumed a normal protein (0⋅8 g/kg/d) weight-loss diet or the same diet in which part carbohydrate and fat content were replaced with isoenergetic whey protein supplements that provided an additional 0⋅4 g/kg/d. Although protein supplementation during the weight loss blunted the initial decline in thigh muscle volume after 5 % weight loss, after 10 % weight loss there was no statistically significant difference in muscle mass loss in the two groups(Reference Smith, Commean and Reeds158). Notably however, the beneficial effect of 10 % weight loss on skeletal muscle insulin sensitivity was eliminated in the women consuming the higher protein intake (1⋅2 g/kg/d), whereas the weight loss resulted in a 25 % improvement in muscle insulin sensitivity in women consuming the normal protein intake(Reference Smith, Yoshino and Kelly159). This implies that higher protein intakes during weight loss could blunt improvements in metabolic function. Nevertheless, in both the higher and normal protein groups, weight loss lowered fasting insulin concentration (without a change in plasma glucose concentration) and endogenous glucose production equivalently, indicating that both groups may have improved hepatic insulin sensitivity. Furthermore, the impact of the two levels of protein intake on plasma glucose homoeostasis remains unknown. Importantly, no exercise intervention was included during weight loss in this study. As the insulin sensitising(Reference Bird and Hawley160) and muscle-sparing(Reference Weinheimer, Sands and Campbell152) effects of exercise are well established, it will be interesting for future studies to examine whether concomitant exercise can ablate the lack of change in muscle insulin sensitivity associated with higher protein intake.
Studies that have included exercise (particularly resistance exercise training) as a co-intervention generally show a greater effect of higher protein intake on lean mass preservation(Reference Mojtahedi, Thorpe and Karampinos155,Reference Layman, Evans and Baum157,Reference Verreijen, Verlaan and Engberink161) . Resistance exercise sensitises the muscle protein synthetic machinery to protein feeding resulting in a greater proportion of the ingested amino acids being incorporated into newly synthesised skeletal muscle protein(Reference Pennings, Koopman and Beelen162). Moreover, resistance exercise attenuates the suppressive effects of energy restriction on the MPS response to protein intake(Reference Murphy, Churchward-Venne and Mitchell163). Verreijen et al.(Reference Verreijen, Verlaan and Engberink161) reported that the consumption of a high whey protein, leucine- and vitamin D-enriched supplement during 13 weeks of energy restriction (−2510 kJ/d) and resistance training (3 d/week) preserved appendicular skeletal muscle mass relative to an isoenergetic control supplement in obese older adults, although both groups improved strength and physical performance to a similar extent. Whey protein is a high quality protein that results in greater stimulation of MPS compared with other protein sources at rest and following resistance exercise in older adults(Reference Yang, Churchward-Venne and Burd164,Reference Pennings, Boirie and Senden165) . This effect of whey is likely attributed to the faster digestion and absorption and the high content of essential amino acids, particularly leucine, an essential amino acid that acts as a key trigger for MPS(Reference Wall, Hamer and de Lange166,Reference Atherton, Smith and Etheridge167) . As such, the muscle preserving effects of supplementation in the intervention group are likely due to a combination of the higher protein quantity and quality consumed during the exercise and weight loss intervention.
Dietary fat quantity and quality can impact sub-acute inflammation(Reference Roche168). In terms of dietary fat composition, we have demonstrated in mice that diets rich in SFA rather than MUFA induce both priming and nod-like receptor protein-3-mediated activation of IL-1β inflammation in equivalently obese adipose tissue(Reference Finucane, Lyons and Murphy169), coincident with alterations in insulin secretion, and greater risk of IR and T2D(Reference Gulseth, Gjelstad and Tiereny170–Reference Yubero-Serrano, Delgado-Lista and Tierney172). As mentioned earlier, IL-1β adversely affects skeletal MPS(Reference Broussard, McCusker and Novakofski121) and inhibits insulin-induced glucose uptake in skeletal myoblasts(Reference Cho and Kang173). Given the impact of dietary saturates on inflammation and the dysfunction to skeletal health caused by IL-1β, there is a putative negative effect of a diet rich in SFA on skeletal muscle metabolism due to IL-1β production. Given the reputed anti-inflammatory and/or pro-resolving attributes of long chain (LC) n-3 PUFA, several researchers have investigated the potential interaction between LC n-3 PUFA and protein intake on skeletal health. In vitro, the combination of protein and LC n-3 PUFA increased MPS and decreased MPB(Reference Kamolrat and Gray174,Reference Jeromson, Mackenzie and Doherty175) . In vivo, Smith and colleagues(Reference Smith, Atherton and Reeds176,Reference Smith, Atherton and Reeds177) demonstrated that LC n-3 PUFA supplementation for 8 weeks increased the mixed MPS response to insulin and amino acid infusion in both healthy younger adults(Reference Smith, Atherton and Reeds177) and older adults(Reference Smith, Atherton and Reeds176), which was at least partly mediated via the mTOR-P70S6K1 signalling pathway. Importantly, another study demonstrated that LC n-3 PUFA supplementation, for 6 months, resulted in clinically relevant gains in muscle mass and strength in older adults(Reference Smith, Julliand and Reeds178). LC n-3 PUFA supplementation has also been reported to enhance some of the strength and functional adaptions to resistance exercise training in older women(Reference Lalia, Dasari and Robinson179,Reference Da Boit, Sibson and Sivasubramaniam180) . However, not all studies have reported a favourable effect of LC n-3 PUFA supplementation on muscle anabolism(Reference Da Boit, Sibson and Sivasubramaniam180,Reference McGlory, Wardle and Macnaughton181) . For example, McGlory et al.(Reference McGlory, Wardle and Macnaughton181) observed no effect of LC n-3 PUFA supplementation for 8 weeks on myofibrillar protein synthesis in response to the consumption of 30 g protein either alone or following a bout of resistance exercise in young men. It is possible that the beneficial effect of LC n-3 PUFA supplementation on muscle anabolism is more pronounced in older individuals experiencing low grade inflammation and AR compared to younger individuals. Further research is required to explore the role of LC n-3 PUFA supplementation, in conjunction with amino acids, leucine and/or protein supplementation, in the prevention and treatment of sarcopenic obesity.
Personalised responses v. non-response in sarcopenic obesity
In human dietary interventions there is often considerable inter-individual variability in responses. In the obesity, IR and T2D field, this paradigm is relatively well accepted, wherein typically up to 40 % of subjects respond to an intervention(Reference Yubero-Serrano, Delgado-Lista and Tierney172,Reference Tierney, McMonagle and Shaw182) . This variability in response may be due to different baseline phenotypes such as hepatic v. skeletal IR(Reference van der Kolk, Kalafati and Adriaens183), wherein different interventions may be appropriate(Reference Trouwborst, Bowser and Goossens184). We and other groups have looked at the impact of different gene–nutrient interactions with respect to identifying the susceptibility to the development of various disease traits(Reference Murphy, Smith and Murphy171,Reference Ortega-Azorin, Coltell and Asensio185,Reference Gomez-Delgado, Alcala-Diaz and Leon-Acuna186) . There is some evidence in relation to the varied responsiveness of the adipokine adiponectin to SFA reduction according to different polymorphisms(Reference Ferguson, Phillips and Tierney187). Other inflammatory genotypes/phenotypes may account for the mixed responsiveness between studies determining the impact of SFA and/or LC n-3 PUFA(Reference Phillips, Kesse-Guyot and Ahluwalia188). This biological phenomenon goes beyond non-compliance. Indeed, we have previously identified that despite equivalent compliance between adolescents, that not everyone responds to an anti-inflammatory nutritional intervention(Reference McMorrow, Connaughton and Magalhaes189), and that response may be determined by baseline metabolic phenotype(Reference Yubero-Serrano, Delgado-Lista and Tierney172). These studies highlight the potential for personalised nutrition approaches to improve outcomes. In the context of sarcopenia or sarcopenic obesity, to date no studies have characterised the inter-individual variability in responses to protein and/or other nutrient(s) supplementation interventions. It will be interesting to understand if the paradigm of response heterogeneity, which seems to be present in obesity and IR(Reference Murphy, Smith and Murphy171,Reference McMorrow, Connaughton and Magalhaes189) , is also present in sarcopenic obesity.
Perspectives for the future
Sarcopenia, obesity and their combined state, sarcopenic obesity, are major health concerns with the ever-growing ageing population. Each of these co-morbidities consequently have an impact on one another with their mechanisms being intertwined and intricate. Deciphering the cross talk between the adipose tissue and the skeletal muscle will enable us to truly identify the processes behind the onset of sarcopenic obesity and will help detect ways to counteract the condition. Treating the condition of sarcopenic obesity is complex as diet-induced weight loss, aimed at reducing fat mass, can accelerate muscle mass loss(Reference Backx, Tieland and Borgonjen-van den Berg154,Reference Porter Starr, Pieper and Orenduff190) . The incorporation of exercise, ideally including both resistance and aerobic exercise training, during weight loss is a potent strategy to minimise weight loss-induced muscle mass in older adults(Reference Colleluori, Aguirre and Phadnis191). In addition, dietary strategies such as higher protein intake may further enhance muscle mass retention. Further work is required to examine combined exercise and nutrition interventions, specifically in individuals with sarcopenic obesity. Importantly, establishing a clinical definition for sarcopenic obesity will enable appropriate participant selection and comparison between studies.
Financial Support
This work was supported by the Department of Agriculture, Food and the Marine Food Institutional Research Measure Grants entitled NUTRIMAL ‘Novel Nutritional Solutions for the Prevention of Malnutrition’ (Grant number 14F822) and IMMUNOMET ‘Dietary manipulation of microbiota diversity for controlling immune function’ (Grant number 14F828); and the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 666010.
Conflict of Interest
None.
Authorship
The authors had joint responsibility for all aspects of preparation of this manuscript.


