


The incidence of inflammatory bowel diseases such as ulcerative colitis and Crohn's disease has risen in the last few years(Reference Rubio and Befrits1). An increment in cyclo-oxygenase-2 (COX-2)-derived PG synthesis has been described during acute and chronic intestinal inflammation, in particular, PGE2 synthesis. In addition, other PGE2 enzyme synthases have been described including the inducible microsomal PGE synthase-1 (mPGES-1), which has been functionally linked to COX-2 and is also overexpressed in some cancers(Reference Rask, Zhu and Wang2, Reference Wang, Hsueh and Lin3).
External stimuli such as inflammatory cytokines or UV irradiation induce the activation of NF-κB transcription factor by phosphorylation of IκB that is degraded by the proteosome allowing NF-κB translocation to the nucleus(Reference Surh, Chun and Cha4). Among the genes regulated by NF-κB are mPGES-1 and COX-2. In fact, 5′-promoter region of COX-2 contains two putative NF-κB-binding sites(Reference Catley, Chivers and Cambridge5). Other factors implicated in the inflammatory response are mitogen-activated protein kinases (MAPK). There are three major classes of MAPK, the extracellular signal-regulated kinase (ERK) and the two stress-activated protein kinase families c-jun N-terminal kinase (JNK) and p38. MAPK regulate NF-κB activation by multiple mechanisms, and they are also implicated in COX-2 and mPGES-1 regulation(Reference Surh, Chun and Cha4, Reference Masuko-Hongo, Berenbaum and Humbert6).
The increasing incidence of inflammatory bowel disease has been associated with changes in dietary pattern(Reference Russel, Engels and Muris7). Although a clear relationship between fruit intake and inflammatory bowel disease has not been established yet, some fruits such as pomegranate have been shown to interact with different pathways involved in inflammation such as NF-κB activation and eicosanoid synthesis pathways and to influence cytokines and matrix metalloproteinase expression(Reference Lansky and Newman8). Some of the properties attributed to pomegranate are probably due to its polyphenol content(Reference Larrosa, Tomás-Barberán and Espín9, Reference Shukla, Gupta and Rasheed10). Recently, the anti-inflammatory effects of a pomegranate extract and one of its major components, ellagic acid (EA), have been reported(Reference Shukla, Gupta and Rasheed11, Reference Papoutsi, Kassi and Chinou12). However, the compounds involved in these effects as well as the main mechanisms of action are not well understood.
Pomegranate ellagitannins (ET) are not absorbed but hydrolysed to produce EA, which is extensively metabolised by the gut microbiota to yield the hydroxy-dibenzopyranone derivatives urolithins (Uro, Fig. 1)(Reference Cerdá, Cerón and Tomás-Barberán13–Reference Cerdá, Periago and Espín15). Uro are bioavailable, and they can reach a high concentration at μm level as glucuronide derivatives in the bloodstream. Nevertheless, a large proportion of Uro reach distal parts of the colon in the aglycone form(Reference González-Sarrías, Azorín-Ortuño and Yáñez-Gascón16). Despite polyphenols being metabolised in vivo, literature dealing with polyphenol-derived metabolites is scarce.
Fig. 1 Structures of the colonic microbiota ellagic acid-derived metabolites urolithin-A and urolithin-B.
We previously studied the anti-inflammatory properties of a pomegranate extract and its main microbiota-derived metabolite Uro-A in a DSS colitis rat model(Reference Larrosa, González-Sarrías and Yañéz-Gascón17). Overall, the present results suggest that Uro-A could play a critical role in the anti-inflammatory properties attributed to pomegranate consumption. These effects included the prevention of colon tissue damage and the decrease of inflammation markers (iNOS, COX-2, mPGES-1 and PGE2 in colonic mucosa). In addition, the G1 to S cell cycle pathway was up-regulated in both groups using microarray analyses. However, a favourable modulation of gut microbiota was also observed (increase of bifidobacteria and lactobacilli and decrease of enterobacteria). Therefore, the microbiota modulation could also be responsible for the observed effects. In addition, the individual effect of EA, Uro-A and Uro-B was not assessed.
In order to shed some light on the above, our main aim was to investigate whether Uro-A could act as an anti-inflammatory compound per se. To this purpose, Uro-A, Uro-B and EA were assayed in a colonic subepithelial fibroblast cell line to study their capacity to inhibit PGE2 production in response to inflammatory stimuli as well as to unravel the potential of the involved mechanisms of action.
Materials and methods
Reagents
EA, resveratrol (Res), quercetin and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide were purchased from Sigma-Aldrich (Steinheim, Germany). Uro-A (3,8-dihydroxy-6H-dibenzo(b,d)pyran-6-one) and Uro-B (3-hydroxy-6H-dibenzo(b,d)pyran-6-one) were chemically synthesised by Kylolab S.A. (Murcia, Spain). IL-1β was purchased from Calbiochem (Milan, Italy). The specific inhibitors PD98059, SB203580, SP600125 and ammonium pyrrolidinedithiocarbamate (PDTC) were also obtained from Sigma-Aldrich.
Cell culture and treatments
The human normal colon fibroblast cell line CCD18-Co was obtained from the American Type Culture Collection (ATCC number CLR-1459; Rockville, IN, USA), and was grown in minimal essential medium supplemented with 10 % v/v fetal bovine serum, 2 mm glutamine, 0·1 mm non-essential amino acids, 1 mm-sodium pyruvate, 0·1 U/ml penicillin and 100 μg/ml streptomycin. Cells were maintained at 37°C under a 5 % CO2 and 95 % air atmosphere at constant humidity. Stocks of EA, Uro-A, Uro-B, quercetin and Res were dissolved in DMSO, and were added to the culture media at the specified concentration. The final concentration of DMSO did not exceed 0·5 %. CCD18-Co cells were seeded and allowed to reach confluence, and subsequently, the medium was replaced with a fresh medium containing 0·1 % fetal bovine serum 24 h after the inflammatory stimulus. Cells were then treated with EA and Uro-A and Uro-B (1 and 10 μm) and were co-stimulated with 1 ng/ml of IL-1β for different times specified below. CCD18-Co cells were used from population doubling level 28 to 35 for all the analyses.
Cell viability
Cells were seeded in 96-well plates and treated as described above. After 18 h of co-incubation with 10 μm of EA, Uro-A or Uro-B and 1 ng/ml of IL-1β, cells were washed three times with PBS, and the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay was performed as described by Morgan(Reference Morgan18).
PGE2 production assay
PGE2 production was assayed as described by Larrosa et al. (Reference Larrosa, Luceri and Vivoli19). Briefly, after serum starvation, cells were treated with EA, Uro-A, Uro-B (1–10 μm) and with Res and quercetin (1–10 μm) as anti-inflammatory polyphenol standards, and were co-stimulated with 1 ng/ml of IL-1β for 18 h. Afterwards, media were recovered and PGE2 levels were measured using an immunoenzymatic method (EIA kit, Cayman Chemical, San Diego, CA, USA) according to the manufacturer's specifications. A dilution of 1:50 was used.
Cyclo-oxygenase inhibitor screening assay
Inhibitory activity of EA, Uro-A and Uro-B on COX-1 (bovine) and COX-2 (human recombinant) enzymes was assayed using the colorimetric commercial kit (COX Inhibitor Screening Assay, Cayman Chemical) according to the manufacturer's protocol. Briefly, 150 μl of assay buffer, 10 μl of haem and 10 μl of enzyme (either COX-1 or COX-2) were carefully mixed with 10 μl of EA (1 and 10 μm), Uro-A or Uro-B (10 and 40 μm), and incubated at 25°C for 5 min. Then, 20 μl of arachidonic acid (substrate solution) were added and incubated at 25°C for 5 min. Afterwards, absorbance was measured at 590 nm using a Fluostar galaxy microplate reader (BMG Labtech, Offenburg, Germany). Res (30 μm) was used as a selective COX1/2 inhibitor positive control(Reference Jang, Cai and Udeani20, Reference Subbaramaiah, Chung and Michaluart21). The COX inhibitor screening assay directly measures PGF2α produced by SnCl2 reduction of COX-derived PGH2. The prostanoid product is quantified via enzyme immunoassay using a broadly specific antibody that binds to all the major PG compounds. All samples were expressed as percentage inhibition of initial activity. The assay was repeated twice.
RNA extraction
Total RNA was isolated using an RNeasy mini kit (Qiagen, Hilden, Germany). RNA was eluted in RNAse-free water and stored at − 80°C. RNA concentration and purity were checked using the Nanodrop spectrophotometer system (ND-1000 3·3 Nanodrop Technologies, Wilmington, DE, USA). Only samples with a ratio between 1·8 and 2·1 were used for quantitative PCR experiments.
Quantitative real-time PCR
Changes in the expression of COX-2 and mPGES-1 genes were assessed by one-step quantitative PCR (Taqman system, Applied Biosystems, ABI, Madrid, Spain). Amplification was performed using a total reaction volume of 25 μl in a MicroAmp Optical 96-well plate covered by optical adhesive covers, and using TaqMan Universal Master Mix. Primers and probes were selected from Assays-on-demand (ABI): PG-endoperoxide synthase-2 (COX-2), Hs00153133_m1; PGE synthase-1, Hs00610420_m1 (_m indicates an assay whose probe spans an exon junction and will not detect genomic DNA). The one-step quantitative PCR were run on the ABI 7500 system following the manufacturer's thermal conditions. The expression levels of target genes were normalised to the levels of glyceraldehyde-3-phosphate dehydrogenase (Hs99999905_m1) using a standard curve method for quantification. All assays were done in triplicate.
Western blot analysis
Cells were homogenised in cold RIPA buffer with protease inhibitor cocktail (Roche, Mannheim, Germany). Protein concentration was measured using Bradford's reagent, and 40 μg of protein/lane was loaded. Antibodies that recognising phosphorylated and total p38 (phosphor-Tyr-182), JNK (phosphor-Thr-183/Tyr-185) and ERK-1/2 (phosphor-Thr-185/Tyr-187) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). COX-2 and mPGES-1 antibodies were purchased from Cayman Chemical (Ann Arbor, MI, USA). Glyceraldehyde-3-phosphate dehydrogenase antibody (Affinity BioReagents™, Golden, CO, USA) is routinely assayed for monitoring total protein load. Proteins were separated by 10–15 % SDS-PAGE, and were transferred to polyvinylidene difluoride membranes (GE Healthcare, Chalfont St Giles, Bucks, UK) by electroblotting. Proteins were detected using ECL plus detection system (GE Healthcare) according to the manufacturer's instructions. Density of the bands was detected using the ImageQuant TL v2005 (GE Healthcare). The Western blot assays were done in triplicate for each treatment.
NF-κB nuclear translocation assay
NF-κB activation was determined with the ELISA TransAM™ NF-κB p65 kit (Active Motif, Rixensart, Belgium), which specifically measures the amount of NF-κB p65 bound to its consensus binding site (5′GGGACTTTCC3′). Nuclear extracts were prepared according to the procedure of Oh & Lee(Reference Oh and Lee22). Protein concentration in the nuclear extracts was determined using Bradford's method. Jurkat nuclear extract (2·5 μg) was used as a positive control.
Cell-based extracellular signal-regulated kinases 1/2, c-Jun N-terminal kinase and p38 mitogen-activated protein kinase phosphorylation ELISA
Changes in the relative amount of ERK1/2, JNK and p38 phosphorylation state in treated CCD18-Co cells were determined using a RayBio Cell-Based ERK1/2, JNK and p38 MAPK ELISA kit (RayBiotech, Inc., Norcross, GA, USA) following the manufacturer's protocol. Briefly, after 24 h of serum starvation, cells were treated with EA, Uro-A or Uro-B (10 μm), and were co-stimulated with 1 ng/ml of IL-1β for 30 min, 2 and 4 h, with or without pre-treatment with the specific inhibitors. The ERK-1/2 inhibitor PD98059 (25 μm), the JNK inhibitor SP600125 (10 μm) and the selective p38 MAPK inhibitor SB203580 (25 μm) were added 1 h before the application of the inflammatory stimulus. Media were discarded at different time points, and the cells were washed and fixed for 20 min at room temperature followed by 1 h blocking step. Anti-phospho-ERK1/2, phospho-JNK or phospho-p38 antibodies or anti-ERK1/2, JNK or p38 antibodies were added to the corresponding wells and incubated for 2 h at room temperature. The cells were then washed and incubated with HRP-conjugated anti-mouse IgG for 1 h. After washing, the substrate 3,3′,5,5′-tetramethylbenzidine was added and incubated for 30 min at room temperature. Absorbance was measured at 450 nm using a Fluostar galaxy microplate reader (BMG Labtech).
Cellular uptake
CCD18-Co cells were seeded at 44 × 103 cm2 density in 9 cm diameter plates with EMEM without phenol red and with the supplements specified above. The presence of metabolites excreted into the medium as well as those located inside the cells was explored. Cells were treated with Uro-A, Uro-B and EA (10 μm), and were co-stimulated or not with 1 ng/ml of IL-1β in order to check whether inflammation conditions could affect cell metabolism. After 18 h, the medium was recovered and processed as described by Larrosa et al. (Reference Larrosa, González-Sarrías and García-Conesa23). Aliquots of 5 μl of media and cell extract samples were analysed by liquid chromatography–MS/MS.
Liquid chromatography–MS/MS analyses
The HPLC–DAD system (1200 series; Agilent Technologies, Waldbronn, Germany) was equipped with an HTC Ultra mass detector in series (Bruker Daltonics, Bremen, Germany). The mass detector was an ion trap mass spectrometer equipped with an electrospray ionisation (capillary voltage, 4 kV; nebuliser 15 psi; dry gas 5 l/min; dry temperature, 350°C) system. Mass scan and MS/MS daughter spectra were measured from m/z 100 to 600 using the Ultra scan mode (26 000 m/z per second). Collision-induced fragmentation experiments were performed in the ion trap using He as the collision gas, and the collision energy was set at 50 %. MS data were acquired in the negative ionisation mode. Chromatographic separations of samples were carried out on a 150 × 0·5 mm internal diameter, 5 μm, reverse phase SB C18 Zorbax column (Agilent) using water–formic acid (99:1, v/v) (A) and acetonitrile (B) as the mobile phases at a flow rate of 10 μl/min. The gradient started with 1 % B in A to reach 60 % B at 30 min, 90 % B at 30 min for 5 min and returned to the initial conditions (1 % B). UV chromatograms were recorded at 280, 305 and 360 nm.
Identification and quantification of Uro-derived and ellagic acid-derived metabolites, Uro and EA were performed as described by Cerdá et al. (Reference Cerdá, Tomás-Barberán and Espín24).
Statistical analysis
All analyses were carried out using the SPSS™ 14.0 software (SPSS Inc, Chicago, IL, USA). All data are expressed as the mean values and standard deviations of independent measurements. Statistical significance was determined by Student's t test using P < 0·05 as the level of significance.
Results
PGE2 production
To test whether EA, Uro-A and Uro-B could modulate PGE2 synthesis, the production of PGE2 by CCD18-Co cells was measured upon stimulation with 1 ng/ml of IL-1β for 18 h. IL-1β produced a significant increase (30-fold) in PGE2 levels (P < 0·05; Fig. 2). Co-treatment with Uro-A (1 and 10 μm) significantly (P < 0·05) decreased PGE2 levels in a dose-dependent manner (2·6-fold and 8-fold, respectively). Uro-B significantly lowered PGE2 production by 1·5-fold at 10 μm, while EA had no effect at any concentration tested (Fig. 2). Res (10 μm) and quercetin (10 μm), used as positive controls, also significantly inhibited PGE2 secretion approximately to the same extent as Uro-A (10 μm) inhibited it. None of the compounds tested affected the cells growth or caused cytotoxicity measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide method (data not shown).
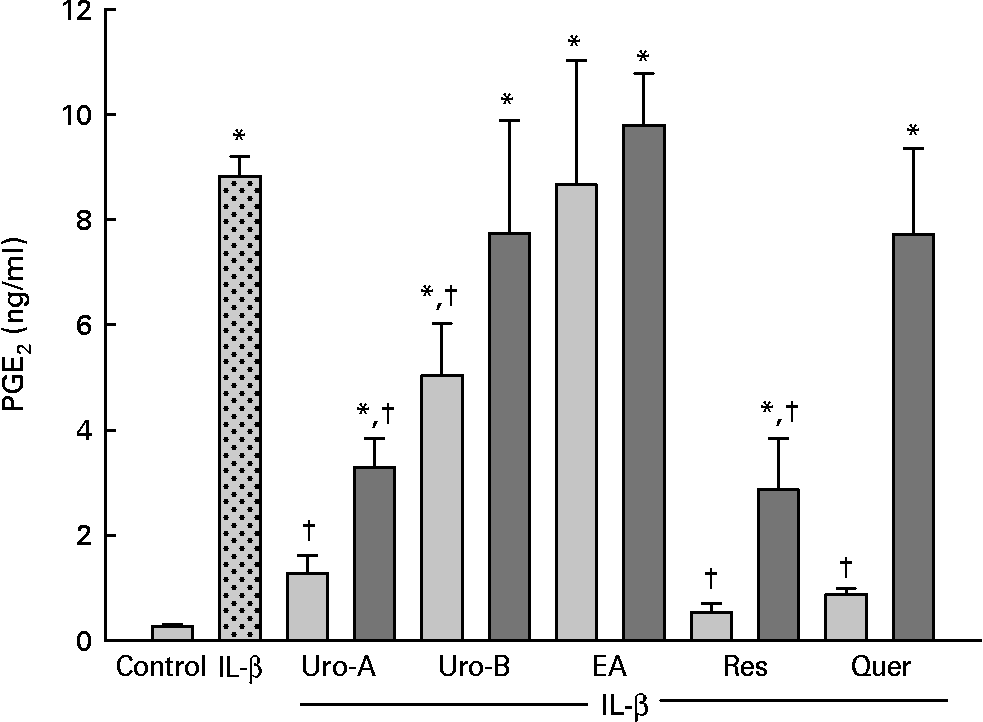
Fig. 2 PGE2 levels in CCD18-Co cells treated with Urolithin (Uro)-A, Uro-B, ellagic acid (EA), resveratrol (Res) and quercetin (Quer; 1–10 μm) and co-stimulated with IL-1β (1 ng/ml) for 18 h. Each bar represents the mean values and standard deviations (n 3). * Mean values were significantly different to the control (P < 0·05). † Mean values were significantly different over IL-1β treatment (P < 0·05). ![]() , 10 μm;
, 10 μm; ![]() , 1 μm.
, 1 μm.
Cyclo-oxygenase-1 and cyclo-oxygenase-2 inhibition assay
Direct inhibition of COX-1 and COX-2 activities by EA (1 and 10 μm), and Uro-A and Uro-B (10 and 40 μm) was assessed with a commercially available COX inhibitory screening assay using purified COX-1 (ovine) and COX-2 (human recombinant) enzymes. Neither EA nor Uro-A nor Uro-B inhibited COX-1 or COX-2- activity at any concentration tested, while Res (30 μm) that was used as a positive control showed significant (P < 0·01) inhibitory effects, 96·44 (sd 1·90) on COX-1 and 57·82 (sd 8·48) COX-2 enzyme activities (results expressed as % inhibition (sd)).
Cyclo-oxygenase-2 and microsomal PGE synthase-1 protein and mRNA levels
As PGE2 levels in an inflammation state are determined by two inducible enzymes, COX-2 and mPGES-1, their expression was analysed by Western blot analysis and quantitative PCR in CCD18-Co cells stimulated with IL-1β and treated simultaneously with EA, Uro-A and Uro-B.
Firstly, changes in protein levels of both COX-2 and mPGES-1 were assessed. COX-2 and mPGES-1 protein expressions were undetectable in control cells, and were noticeably induced (6- and 5-fold, respectively) in cells treated with IL-1β for 18 h (Fig. 3). Uro-A (10 μm) significantly (P < 0·05) lowered COX-2 (3-fold) and mPGES-1 (2-fold) proteins. Neither COX-2 nor mPGES-1 protein levels were changed upon exposure of cells to EA (10 μm) or Uro-B (10 μm).

Fig. 3 Western blot analysis of cyclo-oxygenase-2 (COX-2 (a)) and microsomal PGE synthase-1 (mPGES-1 (b)). Protein was detected in CCD18-Co cells stimulated with 1 ng/ml of IL-1β and treated with 10 μm of Urolithin (Uro)-A, Uro-B or ellagic acid (EA) for 18 h. * Mean values were significantly different in protein levels to the control (P < 0·05). † Mean values were significantly different in protein levels over IL-β treatment (P < 0·05). Results are expressed as mean values and standard deviations (n 3). The Western blot image illustrates the expression change, and the densitometric values are the mean of three independent experiments. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To validate the present results at the mRNA level, COX-2 and mPGES-1 mRNA time-induced expressions by 1 ng/ml IL-1β treatment were analysed. The mRNA levels of COX-2 and mPGES-1 in 1 ng/ml IL-1β-treated cells reached a maximum of 200-fold at 4 h and 20-fold at 8 h of incubation, respectively (data not shown), with the increment being less marked at 18 h (50-fold for COX-2 and 6-fold for mPGES-1; Fig. 4). Accordingly, in subsequent experiments, COX-2 and of mPGES-1 mRNA levels were measured at 4 and 18 h of treatment. Treatment with Uro-A (10 μm) significantly (P < 0·05) decreased mRNA levels of COX-2 at 4 and 18 h (Fig. 4(a)) and mPGES-1 at 4 h but not at 18 h (Fig. 4(b)). Cells treated with Uro-B and EA exhibited mRNA levels of COX-2 and mPGES-1 that were similar to those exhibited by the cells treated with IL-1β (Fig. 4).
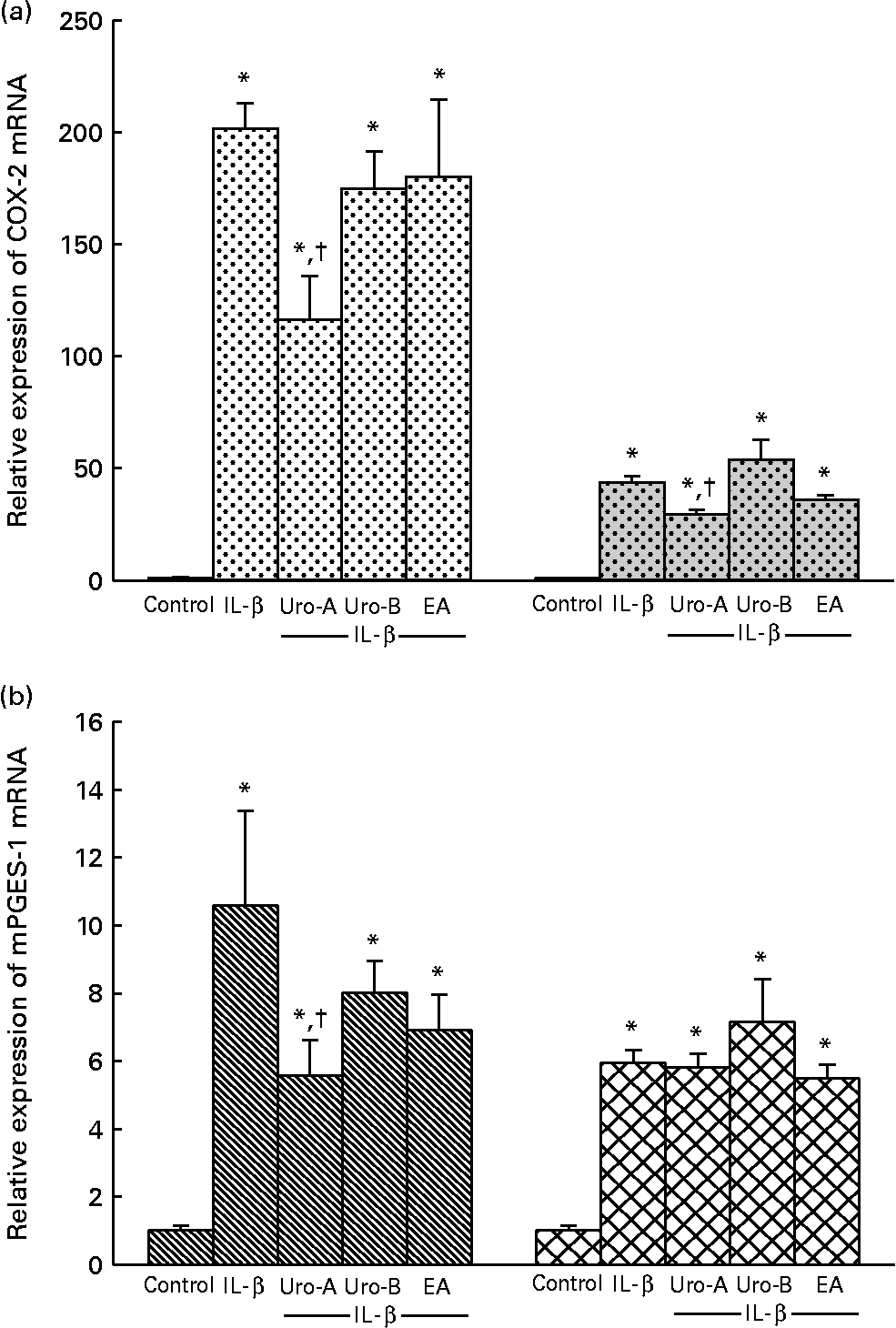
Fig. 4 Quantitative PCR analysis of cyclo-oxygenase-2 (COX-2 (a)) and microsomal PGE synthase-1 (mPGES-1 (b)) expressions in CCD18-Co cells. Data were studied following normalisation to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Results are expressed as mean values and standard deviations (n 6). * Mean values were significantly different to the control (P < 0·05). † Mean values were significantly different over IL-1β treatment (P < 0·05). Uro, urolithin; EA, ellagic acid. (a) ![]() , COX-2 4 h;
, COX-2 4 h; ![]() , COX-2 18 h. (b)
, COX-2 18 h. (b) ![]() , mPGES-1 4 h;
, mPGES-1 4 h; ![]() , mPGES-1 18 h.
, mPGES-1 18 h.
NF-κB p65 DNA-binding assay
In a previous screening, treatment with 1 ng/ml of IL-1β at 30 min, 2 and 4 h resulted in the nuclear translocation of p65 NF-κB and binding to its DNA consensus sequence at 2 and 4 h but not at 30 min (data not shown). Subsequent experiments were done at 2 and 4 h (Fig. 5). Treatments with Uro-A (10 μm) and Uro-B (10 μm) significantly inhibited (P < 0·05) p65-binding activity at both times. EA did not produce any effect on NF-κB (Fig. 5).
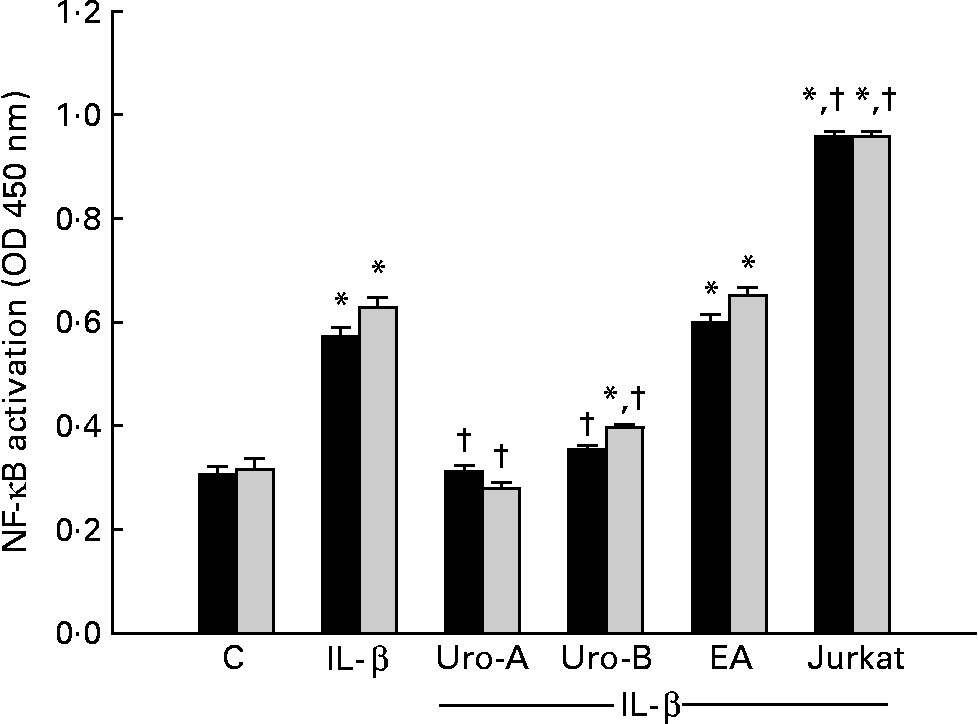
Fig. 5 Effect of ellagic acid (EA), urolithin (Uro)-A and Uro-B on the activation of NF-κB p65. * Mean values were significantly different to the control (P < 0·05). † Mean values were significantly different over IL-1β treatment (P < 0·05). OD, optical density; C, control. ■, 2 h; ![]() , 4 h.
, 4 h.
Involvement of NF-κB in cyclo-oxygenase-2 and microsomal PGE synthase-1 expressions
Cells were treated for 1 h with the NF-κB inhibitor PDTC (100 μm) before IL-1β stimulation. COX-2 and mPGES-1 expressions were checked after 18 h by Western blot analysis. Treatment with PDTC significantly decreased both mPGES-1 and COX-2 expressions (Fig. 6), confirming the participation of NF-κB in the regulation of COX-2 and mPGES-1 expressions.
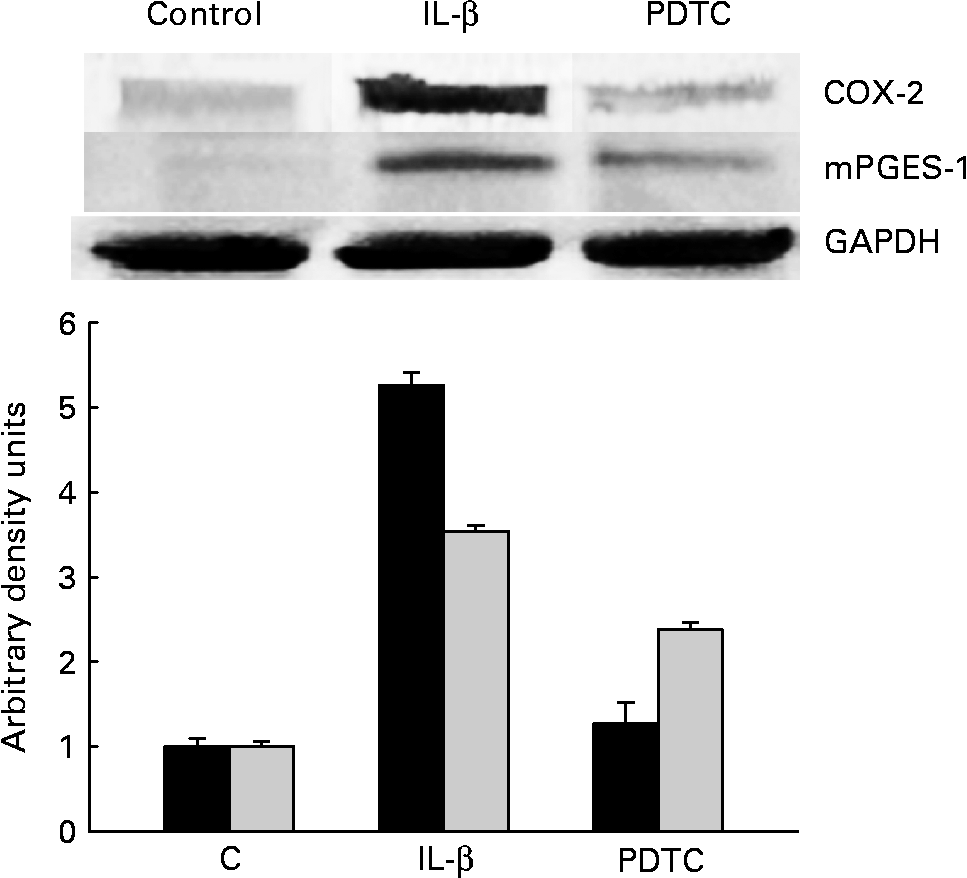
Fig. 6 Inhibition of NF-κB activation prevents cyclo-oxygenase-2 (COX-2) and microsomal PGE synthase-1 (mPGES-1) expression in CCD18-Co cells stimulated with IL-1β. Pyrrolidinedithiocarbamate (PDTC), NF-κB inhibitor. The Western blot image illustrates the expression change, and the densitometric values are the mean of three independent experiments. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. ■, COX-2; ![]() , mPGES-1.
, mPGES-1.
Phosphorylation status of extracellular signal-regulated kinase, c-Jun N-terminal kinase and p38 signalling in CCD18-Co cells
Since MAPK pathways are involved in IL-1β-induced inflammation, the potential influence of MAPK signalling pathways was studied upon Uro-A, Uro-B and EA treatment. Both total and phosphorylated forms of ERK1/2, JNK and p38 in cells stimulated with IL-1β were measured at 30 min, 2 and 4 h using cell-based ELISA. Stimulation of cells with 1 ng/ml of IL-1β produced a significant increase in the phosphorylation status (activation) of the ERK1/2, JNK, and p38 kinases at 30 min (Fig. 7). However, this effect was not maintained at 2 and 4 h for the ERK1/2 and JNK and only at 2 h for p38 (results not shown). Therefore, the influence of EA, Uro-A and Uro-B on these kinases was tested at 30 min. When cells were stimulated with IL-1β and co-treated with Uro-A (10 μm), the phosphorylation state of JNK and p38 kinases was slightly but significantly lowered (P < 0·05), while no effect was observed on the ERK1/2 pathway. Uro-B (10 μm) significantly attenuated p38 activation, whereas no effect (P < 0·05) was observed on JNK pathway (Fig. 7). EA had no effect on ERK1/2 and p38 kinases and it significantly increased the activation of JNK. Pre-incubation for 1 h with the specific inhibitors PD98059 (25 μm), SB203580 (25 μm) and SP600125 (10 μm) as positive controls significantly reduced (P < 0·05) the activation of ERK1/2, p38 and JNK, respectively (Fig. 7). The results obtained with the ELISA kits were further confirmed by Western blot analysis (Fig. 8). Therefore, the slight but statistically significant changes in the phosphorylation state of MAPK indicated their involvement in the activation of the transcription factor NF-κB.
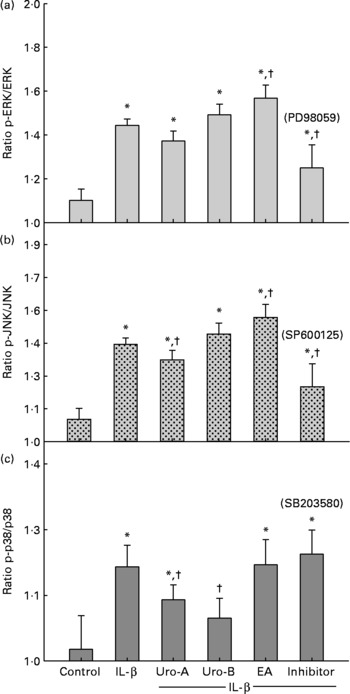
Fig. 7 Effect of ellagic acid (EA), Urolithin (Uro)-A and Uro-B on mitogen-activated protein kinase activation. Extracellular signal-regulated kinase (ERK1/2 (a)), c-Jun N-terminal kinase (JNK (b)) and p38 (c). * Mean values were significantly different to the control (P < 0·05). † Mean values were significantly different over IL-1β treatment (P < 0·05). All experiments were carried out in triplicate. Plotted values represent mean values and standard deviations.
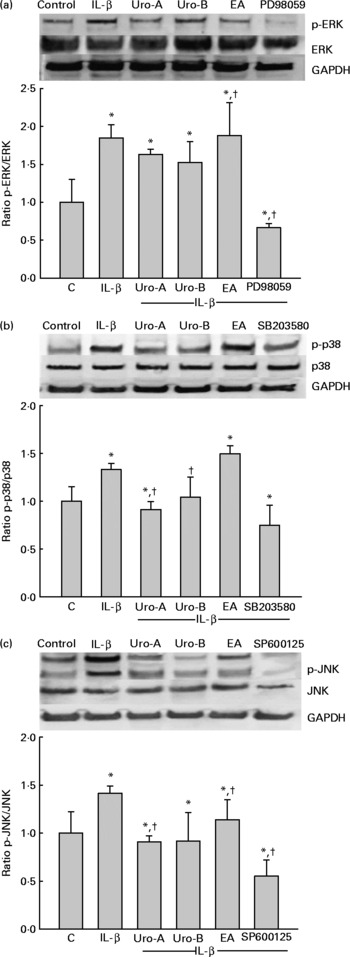
Fig. 8 Western blot analysis of the effect of ellagic acid (EA), Urolithin (Uro)-A and Uro-B on mitogen-activated protein kinase activation. Extracellular signal-regulated kinase (ERK1/2 (a)), c-Jun N-terminal kinase (JNK (b)) and p38 (c). * Mean values were significantly different to the control (P < 0·05). † Mean values were significantly different over IL-1β treatment (P < 0·05). Results are expressed as mean values and standard deviations (n 3). The Western blot image illustrates the expression change, and the densitometric values are the mean of three independent experiments.
Cellular uptake
Cell media and cell extract samples were analysed by LC–MS/MS to explore cell metabolism of EA, Uro-A and Uro-B under normal and inflammation conditions. No metabolites derived from EA, Uro-A or Uro-B were detected in cell media or in cell extracts in both normal and inflammation conditions, and incubation times were assayed. Trace amounts of Uro-A (6·4 pm), Uro-B (3·33 pm) and EA (0·68 pm) were found in the cell extracts.
Discussion
Antioxidant, anti-inflammatory and anticancer properties have been attributed to pomegranate and other ellagitannin-containing foods. Most studies that demonstrated pomegranate properties have been conducted with juices or extracts that contain simple sugars, amino acids, fibre, sterols and a large amount of polyphenols including anthocyanins, flavones, flavonols and mainly ET(Reference Lansky and Newman8). It has been suggested that polyphenols may be partly responsible for the beneficial activities attributed to pomegranate(Reference Faria, Monteiro and Mateus25, Reference Adams, Seeram and Aggarwal26). However, these in vitro studies did not take into account the bioavailability of pomegranate polyphenols. Pomegranate ET are not absorbed as such, but they are hydrolysed to EA and further metabolised by colon microbiota to yield Uro(Reference Cerdá, Cerón and Tomás-Barberán13–Reference Cerdá, Periago and Espín15, Reference Cerdá, Tomás-Barberán and Espín24, Reference Cerdá, Soto and Albaladejo27). Uro are bioavailable, and can reach significant concentrations as glucuronide conjugates in plasma and urine, whereas high Uro aglycone concentrations (above 100 μm) can reach the colon(Reference Espín, González-Barrio and Cerdá28, Reference González-Sarrías, Azorín-Ortuño and Yáñez-Gascón16). Previous results in our research group suggested an in vivo anti-inflammatory effect of the pomegranate-derived metabolite Uro-A(Reference Larrosa, González-Sarrías and Yañéz-Gascón17). However, other potential anti-inflammatory actions mediated by gut microbiota could also be responsible for the effects observed. In this context, the individual role of pomegranate metabolites in colon and their mechanism of action were not deeply studied.
Intestinal fibroblasts play a pivotal role by influencing epithelial function, and amplifying or reducing the inflammatory response by secreting mediators such as PGE2 and metalloproteinases(Reference Mahida, Beltinger and Makh29, Reference Mifflin, Saada and Di Mari30). The present results show that treatment of colonic fibroblast with IL-1β increases PGE2 levels, and that this response is partially attenuated by Uro-A and Uro-B but not by EA. Therefore, we confirmed that Uro-A was able to inhibit PGE2 synthesis, which was in accordance with our previous findings in DSS-treated rats upon oral administration with Uro-A(Reference Larrosa, González-Sarrías and Yañéz-Gascón17).
We next assessed the effect of Uro-A, Uro-B and EA on the activity of COX-1 and COX-2 enzymes to check whether a direct inhibition of these enzymes was the reason for the attenuated PGE2 production. None of the compounds showed inhibitory enzyme activity. There is no literature about the inhibition of COX-1 or COX-2 activity by Uro. Regarding EA, the present results are in agreement with the lack of inhibitory properties of EA for COX-2 activity or PGE2 production(Reference Zhang, DeWitt and Murugesan31, Reference Sadhu, Okuyama and Fujimoto32). This suggests that others instead of EA are responsible for pomegranate COX-2 inhibitory activity, since a recent report has demonstrated that metabolites that become bioavailable in plasma after pomegranate fruit extract ingestion are able to inhibit COX-2 activity(Reference Shukla, Gupta and Rasheed11). PGE2 production in inflammation status is determined by COX-2 and mPGES-1 enzymes. A number of studies have reported that cells overexpressing both COX-2 and mPGES-1 enzymes produce more PGE2, which suggests that mPGES-1 inhibition could improve the adverse effects of PGE2 production(Reference Chell, Kaidi and Williams33). Thus, we further studied protein and mRNA expression of these enzymes. Uro-A reduced COX-2 and mPGES-1 expressions at both mRNA and protein levels. However, the doses assayed for Uro-B and EA had no effect on either COX-2 or mPGES-1 expression. Inhibition of COX-2 expression by pomegranate extract and juice in vitro and after topical application has been reported previously(Reference Adams, Seeram and Aggarwal26, Reference Afaq, Malik and Syed34). Probably, in these studies, ET are the responsible fraction for the observed in inhibition COX-2 expression as the assay conditions were not physiological (i.e. absence of metabolism, gut microbiota, etc.). The results presented here regarding the down-regulation of COX-2 and mPGES-1 expressions match with our previous results in inflamed colonic mucosa of rats fed with pomegranate extract or Uro-A(Reference Larrosa, González-Sarrías and Yañéz-Gascón17).
Stimulation of cells with the inflammatory cytokine IL-1β led to an activation of the pathways of NF-κB transcription factor and MAPK that mediate COX-2 induction in many cell types(Reference Newton, Kuitert and Bergmann35–Reference Yang, Chien and Hsiao38). The treatment with IL-1β activated NF-κB and all the three classes of MAPK (ERK, JNK and p38 MAPK). The present results show that Uro-A and Uro-B inhibited NF-κB activation by preventing the union of the p65 subunit to its consensus binding site, suggesting that inhibition of COX-2 and mPGES-1 expression and therefore PGE2 could be mediated by NF-κB inhibition. A slight inhibition of p38 MAPK activation was detected upon incubation with both Uro-A and Uro-B. In the case of JNK, Uro-A displayed a slight inhibition, whereas no effect was observed with Uro-B treatment. In addition, neither Uro-A nor Uro-B lowered the phosphorylation status of ERK1/2. These results are in contrast with previous findings showing a large decrease in the activated form p-ERK1/2 after incubation with a mixture of Uro and EA in Caco-2 cells for 72 h(Reference González-Sarrías, Espín and Tomás-Barberán39). However, it should be highlighted that the divergence in results could be due to the cell line assayed (cancer epithelial Caco-2 v. normal fibroblastic CCD18-Co), incubation time (72 v. 18 h) and different doses assayed (mixture of 40 μm of Uro-A and Uro-B and 10 μm EA v. 10 μm of each individual compound in the present study) as well as due to the lack of inflammatory stimuli in the study of González-Sarrías et al. (Reference González-Sarrías, Espín and Tomás-Barberán39). We cannot rule out that the effect observed in the previous study in Caco-2 cells was due to the 40 μm Uro-A treatment.
On the contrary, EA did not attenuate NF-κB or MAPK activation in accordance with the lack of anti-inflammatory activity. These data are in agreement with the study done by Masamune et al. (Reference Masamune, Satoh and Kikuta40), who reported the lack of inhibition of NF-κB translocation to nucleus in IL-1β-treated stellate cells upon incubation with EA. However, the inhibition of MAPK activation was observed in that study(Reference Masamune, Satoh and Kikuta40). These contradictory data regarding MAPK activation could be due to the different MAPK activation patterns depending on the cell type(Reference Surh, Chun and Cha4). A slight increase in ERK phosphorylation was detected in cells treated with EA over cells treated with IL-1β (Fig. 7(a)). This activation could be due to PGE2 accumulation, which can activate the ERK pathway in several cell lines(Reference Krysan, Reckamp and Dalwadi41) and whose production was not inhibited by EA (Fig. 2).
The present results show that the anti-inflammatory effects of Uro-A and Uro-B are mediated by the inhibition of NF-κB transcription factor, despite the effects that were observed on the inhibition of MAPK activation being slight. For this reason, the results were confirmed by two experimental techniques (ELISA and Western blot analysis). However, the role of NF-κB and MAPK in the induction of COX-2 expression and PGE2 production after IL-1β treatment is not yet fully understood. For instance, IL-1β stimulation of cardiac myocytes or smooth tracheal cells led to an increased PGE2 production and COX-2 expression that was mediated by MAPK (p38 and ERK) and NF-κB activation but through independent pathways(Reference LaPointe and Isenović37, Reference Yang, Chien and Hsiao38). However, induction of COX-2 expression required the activation of both JNK and p38 MAPK pathways in renal mesangial cells(Reference Guan, Buckman and Miller36). For these reasons, further research is needed to elucidate the mechanisms implicated in the anti-inflammatory effects of these compounds using other cell models and inflammatory stimuli. In addition, other promoters involved in the transcriptional activation of COX-2 such as C/EBP or CRE/ATF(Reference Sirois and Richards42, Reference Inoue, Yokoyama and Hara43), the transcription factor Egr-1 that contributes to mPGES-1 expression(Reference Subbaramaiah, Yoshimatsu and Scherl44), the cytosolic phospholipase A2 or the protein kinase A that has also been described in the IL-1β-mediated PGE2 synthesis(Reference Pascual, Carr and Seeds45), should be studied.
The present results also suggest a receptor-mediated anti-inflammatory effect as no metabolites were found in cell media or cell extracts upon incubation Uro-A or Uro-B using this cell line. Substantial uptake and cell metabolism have been reported recently in Caco-2 cells upon incubation with either Uro-A or Uro-B(Reference González-Sarrías, Azorín-Ortuño and Yáñez-Gascón16). It is of note that fibroblasts are metabolically less active than epithelial cells. In addition, ‘normal’ over ‘cancerous’ phenotype could also have a great influence(Reference Meadows, Kong and Berdichevsky46). In the present study, a trace amount of Uro-A was detected in the cell extracts (6·4 pm), although this could be due to contamination of cell media. Whether the anti-inflammatory effect was due to a competitive binding of Uro with IL-1β receptor or to the very low Uro concentration detected inside the cells remains unanswered.
In summary, Uro-A decreased PGE2 production by down-regulating COX-2 and mPGES-1 expressions, preventing NF-κB activation and decreasing the activated forms of p38 MAPK and JNK. Uro-B was able to inhibit the activation of NF-κB and p38 MAPK. However, attenuation of PGE2 levels was lower than in the case of Uro-A, and no effect on COX-2 and mPGES-1 at the assayed doses was observed. No anti-inflammatory effects were observed for EA under these assay conditions.
Further research into the possible synergy between both Uro and the assay of other inflammation models could help to discern whether the consumption of ET-containing foods (as Uro precursors) could improve our health status by preventing the incidence of inflammation-related diseases, especially those affecting the colon where Uro can reach high concentrations.
Acknowledgements
The present work was supported by the Projects CICYT-BFU2007-60576 and Consolider Ingenio 2010, CSD2007-00063 (Fun-C-Food). A. G.-S. is the holder of a FPI predoctoral grant from MICINN (Spain). M. L. is the holder of a postdoctoral grant from MICINN. A. G.-S. carried out the laboratory experiments: cell culture, quantitative PCR, ELISA and western blot analysis. M. L. carried out the PGE2 measurements, and contributed to the design of the experiments, analysis of data and drafting of the manuscript. F. A. T.-B. and P. D. contributed to drafting and revising of the manuscript critically. J. C. E. contributed to the conception and design of the experiments, interpretation of data, drafting of the manuscript, revising and final approval. The authors declare that there is no conflict of interest.








